一种托法替布中间体的制备工艺的制作方法
本发明属于药物合成技术领域,具体涉及一种托法替布中间体的制备工艺,更具体地,涉及一种托法替布中间体(3r,4r)-4-甲基-3-(甲基-1h-吡咯并[2,3-d]嘧啶-4-氨基)-β-氧-1-哌啶丙腈的制备工艺。
背景技术:
枸橼酸托法替布(tofacitinibcitrate),商品名xeljanz,是作用于细胞内酪氨酸激酶的小分子口服抑制剂,由美国fda于2012年11月批准用于成人的中度至重度活动性ra的治疗。其适应症为治疗成人活动期及对甲氨蝶呤(mtx)反应不佳的中至重度类风湿关节炎患者,可联合甲氨喋呤用于治疗对肿瘤坏死因子抑制剂应答不足的活动性类风湿性关节炎。
化合物(3r,4r)-4-甲基-3-(甲基-1h-吡咯并[2,3-d]嘧啶-4-氨基)-β-氧-1-哌啶丙腈是合成枸橼酸托法替布的关键中间体。其cas号为:477600-75-2,结构式如下:
现有的制备上述中间体的方法存在纯度低,尤其是大剂量制备时,纯度下降明显的问题。例如,文献(org.lett.,vol.11,no.9,2009,p2003)中提到了上述中间体的制备方法,该方法包括使用dbu作为碱,正丁醇作为溶剂,通过n-甲基-n-((3r,4r)-4-甲基哌啶-3-基)-7h-吡咯并[2,3-d]嘧啶-4-胺及氰基乙酸乙酯在40℃反应得到中间体(3r,4r)-4-甲基-3-(甲基-1h-吡咯并[2,3-d]嘧啶-4-氨基)-β-氧-1-哌啶丙腈,采用该方法扩大生产时,中间体的纯度下降较明显。
技术实现要素:
针对现有技术存在的上述不足,本发明的目的是提供一种托法替布中间体的制备工艺。该制备工艺通过控制反应温度、物料的加入次序及方式、氰基乙酸乙酯的加入比例等,使托法替布中间体(3r,4r)-4-甲基-3-(甲基-1h-吡咯并[2,3-d]嘧啶-4-氨基)-β-氧-1-哌啶丙腈收率、纯度均较高,且扩大生产时,中间体的纯度下降量较少。
本发明的发明人研究发现,在中间体(3r,4r)-4-甲基-3-(甲基-1h-吡咯并[2,3-d]嘧啶-4-氨基)-β-氧-1-哌啶丙腈的制备过程中,会出现一种有关物质a,通过常规的重结晶方式以及成盐制备枸橼酸托法替布过程,未见明显减少,而且在放大过程中此有关物质有增多的趋势。此有关物质的检出液相方法如下:
色谱柱:inertsilods-34.6*2505μm
流动相:a:乙腈;b:磷酸盐缓冲液(取磷酸二氢钾1.36g,加1000ml纯化水溶解,用氢氧化钠溶液调节ph至7.5)
柱温:50℃
波长:210nm
进样量:10μl
梯度:时间a相b相
01090
305050
405050
411090
481090
溶解溶剂:乙腈/磷酸盐缓冲液(取磷酸二氢钾1.36g,加1000ml纯化水溶解,用氢氧化钠溶液调节ph至7.5)=30∶70
有关物质a出峰位置在23.8min左右。
通过分离制备,发明人对此有关物质a进行了结构确证,其结构如下:
该有关物质a的存在影响了中间体(3r,4r)-4-甲基-3-(甲基-1h-吡咯并[2,3-d]嘧啶-4-氨基)-β-氧-1-哌啶丙腈的制备纯度,而且随着制备量的加大,有关物质a的含量有所增加,如采用文献org.lett.,vol.11,no.9,2009,p2003中的制备工艺制备中间体时,在百克以内投料量的产品中,其有关物质a含量为0.08%,在公斤级投料量的产品中,其有关物质a含量为0.43%。
在上述发现的基础上,本发明提供了一种托法替布中间体的制备工艺,通过对物料的加入次序及方式、氰基乙酸乙酯的加入比例,以及反应过程的温度等控制实现上述有关物质a含量的减少。
具体地,该制备工艺包括以下步骤:
1)将氰基乙酸乙酯加入正丁醇中,搅拌均匀;
2)在30℃以下,将dbu滴加至步骤1)所得的溶液中;
3)将n-甲基-n-((3r,4r)-4-甲基哌啶-3-基)-7h-吡咯并[2,3-d]嘧啶-4-胺分批加入步骤2)所得的溶液中进行反应;
4)在步骤3)反应完全的体系中滴加水,搅拌、过滤、洗涤,得到托法替布中间体(3r,4r)-4-甲基-3-(甲基-1h-吡咯并[2,3-d]嘧啶-4-氨基)-β-氧-1-哌啶丙腈。
优选地,所述氰基乙酸乙酯与n-甲基-n-((3r,4r)-4-甲基哌啶-3-基)-7h-吡咯并[2,3-d]嘧啶-4-胺的摩尔比为4∶1。与上述文献相比,将氰基乙酸乙酯与n-甲基-n-((3r,4r)-4-甲基哌啶-3-基)-7h-吡咯并[2,3-d]嘧啶-4-胺的比例增大一倍,也显示出了有关物质a的含量减少。
由于dbu在加入过程中剧烈放热,故采用滴加的方式,而且需要缓慢滴加。另外,该步的反应温度控制对有关物质a的含量控制尤为重要。优选地,步骤2)中,在20℃时,将dbu滴加至步骤1)所得的溶液中。
本发明中,原料n-甲基-n-((3r,4r)-4-甲基哌啶-3-基)-7h-吡咯并[2,3-d]嘧啶-4-胺通过后加且分批的方式避免dbu加入过程中的放热,优选地,其分3批加入。
优选地,步骤3)中,反应的条件包括:反应温度为35℃,反应时间为15h。
优选地,步骤4)中,采用1∶1的乙醇/水进行洗涤。
本发明中未加以限定的操作参数均可依据本领域的常规技术手段进行。
与现有技术相比,本发明具有如下有益效果:
采用本发明的制备工艺可有效降低有关物质a在中间体中的含量,保证中间体的纯度,同时扩大制备量时,有关物质a的增量不大,具体地,在百克以内投料量的产品中,其有关物质a未检出,在公斤级投料量的产品中,其有关物质a含量为0.05%。
附图说明
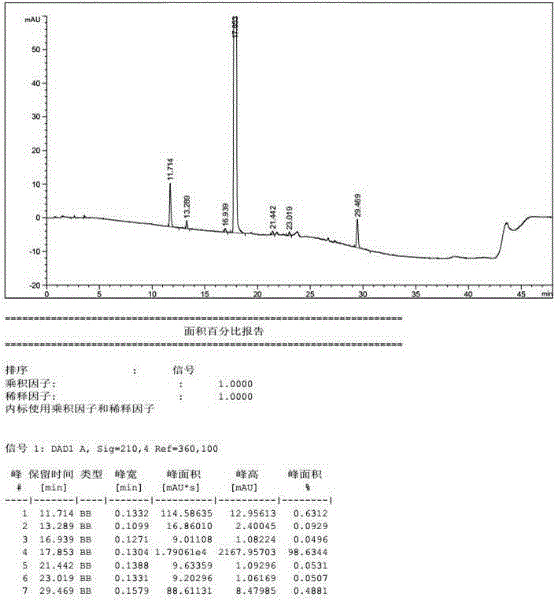
图1:实施例2得到的产品纯度图谱。
图2:对比例2得到的产品纯度图谱。
具体实施方式
下面将结合本发明实施例对本发明的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。
实施例1
将氰基乙酸乙酯(73.8g,0.64mol)加到200ml正丁醇中,搅拌冷却至20℃,缓慢滴加dbu(25g,0.16mol),分3批加入n-甲基-n-((3r,4r)-4-甲基哌啶-3-基)-7h-吡咯并[2,3-d]嘧啶-4-胺(40g,0.16mol),完毕,升温至35℃,反应15h。将200ml水滴加入反应体系中,搅拌0.5h,过滤,得到白色固体,使用100ml乙醇/水(1∶1)洗涤,得到47.5g白色固体,收率93.3%,纯度98.63%,有关物质a未检出。
实施例2
将氰基乙酸乙酯(3.68kg,32.5mol)加到10l正丁醇中,搅拌冷却至20℃,缓慢滴加dbu(1.25kg,8.2mol),分3批加入n-甲基-n-((3r,4r)-4-甲基哌啶-3-基)-7h-吡咯并[2,3-d]嘧啶-4-胺(2.0kg,8.2mol),完毕,升温至35℃,反应15h。将10l水滴加入反应体系中,搅拌0.5h,过滤,得到白色固体,使用2l乙醇/水(1∶1)洗涤,得到2.3kg白色固体,收率90.2%,纯度98.53%,有关物质a含量为0.05%。
对比例1
将n-甲基-n-((3r,4r)-4-甲基哌啶-3-基)-7h-吡咯并[2,3-d]嘧啶-4-胺(40g,0.16mol)中加到200ml正丁醇中,搅拌,加入氰基乙酸乙酯(36.9g,0.32mol),冷却至20℃,缓慢滴加dbu(25g,0.16mol),滴加完毕过后,升温至40℃,反应15h。将200ml水滴加入反应体系中,搅拌0.5h,过滤,得到白色固体,使用100ml乙醇/水(1∶1)洗涤,得到46.3g白色固体,收率91%,纯度98.86%,有关物质a含量为0.08%。
对比例2
将n-甲基-n-((3r,4r)-4-甲基哌啶-3-基)-7h-吡咯并[2,3-d]嘧啶-4-胺2.0kg中加到10l正丁醇中,搅拌,加入1.84kg氰乙酸乙酯,冷却至20℃,缓慢滴加1.25kgdbu,滴加完毕过后,升温至40℃,反应15h。将10l水滴加入反应体系中,搅拌1h,过滤,使用2l乙醇/水(1∶1)洗涤,得到2.3kg白色固体,收率90.2%,纯度98.15%,有关物质a含量为0.43%。
实施例和对比例的结果表明,采用本发明的制备工艺可有效降低有关物质a在中间体中的含量,同时扩大制备量时,有关物质a的增量不大。
以上已经描述了本发明的实施例,上述说明是示例性的,并非穷尽性的,并且也不限于所披露的实施例。在不偏离所说明实施例的范围和精神的情况下,对于本技术领域的普通技术人员来说许多修改和变更都是显而易见的。
- 还没有人留言评论。精彩留言会获得点赞!