一种幽门螺旋杆菌O2血清型O抗原寡糖类化合物的合成的制作方法
本发明涉及一种幽门螺旋杆菌o2血清型o抗原寡糖类化合物的合成,属于有机合成领域。
背景技术:
幽门螺旋杆菌是一种微需氧型的革兰氏阴性菌,全球约有一半人口感染了这种致病菌,因为较低的社会经济地位和较差的医疗卫生条件,在发展中国家中这一比例更是高达80%,据报道在我国城市和农村分别有47%和66%的人口感染了幽门螺旋杆菌。口-口传播和粪-口传播是该致病菌最主要的传播方式,母婴传播也是该致病菌的传播方式之一。
大部分的幽门螺旋菌的感染发生儿童时期,一旦感染,治病菌可能在胃粘膜上存在一生。尽管70%的被感染者在临床上是无症状的,但是这种致病菌已经被证实与一系列胃肠道疾病有关,15%-20%的长期感染者将会引发胃炎,胃和十二指肠溃疡等一系列胃肠道疾病,另外还有1%-2%的长期感染者将发展成胃癌。胃癌已成为全球发病率第五高(~952000例,占癌症病人的6.2%,2012年who数据)和死亡率第三高的癌症(~723000例,占癌症病人的8.8%,2012年who数据)1994年who下属的国际癌症研究机构(iacr)将幽门螺旋杆菌分类为i类致癌物。
目前针对幽门螺旋杆菌感染相关的胃肠道疾病的主要治疗方法是一种利用两种抗生素(甲硝咪唑和克拉霉素)和一种质子泵抑制剂的联合治疗方法,称之为三联疗法。这种方法在治疗幽门螺旋杆菌上取得了一定的效果,但是仍有不少不足之处,主要限制表现在:1)抗生素在胃肠道中的代谢时间比较短,2)抗生素在胃这种特殊ph低环境下相对不稳定,3)幽门螺杆菌一般寄居在胃黏膜处,而抗生素浓度在黏膜浓度很低,4)更重要的是随着耐药性细菌的出现抗生素的治疗效果有逐渐减弱的趋势。基于以上缺陷,有报道使用三联疗法治疗后的幽门螺旋杆菌的一年复发率已达15%-30%。
因此,为了克服这一缺陷,研究针对这一致病菌的疫苗相关研究越来越受到重视。近年来,针对幽门螺旋杆菌疫苗的研究表现在的毒力因素抗原上,如尿酶b,细胞毒素相关毒素(caga),空泡毒素(vaca),鞭毛,热休克蛋白,黏附素,中性粒细胞活性蛋白等。另外,针对灭活全菌细胞和菌溶产物的研究也是疫苗的热点之一。
与其他革兰氏阴性菌一样,幽门螺旋杆菌的细胞表面主要是有脂多糖组成,脂多糖对该细菌的生物膜结构和功能的完整性具有相当重要的作用,也存在于细菌的分泌物中,是该细菌的致病因素和毒力因素之一。
monteiro系统地研究了各类幽门螺旋杆菌的脂多糖结构发现其结构与典型的革兰氏阴性菌一致,包含三个不同的结构单元,分别是多聚糖o糖链,核心寡糖链和富含脂肪酸的脂质a。在幽门螺旋杆菌中,核心寡糖链结构相对保守而多聚糖o糖链表现出特异性的表型。根据多聚糖o糖链中具体单糖种类和连接方式的不同可以将幽门螺旋杆菌分类6种不同的血清型(o1-o6)。2015年monteiro报道了幽门螺旋杆菌的o2血清型o聚糖链的具体结构,该血清型的o-抗原是由葡萄糖残基依次通过α-(1→2)-和α-(1→3)-糖苷键依次连接而成的重复单元结构,最长可达12个重复单元的24糖链。
目前针对幽门螺旋杆菌脂多糖的研究主要是通过从灭活细菌中提取,这一方法的不足在于一次提取得到的产物极少,另外受细菌基因表达和修饰的特性,提取得到的脂多糖也存在结构不够专一,并且容易附带结构类似杂质的特点,实验重复性差,对研究存在一定的干扰。我们利用化学合成方法,以幽门螺旋杆菌o2血清型o抗原寡糖为目标化合物,通过糖基砌块的设计和构建以及糖基化反应的选择和优化得到o抗原二糖,四糖和六糖,并且所合成的二糖,四糖和六糖还原端均组装有氨基连接臂,可以与载体蛋白连接成糖缀合物,开展进一步的免疫学研究。
技术实现要素:
本发明所要解决的技术问题是通过化学方法合成幽门螺旋杆菌o2血清型o抗原寡糖片段。α-1,2顺式糖苷键的构建是合成目标结构的关键步骤。本发明利用二丙酮葡萄糖为唯一起始原料通过糖基砌块的构建和糖基化反应条件的优化最终得到保护的全α连接的保护二,四,六糖,最终脱去保护得到最后的目标化合物组装有氨基连接臂的幽门螺旋杆菌o2血清型o抗原二糖,四糖和六糖。同时,用该方法合成得到的二糖,四糖和六糖还原端均组装有氨基连接臂,可以和载体蛋白结合用于免疫学研究。
其中m为二糖片段的重复单元数,可以为2~12;
r为-(ch2)n-n-y1y2或者-(ch2)n-n-y1y2(linker),n=1~10,n为氮y1为氢(h)或者烷氧基如苄基(bn),y2为氢(h)或者烷氧羰基如苄甲氧羰基(cbz),r1,r2,r3,r4,r5,r6为氢或者醚类基团如苄基(bn)。
本发明的第一个目的是提供一种幽门螺旋杆菌o2血清型o抗原寡糖类化合物的制备方法,所述方法利用糖砌块a、b、c构建幽门螺旋杆菌o2血清型o抗原寡糖类化合物,包括构建糖砌块之间的1,2-α-顺式-糖苷键和1,3-α-顺式-糖苷键;
其中糖砌块a、b、c分别为结构式ii、iii所示的化合物:
其中,lg为糖基化反应离去基团,包括三氯乙酰亚胺基、对甲基苯硫基;linker包括-(ch2)n-n-y1y2或者-(ch2)n-n-y1y2(linker),y1为氢(h)或者烷氧基如苄基(bn),y2为氢(h)或者烷氧羰基如苄甲氧羰基(cbz);
r1,r2,r3,r4,r5,r6为氢或者醚类基团;r7为氢或者酰基类基团。
在本发明的一种实施方式中,所述方法中1,3-α-顺式-糖苷键的构建是在二氯甲烷和乙醚的混合溶剂中进行的。
在本发明的一种实施方式中,所述混合溶剂中二氯甲烷与乙醚的体积比为1:(2.5-5)。
在本发明的一种实施方式中,所述方法中1,3-α-顺式-糖苷键的构建还需要引入添加剂。
在本发明的一种实施方式中,所述添加剂包括噻吩。
在本发明的一种实施方式中,所述添加剂与糖切块的摩尔比为(80-120):1。
在本发明的一种实施方式中,所述方法中1,2-α-顺式-糖苷键的构建是在二氯甲烷溶剂中进行的。
在本发明的一种实施方式中,1,3-葡萄糖α糖苷键的构建方法优选:糖基供体和糖基受体在甲苯中共沸,加入二氯甲烷和乙醚,反应底物的浓度为0.01~0.1m,以分子筛作为干燥剂,加入相当于糖基供体当量100倍的噻吩,在室温下搅拌30min后冷却至0℃,反应时间为12h,用吡啶终止反应。
在本发明的一种实施方式中,所述方法包括以下步骤:
(1)合成葡萄糖单糖砌块a、b、c;
(2)合成二糖化合物:构建糖砌之间的1,2-α-顺式-糖苷键;
(3)合成四糖化合物:构建步骤(2)所得二糖化合物之间的1,3-α-顺式-糖苷键。
在本发明的一种实施方式中,所述方法还包括:参照步骤(3),构建多糖化合物与多糖化合物之间的1,3-α-顺式-糖苷键,制备得到幽门螺旋杆菌o2血清型o抗原寡糖类化合物;所述多糖化合物为式iv所示的化合物;
其中,m为二糖片段的重复单元数,为2~12;
linker包括-(ch2)n-n-y1y2或者-(ch2)n-n-y1y2(linker),y1为氢(h)或者烷氧基如苄基(bn),y2为氢(h)或者烷氧羰基如苄甲氧羰基(cbz);
r1,r2,r3,r4,r5,r6为氢或者醚类基团;r7为氢或者酰基类基团。
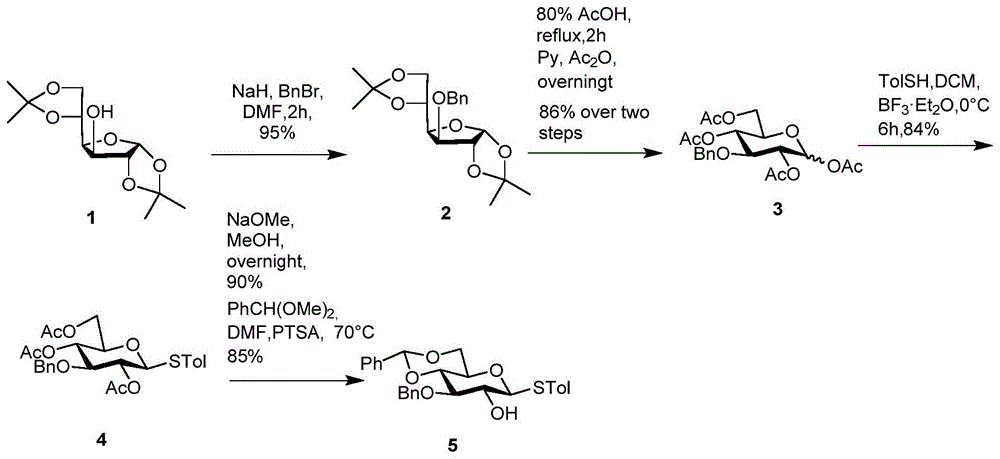
在本发明的一种实施方式中,所述步骤(1)中单糖砌块a的合成方法包括:以二丙酮葡萄糖为起始原料,在氢化钠(nah)和溴化苄(bnbr)的条件下生成3-苄基-二丙酮葡萄糖,随后用酸水解掉两个丙叉环;得到的3-苄基化合物用乙酰基保护裸露的4个羟基,接着在对甲基苯硫酚和路易斯酸(bf3·e2to)的作用下给端基位用苯硫基保护;下一步在碱性环境下水解掉剩余的2,4,6号位上的乙酰基,随后用苄叉基同时保护住4,6号位羟基,空出2-oh得到糖砌块a。
在本发明的一种实施方式中,所述步骤(1)中单糖砌块a的合成方法具体包括:以市售的二丙酮葡萄糖为起始原料,在氢化钠(nah)和烯丙基溴(allbr)的作用下在3号位用烯丙基保护住,随后用烯丙醇(alloh)和盐酸将3-烯丙基化合物的两个丙叉基脱去,同时在一号位也用烯丙基保护。烯丙基葡萄糖随用在氢化钠(nah)和溴化苄(bnbr)作用下将葡萄糖剩余的三个羟基用苄基保护。下一步用氯化钯(pdcl2)和甲醇作用下将葡萄糖1,3号位的两个烯丙基脱除,接着在吡啶和醋酸酐的条件下用乙酰基保护住1.3号位。最后用醋酸肼选择性的脱除葡萄糖1号位的乙酰基,得到的1-oh化合物用三氯乙腈和dbu做成糖基供体,即为糖砌块a。
在本发明的一种实施方式中,单糖砌块a的合成是在有机溶剂中进行的;所述有机溶剂包括二氯甲烷、乙酸乙酯、甲醇,n,n-二甲基甲酰胺、四氢呋喃,吡啶、氯仿中的一种或者几种。
在本发明的一种实施方式中,单糖砌块a的合成反应中,底物浓度为0.02~0.5m。
在本发明的一种实施方式中,酸为盐酸,乙酸或者对甲苯磺酸。
在本发明的一种实施方式中,反应温度为-78℃~溶剂回流温度,反应时间为1-48h。
糖砌块a的结构式如式ii所示,其端基位为苯硫基,是一种离去基团。
在本发明的一种实施方式中,所述步骤(1)中单糖砌块b的合成方法包括:将上述的单糖砌块a的2-oh用乙酰基保护,得到的2-oac化合物与连接臂反应,得到端基位有连接臂的葡萄糖化合物,最后在甲醇和甲醇钠中脱去2-oac得到葡萄糖砌块b。
在本发明的一种实施方式中,所述步骤(2)中的二糖化合物包括如式v所示的二糖供体d和二糖受体e:
其中,linker包括-(ch2)n-n-y1y2或者-(ch2)n-n-y1y2(linker),y1为氢(h)或者烷氧基如苄基(bn),y2为氢(h)或者烷氧羰基如苄甲氧羰基(cbz);
r1,r2,r3,r4,r5,r6为氢或者醚类基团;r7为氢或者酰基类基团。
在本发明的一种实施方式中,所述步骤(2)包括:以糖砌块a和c作为原料,以二氯甲烷为溶剂,加入干燥剂,在-78℃下进行,制备得到二糖供体d;
利用糖砌块b和c合成二糖受体e,糖苷化反应方法和合成二糖供体d一致,糖苷化反应完成,产物纯化后用甲醇钠溶液脱去3’-oac得到二糖受体e。
所述的干燥剂为
所述的干燥剂与反应物的质量比为1.0~4.0。
在本发明的一种实施方式中,所述步骤(2)中反应用tlc板监测反应进程反应结束后用吡啶终止反应,产物用硅胶柱纯化。
在本发明的一种实施方式中,所述步骤(3)包括:以二糖供体d和二糖受体e合成四糖化合物。,
在本发明的一种实施方式中,所述四糖化合物包括如式vi所示的四糖供体f和四糖受体g,
在本发明的一种实施方式中,所述步骤(3)包括:在dcm、et2o混合溶剂中,加入添加剂噻吩,使用
四糖e在甲醇钠溶液中脱去3”’-oac得到四糖g。
在本发明的一种实施方式中,制备得四糖f时还可以加入促进剂。
在本发明的一种实施方式中,促进剂包括n-碘代丁二酰亚胺nis、三氟甲基磺酸三甲基硅酯tmsotf、三氟甲磺酸tfoh、三氟甲烷磺酸银agotf中的一种或者几种。
在本发明的一种实施方式中,混合溶剂中的dcm、et2o的体积比优选1:3。
在本发明的一种实施方式中,参照四塘的构建方法,还可以进一步合成八糖,十糖,十二糖等一系列幽门螺旋杆菌o2-血清型寡糖衍生物。
在本发明的一种实施方式中,寡糖链可以以结合二糖的模式由还原端向非还原端延伸,包括两个步骤:1)还原端带有连接臂的寡糖脱去非还原端的脂类基团;2)与二糖供体16的糖基化反应。
在本发明的一种实施方式中,利用二糖和四糖构建1,3-α-顺式-糖苷键合成六糖化合物;所述六糖化合物包括式vii所示的化合物;
在本发明的一种实施方式中,二糖化合物的合成条件:以单糖砌块a和b(lg/linker)为底物,反应溶剂为二氯甲烷,使用干燥剂,反应温度为-78℃,在路易斯酸的催化下进行糖基化反应。反应过程都为惰性气体保护,反应原料消失后,用吡啶或者三乙胺淬灭反应,过滤,洗涤,萃取,干燥,产物用硅胶柱纯化分别得到二糖i/ii。
在本发明的一种实施方式中,四糖的组装方法:以二糖化合物i为供体,二糖化合物ii为受体组装四糖,反应条件为以二糖化合物i和ii为底物,反应溶剂为二氯甲烷和乙醚的混合溶剂,同时添加噻吩。使用干燥剂,反应温度为室温。反应在路易斯酸的催化下进行。反应过程都为惰性气体保护,反应原料消失后,过滤,洗涤,萃取,干燥,产物用硅胶柱纯化分别得到四糖。
在本发明的一种实施方式中,六糖组装的条件:四糖化合物在碱性条件下脱去非还原酯基,然后和二糖化合物i作为底物组装六糖化合物。反应条件与组装四糖化合物时相同。
在本发明的一种实施方式中,所述方法还包括脱保护。
在本发明的一种实施方式中,所述脱保护是利用pd/c,在氢气下反应24h,脱除所有的芳香基团,经c18反相柱纯化得到幽门螺旋杆菌o2血清型o抗原寡糖类化合物。
在本发明的一种实施方式中,所述脱保护具体包括:1)
1)在碱性条件下将酰基脱除,所用溶剂为甲醇,四氢呋喃,乙酸乙酯中的一种或者几种组成的混合溶剂;反应结束用h+树脂中和,反应温度为室温~40℃,反应时间为2h~2d得到半脱保护的糖类化合物;
2)利用钯/碳、氢气对半脱保护产物进行全脱保护,反应溶剂为四氢呋喃,二氯甲烷,乙酸乙酯,叔丁醇,水,甲醇,醋酸中的一种或者多种,所用钯碳为10%的钯碳,钯碳与反应物的质量比为0.1:1~0.5:1,脱苄基和苄叉基所用的氢气压力为1~100atm,所述的反应温度为常温,反应时间为2~48h。
本发明的第二个目的是提供一种组装有氨基连接臂的幽门螺旋杆菌o2血清型o抗原寡糖类化合物,其结构式如式vii所示,所述化合物是利用上述方法制备得到的;
在本发明的一种实施方式中,一种组装有氨基连接臂的幽门螺旋杆菌o2血清型的o-抗原寡糖的结构为:
α-d-glc-(1-2)-α-d-glc-(1-3)-α-d-glc-(1-2)-α-d-glc-(1-3)-α-d-glc-(1-2)-α-d-glc-linker。
本发明的第三个目的是提供一种糖-蛋白缀合物的制备方法,所述方法是利用上述的组装有氨基连接臂的幽门螺旋杆菌o2血清型o抗原寡糖类化合物。
本发明的第四个目的是将上述的组装有氨基连接臂的幽门螺旋杆菌o2血清型o抗原寡糖类化合物应用于在开发或制备疗幽门螺旋杆菌疫苗或者疗幽门螺旋杆菌感染导致的疾病的药物中的应用。
本发明有益效果:
本发明是通过化学合成得到幽门螺旋杆菌o2血清型o-抗原二糖,四糖和六糖。本发明通过保护剂策略,温度效应,溶剂效应和添加剂效应,发现了一种非常有利于葡萄糖α糖苷键生成的方法,并且将此方法应用在幽门螺旋杆菌o2血清型o-抗原二糖,四糖和六糖的合成之中。合成得到的幽门螺旋杆菌o2血清型o-抗原二糖,四糖和六糖的还原端均组装有氨基连接臂,可以与载体蛋白制成糖缀合物,用于免疫学研究,对发展预防和治疗幽门螺旋杆菌具有重要作用。
附图说明
图1:单糖砌块5的合成;
图2:单糖砌块8的合成;
图3:单糖砌块15的合成;
图4:二糖化合物16和18的合成;
图5:四糖化合物19的合成;
图6;六糖化合物21的合成;
图7:二糖、四糖和六糖的脱保护。
具体实施方式
下面将结合实施例对本发明的实施方案进行详细描述,但是本领域技术人员将会理解,下列实施例仅用于说明本发明,而不应视为限定本发明的范围。实施例中未注明具体条件者,按照常规条件或制造商建议的条件进行。所用试剂或仪器未注明生产厂商者,均为可以通过市购获得的常规产品。
实施例1
糖砌块5的合成如图1:
如图1所示,以市售双丙酮葡萄糖1为起始原料,在氰化钠和作用下与溴化苄反应生成3-obn葡萄糖化合物2。丙叉基在80%醋酸溶液中脱除后,在吡啶和醋酸酐的的作用下用乙酰基保护4个新生成的羟基得到化合物3。随后化合物3在三氟化硼·乙醚(bf3·et2o)的的作用下与苯硫酚反应,生成1-硫苷化合物4。用甲醇甲醇钠溶液脱去化合物4的三个乙酰基,接着在苯甲醛二甲缩醛和对甲基苯环酸的作用下用苄叉基保护4,6号位羟基,留出2号位裸露羟基得到糖砌块5。
具体试验操作和步骤。
化合物2:将二丙酮葡萄糖(50g,192mmol)溶于dmf(480ml)中,在0℃下搅拌20min,缓缓加入氰化钠(15.4g,284mmol),继续在0℃下搅拌30min,接着向反应液中加入溴化苄(34.2ml,288mmol),反应恢复到室温,继续搅拌3h。用tlc检测反应液待原料反应完全后,用冰水淬灭反应,用二氯甲烷和饱和食盐水萃取,分离有机相,经无水硫酸钠干燥后浓缩,用硅胶柱纯化(石油醚/乙酸乙酯,15:1)得到化合物2(67g,183mmol,95%)。1hnmr(400mhz,chloroform-d)δ7.55–7.04(m,5h,ar-h),5.90(d,j=3.7hz,1h,h-1),4.68(d,j=11.8hz,1h,bn-h),4.63(d,j=11.8hz,1h,bn-h),4.58(d,j=3.7hz,1h,h-2),4.37(dt,j=7.7,6.1hz,1h,h-5),4.15(dd,j=7.8,3.2hz,1h,h-6a),4.11(dd,j=9.1,6.7hz,1h,h-6b),4.01-3.99(m,2h,h-3,h-4),1.49(s,3h,me-h),1.43(s,3h,me-h),1.37(s,3h,me-h),1.31(s,3h,me-h)。13cnmr(101mhz,chloroform-d)δ137.65,128.40(2c),127.84,127.65(2c),111.79,108.98,105.30,82.67,81.72,81.33,72.54,72.39,67.40,26.85,26.79,26.26,25.45。
化合物3:将化合物2(67g,183mmol)溶解在1m盐酸溶液(480ml)中,80℃下搅拌2h,用tlc检测反应,待原料反应完全后,用饱和碳酸氢钠溶液调节反应液至中性后,用旋蒸旋干溶剂,得到的产物用甲苯共沸三次,在油泵上抽真空过夜。
将上一步得到的新化合物溶于醋酸酐(360ml),同时加入无水乙酸钠(18.9g),反应液在80℃下搅拌4h,用tlc检测原料反应完全之后,将反应液倒入冰水中,接着用乙酸乙酯和饱和碳酸氢钠萃取,分离有机相,用无水硫酸钠干燥之后浓缩。浓缩液用硅胶柱纯化(石油醚/乙酸乙酯,4:1)得到化合物3(69g,157mmol,86%)。-构型:1hnmr(400mhz,chloroform-d)δ7.36–7.19(m,5h,ar-h),6.31(d,j=3.6hz,1h,1-h),5.16(t,j=9.8hz,1h,4-h),5.05(dd,j=10.0,3.7hz,1h,2-h),4.71(d,j=11.8hz,1h,bn-h),4.63(d,j=11.8hz,1h,bn-h),4.24–4.16(m,1h,6a-h),4.09–3.92(m,3h,3-h,5-h,6b-h),2.16(s,3h,me-h),2.07(s,3h,me-h),1.99(s,3h,me-h),1.97(s,3h,me-h)。13cnmr(101mhz,chloroform-d)δ170.58(d,j=2.1hz),169.56,169.25,168.73,137.94,128.39(2c),127.78,127.48(2c),89.41,76.97,74.81,71.54,70.24,69.14,61.82,20.79(2c),20.63,20.50。-构型1hnmr(400mhz,chloroform-d)δ7.33–7.13(m,5h,ar-h),5.63(d,j=8.2hz,1h,1-h),5.12(td,j=9.2,4.2hz,2h,2-h,4-h),4.59(s,2h,2bn-h),4.19(dd,j=12.5,5.0hz,1h,6a-h),4.05(dd,j=12.4,2.3hz,1h,6b-h),3.76–3.66(m,2h,3-h,5-h),2.05(s,3h,me),2.02(s,3h,me),1.94(s,6h,2me)。13cnmr(101mhz,chloroform-d)δ170.55,169.26,169.08,169.02,137.59,128.43,127.89,127.76,91.92,79.90,74.19,72.89,71.54,69.09,61.78,20.75,20.62.
化合物4:将化合物3(42g,96mmol)溶于无水dcm(240ml)中,加入对甲基苯硫酚(17.9g,144mmol)和适量新鲜活化的分子筛,室温下搅拌30min,随后降温至0℃。继续搅拌20min后缓缓加入三氟化硼乙醚(60ml)。反应液在室温下搅拌24h,用tlc监测反应进程,待原料完全反应之后,用二氯甲烷和饱和碳酸氢钠溶液萃取反应液。分离有机相,经无水硫酸钠干燥后浓缩,浓缩液使用硅胶柱纯化(石油醚/乙酸乙酯,4:1)得到化合物4。(40.5g,81mmol)。1hnmr(400mhz,chloroform-d)δ7.45–6.91(m,9h,ar-h),5.05(t,j=9.7hz,1h,4-h),5.02(t,j=9.5hz,1h,2-h),4.64–4.51(m,3h,1-h,2bn-h),4.16(t,j=3.4hz,2h,6ab-h),3.71(t,j=9.2hz,1h,3-h),3.60(ddd,j=9.9,5.0,3.1hz,1h,5-h),2.33(s,1h,me-h),2.07(s,3h,me-h),2.04(s,3h,me-h),1.95(s,3h,me-h)。13cnmr(101mhz,chloroform-d)δ170.59,169.32,169.17,138.38,137.73,133.26,129.60,128.44,127.85,127.76,86.41,81.59,77.46,77.14,76.82,76.06,74.26,71.40(d,j=3.1hz),69.71,62.56,21.14,20.96,20.75,20.72。
化合物5:化合物4(27g,54mmol)溶于甲醇(150ml),室温下加入5m甲醇钠(22ml),反应液在室温下继续搅拌2h。用tlc监测反应,待原料彻底反应完全之后用amberliteir120h+树脂中和反应液至中性。滤去树脂,旋干滤液得到新化合物(~20g),油泵上抽真空过夜。
将新化合物(~20g,53mmol)溶于无水dmf,依次加入苯甲醛二甲缩醛(8.3ml)和对甲基苯磺酸(1.0g)。反应液在70℃下搅拌6h,用tlc监测反应,待原料反应完全之后用三乙胺中和反应液。反应液浓缩后用二氯甲烷和饱和食盐水萃取,分离有机相,用无水硫酸钠干燥后浓缩,浓缩液经硅胶柱纯化得到化合物5(19.1g,41mmol)。1hnmr(400mhz,chloroform-d)δ7.68–7.21(m,13h,ar-h),7.13(d,j=7.9hz,2h,ar-h),5.56(s,1h,phch-h),4.94(d,j=11.5hz,1h,bn-h),4.78(d,j=11.5hz,1h,bn-h),4.56(d,j=9.7hz,1h,1-h),4.38(dd,j=10.5,4.9hz,1h,6a-h),3.78(t,j=10.3hz,1h,6b-h),3.72–3.65(m,1h,3-h),3.63(t,j=9.0hz,1h,4-h),3.47(td,j=9.3,9.0,5.8hz,2h,2-h,5-h),2.56–2.49(m,1h,2oh-h),2.34(s,3h,me)。13cnmr(101mhz,chloroform-d)δ138.75,138.20,137.20,133.83,129.81,129.00,128.45,128.25,128.11,127.86,127.20,125.99,101.24,88.58,81.61,81.13,74.81,72.13,70.73,68.64,21.17。
实施例2
糖砌块8的合成
糖砌块8的合成如图2
如图2所示,化合物8是化合物5还原端组装有氨基连接臂的类似物。从化合物5出发,先用乙酰基保护2-oh得到化合物6,接着以对甲基苯硫基作为离去基团,在促进剂nis和tmsotf下与五碳氨基连接臂反应得到化合物7。最后,用甲醇和甲醇钠脱去化合物7的2-oac得到糖砌块8。
具体试验操作和步骤:
化合物6:将化合物5(9.7g,21mmol)溶于干燥吡啶(60ml)中,冰浴下缓缓加入乙酸酐(40ml)。反应液在室温下搅拌6h,用tlc检测原料反应完全之后,用二氯甲烷稀释反应液,用1m盐酸溶液洗涤,饱和碳酸氢钠溶液和二氯甲烷萃取,分离有机相,用无水硫酸钠干燥后浓缩。浓缩液经硅胶柱(石油醚/乙酸乙酯,7:1)纯化得到化合物6(7.3g,14.4mmol)。1hnmr(400mhz,chloroform-d)δ7.54–7.19(m,12h,ar-h),7.11(d,j=7.9hz,2h,ar-h)5.56(s,1h,phch-h),4.99(dd,j=10.0,8.6hz,1h,2-h),4.85(d,j=12.0hz,1h,bn-h),4.68–4.60(m,2h,1-h,bn-h),4.37(dd,j=10.5,5.0hz,1h,6a-h),3.79(t,j=10.3hz,1h,6b-h),3.75–3.69(m,2h,3-h,4-h),3.47(dq,j=7.9,5.0hz,1h,5-h),2.33(s,3h,me),2.03(s,3h,me)。13cnmr(101mhz,chloroform-d)δ169.22,138.26,137.12,133.39(2c),129.65(2c),128.99,128.29(2c),128.24(2c),128.21,127.87(2c),127.66,125.96(2c),101.19,86.96,81.30,79.77,74.31,71.35,70.46,68.53,21.13,20.95。
化合物7:将化合物6(7.3g,14.4mmol)和氮-苄基-氮-甲酸苄酯-戊醇(8.62g,28.8mmol)溶于无水dcm(100ml)中,加入适量新鲜活化的分子筛,室温下搅拌30min,随后转移至冰浴。30min之后往反应液中一次加入nis(5.6g,17.3mmol)和tmsotf(0.3ml,1.7mmol)。继续冰浴下反应10h,用tlc监测反应进程,待原料反应完全之后,依次用10%的硫代硫酸钠,饱和碳酸氢钠溶液和二氯甲烷萃取反应液,分离有机相,用无水硫酸钠干燥之后浓缩。浓缩液经硅胶柱纯化(石油醚/乙酸乙酯,3:1)得到化合物7(7.5g,11.1mmol)。
化合物8:将化合物7(7.0g,10.3mmol)溶于甲醇(50ml)中,室温下加入甲醇钠(110mg),反应液在室温下搅拌8h,用tlc监测反应,待原料全部反应完毕,用amberliteir120h+树脂中和反应液至中性。滤去树脂,浓缩后经硅胶柱纯化(石油醚/乙酸乙酯,3:1)得到化合物8(4.6g,8.45mmol)。
实施例3
糖砌块15的合成
如图3所示,其二丙酮葡萄糖1为起始原料,在nah的作用下与烯丙基溴反应得到3-烯丙基化合物9。化合物9在加有盐酸的烯丙醇中进行开环反应,得到1,3-二烯丙基化合物10。接下来用苄基保护葡萄糖剩下的三个裸露羟基得到化合物11。化合物11在干燥甲醇中利用氯化钯专属性地脱去1,3号位的烯丙基得到1-oh化合物12。用乙酰基保护前一步反应生成的两个羟基得到化合物13。在醋酸肼的作用下选择性脱除1-oac基,得到1-oh化合物14。最后用三氯乙腈和dbu与化合物14反应得到三氯乙酰亚胺酯糖基砌块15。
具体试验操作和步骤:
化合物9:将市售的二丙酮葡萄糖1(50g,191mmol)溶于无水dmf(380ml),冰浴下搅拌30min,随后分批加入氢化钠(23g)和烯丙基溴(34ml)。反应在冰浴下搅拌2h,用tlc监测反应,待原料反应完毕之后用冰水淬灭反应,用二氯甲烷和饱和食盐水萃取反应液,用无水硫酸钠干燥后浓缩,浓缩液经硅胶柱纯化(石油醚/乙酸乙酯,20:1)得到化合物2(57.1g,190mmol)。1hnmr(400mhz,cdcl3):δ=5.95–5.77(m,2h,1-h,ally-h),5.31(dd,j=17.2,1.7hz,1h,ally-h),5.20(dt,j=10.4,1.5hz,1h,ally-h),4.54(d,j=3.7hz,1h,2-h),4.31(dt,j=7.7,6.0hz,1h,5-h),4.34-4.07(m,4h,4-h,6a-h,2ally-h),4.00(dd,j=8.6,5.8hz,1h,6b-h),3.94(d,j=3.0hz,1h,3-h),1.50(s,3h,me),1.43(s,3h,me),1.35(s,3h,me),1.31(s,3h,me)。13cnmr(101mhz,chloroform-d)δ134.28,117.41,111.88,109.07,105.36,82.91,81.53,81.30,72.62,71.47,67.41,26.96,26.93,26.37,25.54.
化合物10:将化合物9(52.8g,176mmol)溶于烯丙醇(350ml)中,加入浓盐酸(10.5ml)。反应液在80℃下搅拌2h,用tlc监测反应,待原料反应完全之后,用饱和碳酸氢钠溶液中和反应液至中性。浓缩反应液,经硅胶柱纯化(二氯甲烷/甲醇,25:1)得到化合物10(40.3g,155mmol)。1hnmr(400mhz,chloroform-d)δ6.01–5.74(m,2h,2ally-h),5.24(dt,j=17.3,1.7hz,2h,2ally-h),5.15(ddt,j=13.5,10.4,1.4hz,2h,2ally-h),4.83(d,j=3.7hz,1h,h-1),4.38(ddt,j=12.7,5.7,1.5hz,1h,ally-h),4.22(ddt,j=12.7,6.0,1.4hz,1h,ally-h),4.15(ddt,j=12.8,5.3,1.5hz,1h,ally-h),3.97(ddt,j=12.8,6.3,1.4hz,1h,ally-h).3.75(d,j=3.6hz,2h,h-6ab),3.59(dt,j=9.2,3.6hz,1h,h-5),3.53(dd,j=9.1,3.8hz,1h,h-2),3.51–3.42(m,2h,h-3,h-4)。13cnmr(101mhz,chloroform-d)δ135.13,133.49,118.12,117.33,97.73,82.36,73.93,72.52,71.38,69.84,68.60,62.04。
化合物11:将化合物10(38g,146mmol)溶于无水dmf(365ml)中,冰浴下搅拌30min,分批缓缓加入氢化钠(35g),继续冰浴下搅拌30min,加入溴化苄(61ml)。反应液在冰浴下继续搅拌5h,用tlc监测反应。待原料全部反应后,用冰水淬灭反应,用饱和食盐水和二氯甲烷萃取,分离有机相,用无水硫酸钠干燥后浓缩,浓缩液经硅胶柱纯化得到化合物11(67.8g,127.7mmol)。1hnmr(400mhz,chloroform-d)δ7.37(ddt,j=21.5,14.3,6.6hz,13h,ar-h),7.24(d,j=7.1hz,2h,ar-h),6.01(dddt,j=34.9,17.4,11.7,5.6hz,1h,ally-h),5.35(dt,j=16.1,7.7hz,1h,ally-h),5.23(t,j=10.2hz,1h,ally-h),4.87(td,j=14.4,12.4,6.9hz,2h,1-h,bn-h),4.79(d,j=6.0hz,1h,bn-h),4.73–4.56(m,2h,2bn-h),4.50(t,j=11.7hz,3h,ally-h,2bn-h),4.35(td,j=15.1,13.9,5.8hz,0h)4.19(dd,j=13.2,5.6hz,ally-h),4.05(dd,j=12.9,6.8hz,ally-h),3.91(q,j=7.8,6.1hz,1h,3-h),3.85–3.71(m,2h,5-h,6a-h),3.65(dt,j=14.7,7.7hz,2h,4-h,6b-h),3.60–3.51(m,1h,2-h)。13cnmr(101mhz,cdcl3)δ138.54,138.35,138.24,137.99,135.40,135.21,134.12,133.81,128.43,128.39,128.37,128.25,128.09,128.05,128.03,127.95,127.93,127.83,127.80,127.72,127.62,118.15,117.20,116.74,116.54,102.69,95.82,84.40,82.19,81.81,79.65,77.83,77.68,77.42,77.33,77.09,77.01,76.78,76.70,75.12,75.05,74.93,74.85,74.52,74.42,73.50,73.22,70.32,70.20,69.01,68.48,68.19.
化合物12:将化合物11(10.2g,19.3mmol)溶于无水甲醇(644ml)中,加入氯化钯(684.5mg),反应液在40℃下搅拌20min,用tlc监测反应,待原料完全反应之后,用饱和碳酸氢钠和二氯甲烷萃取反应液,分离有机相,用无水硫酸钠干燥之后浓缩,浓缩液经硅胶柱纯化(石油醚/乙酸乙酯,3:1)得到化合物12(6.5g,14.4mmol)。1hnmr(400mhz,chloroform-d)δ7.31–7.15(m,15h,ar-h),5.18(d,j=3.5hz,1h,1-h),4.92(d,j=11.5hz,1h,bn-h),4.77(d,j=11.3hz,1h,bn-h),4.65(d,j=11.8hz,1h,bn-h),4.59(d,j=11.8hz,1h,bn-h),4.54(d,j=12.2hz,1h,bn-h),4.43(d,j=12.2hz,1h,bn-h),4.04(t,j=9.2hz,1h,3-h),3.96(ddd,j=10.1,3.9,2.5hz,1h,5-h),3.66–3.58(m,2h,6ab-h),3.46(dd,j=10.0,8.9hz,1h,4-h),3.36(dd,j=9.5,3.5hz,1h,2-h)2.91(s,2h,oh-h)。13cnmr(101mhz,cdcl3)δ138.36,137.80,137.74,128.62,128.58,128.44,128.42,128.20,128.13,128.10,128.05,128.00,127.95,127.91,127.81,127.78,127.76,97.11,90.71,82.19,79.54,77.65,77.45,77.41,77.29,77.09,76.77,76.56,74.60,74.52,74.29,73.50,73.27,72.83,69.78,68.72.
化合物13:将化合物12(6.1g,14mmol)溶于吡啶(40ml),冰浴下搅拌30min,缓缓加入乙酸酐(30ml)。反应液在冰浴下搅拌过夜。用tlc监测反应进程,待原料全部反应完全后,用1m盐酸溶液淬灭反应。用饱和碳酸氢钠溶液和乙酸乙酯萃取反应液,分离有机相,用无水硫酸钠干燥后浓缩,浓缩液经硅胶柱纯化(石油醚/乙酸乙酯,6:1)得到化合物13。(6.77g,12.66mmol)。1hnmr(400mhz,chloroform-d)δ7.52–7.15(m,13h,ar-h),7.12(dd,j=7.6,1.9hz,2h,ar-h),5.63(d,j=8.1hz,1h,h-1),5.26(t,j=9.4hz,1h,h-3),4.69(d,j=12.0hz,1h,bn-h),4.65(d,j=12.0hz,1h,bn-h),4.58(d,j=12.0hz,1h,bn-h),4.47(t,j=6.0hz,3h,bn-h),3.80–3.67(m,3h,h-4,h-6a,h-6b),3.60(dt,j=9.9,2.5hz,1h,h-5),3.51(dd,j=9.5,8.1hz,1h,h-2),2.07(s,3h,me),1.84(s,3h,me)。13cnmr(101mhz,chloroform-d)δ169.80,168.98,137.88,137.79,128.43,128.11,127.88,127.85,127.83,94.07,78.45,75.61,75.53,75.34,74.38,74.31,73.64,67.85,21.07,20.96。
化合物14:将化合物13(1.6g,3.0mmol)溶于无水dmf(30ml),加入乙酸肼(305mg)。反应液在40℃下搅拌2h,用tlc监测反应,待原料反应完毕以后将反应液浓缩。用饱和食盐水和dcm萃取,分离有机相,经无水硫酸钠干燥后浓缩,浓缩液用硅胶柱纯化(石油醚/乙酸乙酯,2:1)得到化合物14(1.36g,2.76mmol)。1hnmr(400mhz,chloroform-d)δ7.37–7.21(m,13h,ar-h),7.13(d,j=7.2hz,2h,ar-h),5.50(t,j=9.6hz,1h,3-h),5.27(d,j=3.6hz,1h,1-h),4.67–4.37(m,6h,bn-h),4.07(d,j=10.0hz,1h,6a-h),3.78(s,1h,1-oh),3.73–3.58(m,3h,4-h,5-h,6b-h),3.48(dd,j=9.8,3.4hz,1h,2-h),1.91(s,3h,me)。13cnmr(101mhz,cdcl3)δ170.14,137.96,137.77,137.64,128.53,128.46,128.43,128.34,128.13,128.12,128.03,128.00,127.95,127.90,127.86,127.78,90.86,77.58,77.50,77.18,76.86,76.10,74.36,74.28,73.90,73.58,73.55,73.45,72.56,69.91,68.37,21.12.
化合物15:将化合物14(300mg,0.61mmol)溶于无水dcm(6ml)中,冰浴下加入三氯乙腈(0.6ml)和dbu(91μl)。反应液在冰浴下搅拌2h,用tlc监测反应,待原料全部反应完毕,用饱和食盐水和二氯甲烷萃取反应液,分离有机相,经无水硫酸钠干燥后浓缩,浓缩液用硅胶柱纯化得到化合物15(357mg,0.56mmol)。1hnmr(400mhz,chloroform-d)δ8.59(s,1h,nh-h),7.28(dt,j=19.8,5.5hz,13h,ar-h),7.19–7.14(m,2h,ar-h),6.54(d,j=3.5hz,1h,h-1),5.59(t,j=9.7hz,1h,h-3),4.65(d,j=12.4hz,1h,bn-h),4.60(d,j=11.9hz,1h,bn-h),4.55–4.48(m,3h,bn-h),4.46(d,j=11.8hz,1h,bn-h),4.04(dt,j=10.1,2.5hz,1h,h-6a),3.84–3.73(m,2h,h-6b,h-4),3.67(dd,j=10.0,3.6hz,2h,h-2,h-5),1.91(s,3h,me)。13cnmr(101mhz,chloroform-d)δ169.87,161.34,137.82,137.77,137.74,93.96,91.20,76.51,75.48,74.66,73.67,73.19,72.99,72.53,67.90,21.12。
实施例4
二糖化合物的16和18合成
如图4所示,二糖化合物16由单糖供体15和单糖受体5在tmsotf作为促进剂的条件下合成。以单糖供体15和单糖受体8用类似的方法合成二糖化合物17,随后17在甲醇甲醇钠的条件下脱去3’-oac得到二糖化合物18。
化合物16:将糖基供体15(3.1g,4.8mmol)和糖基受体(2.8g,6.1mmol)混合,在氮气保护下用甲苯共沸3次,在油泵下抽真空过夜。加入二氯甲烷(100ml)和适量分子筛,室温下搅拌30min后冷却至-78℃,继续搅拌下30min,加入促进剂tmsotf(100μl),反应在低温下搅拌5h。用tlc监测反应进程,待糖基供体全部反应消耗之后,用三乙胺淬灭反应。用二氯甲烷和饱和碳酸氢钠溶液萃取反应液,分离有机相,用无水硫酸钠干燥后浓缩。浓缩液用硅胶柱纯化(石油醚/乙酸乙酯,10:1)得到化合物16(2.9g,3.2mmol)。1hnmr(400mhz,chloroform-d)δ7.52–7.45(m,2h,ar-h),7.41–7.23(m,21h,ar-h),7.14(td,j=7.5,1.6hz,3h,ar-h),7.07(d,j=7.9hz,2h,ar-h),7.02(t,j=7.6hz,2h,ar-h),5.99(d,j=3.7hz,1h,1’-h),5.60(s,1h,phch),5.55(t,j=9.7hz,1h,3’-h),4.99(d,j=10.1hz,1h,bn-h),4.90(d,j=8.9hz,1h,1-h),4.85(d,j=12.2hz,1h,bn-h),4.64(d,j=10.2hz,1h,bn-h),4.61–4.51(m,2h,2bn-h),4.47(d,j=11.2hz,1h,bn-h),4.41–4.35(m,1h,6a-h),4.34(d,j=11.0hz,1h,bn-h),4.26(dd,j=11.1,5.3hz,2h,5’-h,bn-h),3.93–3.83(m,2h,2-h,3-h),3.80(d,j=10.3hz,1h,6a-h),3.74(dd,j=12.3,3.3hz,1h,4-h),3.71–3.66(m,1h,4’-h),3.56(dd,j=10.3,3.6hz,1h,2’-h),3.51(dd,j=9.6,5.0hz,1h,5-h),3.17–3.06(m,2h,6’-h),2.35(s,3h,me),1.96(s,3h,me)。13cnmr(101mhz,cdcl3)δ192.42,170.09,163.32,138.36,138.26,138.17,138.15,137.86,137.83,137.77,137.47,137.19,136.42,134.48,132.49,132.33,129.85,129.81,129.76,129.02,128.56,128.44,128.37,128.34,128.32,128.29,128.25,128.19,127.85,127.82,127.76,127.75,127.68,127.55,125.99,101.28,95.59,88.02,85.23,81.98,81.09,78.88,76.04,75.95,75.58,74.84,74.42,74.39,74.32,73.59,73.47,72.48,72.36,71.33,70.10,69.92,69.78,68.67,67.73,67.41,62.62,21.14,21.12。
化合物17:将糖基供体15(1.42g,2.3mmol)和带有氨基连接臂的糖基受体8(1.11g,1.6mmol)混合,在氮气保护下用甲苯共沸3次,在油泵下抽真空过夜。之后加入无水dcm(100ml)和适量分子筛,室温下搅拌30min后降温至-78℃,继续搅拌30min后,加入促进剂tmsotf(37μl),反应液在低温下继续搅拌6h。用tlc监测反应,待糖基供体完全消耗之后,用三乙胺淬灭反应。用二氯甲烷和饱和碳酸氢钠溶液萃取反应液,分离有机相,用无水硫酸钠干燥后浓缩。浓缩液用硅胶柱纯化(石油醚/乙酸乙酯,4:1)得到化合物17(1.35g,1.2mmol)。1hnmr(400mhz,chloroform-d)δ7.48–6.97(m,35h,ar-h),5.64(s,1h,1’-h),5.55(d,j=7.7hz,2h,phch,3’-h),5.16(d,j=10.3hz,2h,2bn-h),4.92(d,j=10.2hz,1h,bn-h),4.65(t,j=12.3hz,2h,2bn-h),4.55(t,j=10.5hz,2h,1-h,bn-h),4.50–4.40(m,4h,2cbz-h,2bn-h),4.32(d,j=10.8hz,1h,6a-h,bn-h),4.23(t,j=9.4hz,2h,5’-h,bn-h),3.73(ddt,j=37.9,19.0,9.3hz,6h,2-h,4-h,5-h,6b-h,4’-h,linker-och),3.50(d,j=9.7hz,1h,2’-h),3.41(q,j=8.7hz,2h,3-h,linker-och),3.18(q,j=11.6,10.7hz,4h,6’ab-h,linker-nch2),1.91(s,3h,me),1.70–1.34(m,4h,linker-ch2),1.22(d,j=27.7hz,2h,linker-ch2)。
化合物18:将化合物17(1.1g,0.96mmol)溶于无水甲醇(20ml),加入甲醇钠(100mg)。反应液在40℃下搅拌24h,用tlc监测反应,待原料全部反应完全之后用amberliteir120h+树脂中和反应液至中性。滤去树脂,浓缩后经硅胶柱纯化(石油醚/乙酸乙酯,3:1)得到化合物18(898mg,0.82mmol)。1hnmr(400mhz,chloroform-d)δ7.67–6.95(m,35h,ar-h),5.68(s,1h,1’-h),5.62(s,1h,phch-h),5.21(d,j=11.2hz,2h,bn-h),4.94(d,j=10.1hz,1h,bn-h),4.83(d,j=11.3hz,2h,bn-h),4.60(t,j=11.1hz,4h,1-h,1bn-h,2cbz-h),4.50(t,j=10.5hz,3h,3bn-h),4.39(d,j=9.8hz,1h,6a-h),4.29(d,j=12.1hz,1h,bn-h),4.25–4.08(m,2h,3’-h,5’-h),3.79(dq,j=18.3,9.5hz,5h,2-h,3-h,4-h,6b-h,linker-och),3.65(t,j=9.5hz,1h,4’-h),3.57–3.37(m,3h,5-h,2’-h,linker-och),3.34–3.14(m,4h,6’ab-h,linker-nch2),2.50(s,1h,3’-oh),1.52(ddt,j=22.4,15.3,7.9hz,4h,linker-ch2),1.41–1.14(m,2h,linker-ch2)。13cnmr(101mhz,cdcl3)δ156.72,156.22,138.90,138.05,137.93,137.66,137.30,129.03,128.59,128.49,128.32,128.28,128.26,128.14,127.98,127.89,127.73,127.68,127.62,127.54,127.34,125.99,104.00,101.23,95.10,82.28,79.60,79.03,77.63,77.42,77.31,77.10,76.79,75.65,75.57,74.63,73.51,73.40,71.97,69.62,69.49,68.81,67.86,67.21,65.95,50.53,50.24,47.05,46.15,29.74,29.63,27.96,27.54,23.33。
实施例5
四糖19的合成如图5所示。
化合物19:将二糖糖基供体15(287mg,0.3mmol)和二糖糖基受体18(230mg,0.12mmol)混合,在氮气保护下用甲苯共沸3次,在油泵下抽真空过夜。之后加入无水dcm(2ml)乙醚(4ml)和噻吩(12.6mmol)以及适量分子筛,室温下搅拌30min后降温至0℃,继续搅拌30min后,加入促进剂nis(135mg)和agotf(26mg),反应液在冰浴下继续搅拌12h。用tlc监测反应,待糖基供体完全消耗之后,用10%硫代硫酸钠溶液和二氯甲烷萃取反应液,分离有机相,用无水硫酸钠干燥后浓缩。浓缩液用硅胶柱纯化(石油醚/乙酸乙酯,4:1)得到化合物19(121mg,0.064mmol)。1hnmr(400mhz,chloroform-d)δ5.81(s,1h,a1-h),5.73(t,j=9.7hz,1h,c3-h),5.63(s,1h,b1-h),5.55(d,j=5.6hz,2h,2phch),5.19(d,j=8.9hz,3h,1c-h,2bn-h),4.90(d,j=10.9hz,1h,bn-h),4.79(d,j=15.8hz,3h,3bn-h),4.54(ddt,j=47.2,27.9,9.9hz,13h,d1-h,a3-h,2cbz-h,9bn-h),4.38–4.14(m,8h,d3-h,b5-h,c5-h,c6-h2,3bn-h),4.03(t,j=9.3hz,1h,b3-h),3.74(td,j=30.8,27.3,8.7hz,9h,a6-h2,a2-h,b4-h,d2-h,d5-h,c4-h,a5-h,linker-och),3.49(ddd,j=42.6,21.7,9.6hz,6h,d6-h2,d4-h,b2-h,a2-h,a4-h),3.20(d,j=34.7hz,5h,b6-h2,linker-nh2,linker-och),1.89(s,3h,me)。13cnmr(101mhz,cdcl3)δ169.61,138.61,138.32,138.16,138.02,137.96,137.68,137.61,137.34,136.87,130.92,128.96,128.80,128.75,128.69,128.56,128.48,128.38,128.32,128.26,128.21,128.09,128.03,127.95,127.86,127.73,127.66,127.62,127.59,127.56,127.51,127.47,127.25,126.18,125.99,103.93,101.22,95.04,94.72,94.50,82.61,82.21,79.44,78.54,78.31,77.36,77.04,76.72,76.25,75.72,75.49,75.37,75.18,74.15,73.44,73.38,73.16,72.13,71.40,69.92,69.43,69.17,68.91,68.81,68.16,67.93,67.18,65.80,65.58,62.36,50.48,47.05,46.15,29.72,29.55,27.89,27.49,23.20,21.09,19.20,13.74。
实施例61,3-α-顺式糖苷键构建体系的优化:
1)溶剂对1,3-α-顺式糖苷键构建的影响:
参照实施例5,经溶剂分别替换为表1中所示的溶剂,测定所得产物中1,3-α-顺式糖苷键的含量。结果见表1。
表1不同溶剂和添加剂体系构建1,3-α-顺式糖苷键
2)促进剂对1,3-α-顺式糖苷键构建的影响:
参照实施例5,将促进剂分别替换为表2中所示的添加剂,测定所得产物中1,3-α-顺式糖苷键的含量。结果见表2。
表2不同促进剂条件构建1,3-α-顺式糖苷键
实施例7
六糖21的合成如图6所示。
四糖化合物19在甲醇和甲醇钠的条件下反应两天脱去3””-oac得到四糖化合物20。随后以化合物20为糖基受体与二糖供体16在路易斯酸促进剂的催化下反应得到六糖化合物21。
化合物20:将化合物19(90mg,0.047mmol)溶于甲醇(9ml)和乙酸乙酯(1ml)的混合溶剂中,加入甲醇钠(30mg)。反应在40℃下搅拌2d,用tlc监测反应,用待原料全部反应完全之后用amberliteir120h+树脂中和反应液至中性。滤去树脂,浓缩后经硅胶柱纯化(石油醚/乙酸乙酯,3:1)得到化合物21(68mg,0.037mmol)。1hnmr(400mhz,chloroform-d)δ5.79(d,j=3.4hz,1h,a1-h),5.68(d,j=5.3hz,1h,b1-h),5.55(d,j=2.7hz,2h,2phch),5.19(d,j=9.2hz,2h,2bn-h),5.06(d,j=3.4hz,1h,c1-h),4.97(d,j=11.1hz,1h,bn-h),4.79(ddd,j=21.1,11.9,7.4hz,4h,4bn-h),4.68–4.44(m,11h,d1-h,b3-h,a4-h,6bn-h,2cbz-h),4.46–4.37(m,2h,2bn-h),4.36–4.23(m,6h,d3-h,a5-h,2bn-h,6-h2),4.17(t,j=9.3hz,1h,c3-h),4.12–4.01(m,2h,c4-h,a3-h),3.87–3.68(m,6h,d2-h,b4-h,c5-h,6-h2,linker-och),3.68–3.62(m,2h,a2-h,d5-h),3.60(d,j=9.4hz,2h,b2-h,b5-h),3.45(dd,j=11.1,3.1hz,1h,6-h),3.40–3.33(m,2h,c2-h,d4-h),3.30–3.11(m,6h,linker-nch2,linker-och,6-h,6-h2),1.56–1.34(m,4h,2linker-ch2),1.23–1.06(m,4h,linker-ch2)。13cnmr(101mhz,cdcl3)δ156.70,156.18,138.86,138.70,138.24,138.20,138.14,138.07,138.01,137.93,137.78,137.62,137.44,137.31,136.91,129.79,129.23,129.01,128.81,128.71,128.69,128.65,128.59,128.50,128.42,128.32,128.29,128.25,128.22,128.20,128.16,128.15,128.02,127.97,127.88,127.84,127.75,127.66,127.62,127.60,127.56,127.48,127.42,127.35,126.16,126.00,103.92,102.01,101.26,101.21,95.76,95.36,94.59,82.79,82.11,81.12,79.58,79.43,78.86,78.25,78.07,77.40,77.28,77.08,76.76,75.35,75.31,74.79,74.67,74.47,73.85,73.57,73.45,73.39,73.29,71.93,71.75,69.92,69.23,68.77,68.21,67.90,67.20,65.81,62.70,50.50,50.20,47.05,46.13,29.74,29.58,27.91,27.50,23.23.
化合物21:将二糖供体15(60mg,0.064mmol)和四糖受体20(50mg,0.027mmol)在氮气保护下用甲苯共沸3次,在油泵下抽真空过夜。之后加入无水dcm(0.5ml)乙醚(1ml)和噻吩(0.6ml)以及适量分子筛,室温下搅拌30min后降温至0℃,继续搅拌30min后,加入促进剂nis(29mg)和agotf(3mg),反应液在冰浴下继续搅拌12h。用tlc监测反应,待糖基供体完全消耗之后,用10%硫代硫酸钠溶液和二氯甲烷萃取反应液,分离有机相,用无水硫酸钠干燥后浓缩。浓缩液用硅胶柱纯化(石油醚/乙酸乙酯,4:1)得到化合物21(33.4mg,0.012mmol)。1hnmr(400mhz,chloroform-d)δ5.91(s,1h,a1-h),5.75(d,j=12.4hz,2h,b1-h,b3-h),5.58(s,2h,c1-h,phch-h),5.52(s,1h,phch),5.50(s,1h,phch),5.36(s,1h,d1-h),5.22(s,3h,e1-h,2bn-h),4.94–4.72(m,7h,7bn-h),4.53(ddt,j=44.2,17.1,10.5hz,18h,f1-h,c3-h,d3-h,2cbz-h,13bn-h),4.34(dt,j=23.0,10.9hz,7h,f4-h,4bn-h,6-h2),4.22(d,j=11.1hz,2h,f5-h,b5-h),4.08(q,j=11.8,9.3hz,3h,a3-h,e3-h,6-h),3.90(d,j=9.6hz,1h,a2-h,6-h),3.88–3.64(m,10h,f2-h,f3-h,b4-h,d4-h,e4-h,a5-h,c3-h,6-h2,linker-och),3.53(dp,j=32.8,11.4,10.7hz,10h,a4-h,c2-h,b2-h,e2-h,c5-h,d2-h,d5-h,e5-h,6-h2),3.36–3.10(m,5h,b6-h2,linker-nch2,linker-och),1.93(s,3h,me),1.57–1.40(m,4h,2linker-ch2),1.23–1.11(m,2h,linker-ch2)。13cnmr(176mhz,cdcl3)δ206.98,192.41,169.83,169.74,156.70,138.41,137.94,137.90,137.71,137.29,136.90,136.44,134.48,130.93,129.77,129.02,128.86,128.84,128.62,128.57,128.50,128.43,128.38,128.34,128.28,128.24,128.05,128.01,127.91,127.87,127.75,127.70,127.65,127.59,127.56,127.53,127.50,127.47,127.45,127.40,127.37,127.33,127.29,126.24,126.21,126.16,126.01,103.95,101.28,101.18,94.82,94.36,94.12,93.96,82.53,82.21,79.40,78.67,77.22,77.04,76.86,76.16,75.78,75.59,75.38,75.20,74.89,74.27,74.18,73.45,73.42,73.39,73.27,72.97,72.13,71.17,70.51,70.07,69.81,69.24,68.98,68.80,68.60,68.15,67.83,67.20,65.83,62.13,50.47,50.16,47.03,46.10,30.96,29.73,29.54,27.88,27.47,23.20,22.72,21.13,21.11,14.15。
依照此方法可以合成八糖,十糖,十二糖等一系列幽门螺旋杆菌o2-血清型寡糖衍生物。
寡糖链可以以+2糖的模式由还原端向非还原端延伸,包括两个步骤。1)还原端带有连接臂的寡糖脱去非还原端的脂类基团;2)与二糖供体16的糖基化反应。
脱去寡糖非还原端脂类基团的方法:将化合物(~50mg)溶于甲醇(10~15ml)中,可以适量添加少量乙酸乙酯或者四氢呋喃增加溶解性。加入甲醇钠(20~30mg),反应液在40℃下搅拌12h~2d。用tlc监测反应进程,待原料全部反应完全之后用amberliteir120h+树脂中和反应液至中性。滤去树脂,浓缩后经硅胶柱纯化。
以二糖16为糖基供体进行糖苷化反应的方法:将糖基供体和糖基受体在氮气保护下用甲苯共沸三次,在油泵上抽真空过夜。将反应物溶于二氯甲烷和乙醚(1:1)的混合溶剂中,同时加入相对于糖基供体100当量的噻吩和适量分子筛。加入nis和agotf,tmsotf和tfoh中的一种或者几种作为促进剂。反应浓度为0.02m(相对于糖基供体),反应时间为12~24h。用tlc监测反应进程,待糖基供体反应完全之后用10%硫代硫酸钠、二氯甲烷和饱和碳酸氢钠溶液萃取反应液,分离有机相,用无水硫酸钠干燥,浓缩后经硅胶柱纯化得到新的寡糖化合物。
实施例7:二糖,四糖和六糖的脱保护
如图7所示,二糖,四糖和六糖化合物的脱保护分为两个步骤。第一,在碱性条件下脱除非还原端的乙酰基;第二,在钯碳的催化下氢化掉所有的芳香基团,得到还原端组装有五碳氨基连接臂的二糖、四糖和六糖。
脱除非还原端二糖乙酰基的方法:将化合物溶于甲醇和乙酸乙酯(9:1)的混合溶剂中,浓度为0.02m,加入相对于原料0.5当量的甲醇钠。反应液在40℃下搅拌12h~2d。用tlc监测反应进程。待原料全部反应完全之后用amberliteir120h+树脂中和反应液至中性。滤去树脂,浓缩后经硅胶柱纯化。
氢化反应的方法:将化合物(6~10mg)溶于异丁醇(3ml)、甲醇(1ml)和水(1ml)的混合溶液中,加入冰乙酸(0.1ml)和钯碳(50mg)。反应液在中压氢气反应釜中进行,反应压力为0.4mpa,反应时间为12h。待原料反应完全之后用硅藻土过滤反应液,浓缩后经c18小柱纯化得到目标化合物。
具体试验和操作步骤:
化合物22:以二糖化合物17为原料,经过脱乙酰基和氢化两个步骤得到脱保护的二糖化合物22。1hnmr(400mhz,deuteriumoxide)δ5.37(d,j=3.8hz,1h,1’-h),4.61(d,j=7.9hz,1h,1-h),4.05(dt,j=10.2,3.3hz,1h,5’-h),4.00–3.89(m,2h,6-h,linker-och),3.81(d,j=3.4hz,2h,6’-h2),3.77–3.67(m,3h),3.58(t,j=9.0hz,1h,4-h),3.52(dd,j=9.9,3.9hz,1h,2’-h),3.49–3.36(m,4h,4’-h,2-h,3-h,5-h),3.02(t,j=7.6hz,2h,linker-nch2),1.70(q,j=7.2hz,4h,2linker-ch2),1.57–1.39(m,2h,linker-ch2)。13cnmr(101mhz,d2o)δ102.77,97.61,77.02,75.71,74.50,72.91,71.63,71.43,70.18,69.84,69.30,60.81,60.23,39.37,28.28,26.43,22.28。
化合物23:以四糖化合物19为起始原料,经过脱乙酰基和氢化两个步骤得到脱保护的四糖化合物23。1hnmr(400mhz,deuteriumoxide)δ5.44(d,j=3.6hz,1h,a1-h),5.28(d,j=3.9hz,1h,b1-h),5.08(d,j=3.7hz,1h,c1-h),4.53(d,j=8.1hz,1h,d1-h),3.96(d,j=9.6hz,2h,a3-h,c4-h),3.92–3.65(m,12h,linker-och,b3-h,c3-h,d3-h,b4-h,d5-h,6*6-h),3.65–3.44(m,7h,linker-och,a2-h,a5-h,b2-h,c2-h,2*6-h),3.43–3.22(m,5h,a4-h,b5-h,d2-h,d4-h,c5-h),2.90(t,j=7.6hz,2h,linker-nch2),1.58(q,j=7.4hz,4h,linker-ch2),1.36(q,j=8.0hz,2h,linker-ch2)。13cnmr(101mhz,d2o)δ102.75,97.71,96.49,95.98,79.83,76.87,75.74,75.37,74.50,72.84,71.82,71.54,71.28,70.21,69.88,69.51,69.29,60.84,60.32,39.38,28.30,26.45,22.29。
化合物24:以六糖化合物21为起始原料,经过脱乙酰基和氢化两个步骤得到脱保护的四糖化合物24。1hnmr(400mhz,deuteriumoxide)δ5.51(d,j=3.7hz,1h,a1-h),5.48(d,j=3.7hz,1h,b1-h),5.31(d,j=3.9hz,1h,c1-h),5.13(d,j=3.8hz,1h,d1-h),5.11(d,j=3.7hz,1h,e1-h),4.55(d,j=8.0hz,1h,f1-h),4.09–3.25(m,38h,6*2-h,6*3-h,6*4-h,6*5-h,6*6-h2,linker-och2),2.93(t,j=7.6hz,2h,linker-nch2),1.61(h,j=6.9,6.2hz,4h,linker-ch2),1.43–1.30(m,2h,linker-ch2)。13cnmr(101mhz,d2o)δ102.77,96.58,95.99,79.76,79.48,76.78,75.76,75.34,75.03,74.51,72.85,71.82,71.63,71.29,71.14,70.22,70.04,69.87,69.72,69.54,69.31,60.84,60.58,60.33,60.05,39.38,28.32,26.46,22.31。
虽然本发明以较佳实例公开如上,但其并非用以限定本发明,任何熟悉此技术的人在不脱离本发明的精神和范围内都可作各种改动与修饰,因此本发明的保护内容应该以权利要求书所限定的为准。
- 还没有人留言评论。精彩留言会获得点赞!