一种头孢烯类鎓盐化合物、制备方法及以该化合物合成头孢喹肟的方法与流程
本发明涉及一种头孢烯类鎓盐化合物、制备方法及以该化合物合成头孢喹肟的方法,属于医药化工领域。
背景技术:
头孢喹肟(cefquinome),其化学名为1-[[(6r,7r)-7-[[(2z)-(2-氨基-4-噻唑基)(甲氧亚胺基)乙酰基]氨基]-2-羧基-8-氧代-5-硫杂-1-氮杂二环[4,2,0]辛-2-烯-3-基]甲基]-5,6,7,8-四氢喹啉内鎓盐。其硫酸盐(cefquinomesulfate)是德国hoechst公司研发的首个动物专用第四代头孢菌素类抗生素,1993年在德国上市,目前已被欧盟兽用药品委员会(cvmp)批准用于动物的呼吸系统疾细菌感染、奶牛乳房炎、母猪产后综合症以及家禽细菌性疾病的治疗,治疗效果显著且治疗后不易复发。
头孢喹肟是一种全新的氨基噻唑酮氧化亚胺头孢菌素,它的c3位上的4价氮-5,6,7,8-四氢喹啉基团使其拥有两性离子特性,具有高度的水溶性,这种分子结构能使头孢喹肟快速穿透、快速渗透革兰氏阴性菌外膜带负电的微孔通道,最大限度地与青霉素果蛋白(pbps)结合,β-内酰胺开环与细菌发生酰化作用,抑制粘肽转肽酶,使其催化的转肽反应不能进行,从而阻碍细胞壁的形成,使菌体失去保护屏障,在低渗透环境中大量水分渗入菌体内,使细胞肿胀、变形、破裂及溶解而亡。同时抵御多种质粒和染色体介质的β-内酰胺的水解作用,表现为β-内酰胺酶的低亲和力和高稳定性。
头孢喹肟作为一种动物专用抗生素,具有抗菌活性强、药代动力学特征优良、毒副作用小、残留低、生物利用度高、抗菌谱广的优点,其独特的特性已受到人们普遍关注。作为一种治疗动物细菌感染的新药,头孢喹肟在我国兽医临床上有广阔的应用前景。
关于头孢喹肟的合成报道国内外有较多文献资料。合成头孢喹肟的主要原料有7-aca或gcle、5,6,7,8-四氢喹啉和ae活性酯,根据文献资料,头孢喹肟的合成路线主要有三条。
合成路线一:以7-aca和ae活性酯为原料先合成头孢噻肟酸,然后与5,6,7,8-四氢喹啉反应生成头孢喹肟。
合成路线二:以7-aca为起始原料,首先与5,6,7,8-四氢喹啉反应生成母核中间体,然后再与ae活性酯反应得到终产物头孢喹肟。
合成路线一、二的反应式见式ii。
合成路线三:以gcle代替7-aca为起始原料合成头孢喹肟。gcle分子结构中的c3位氯甲基与5,6,7,8-四氢喹啉进行取代反应制得中间体,再脱去c7位氨基和c4位羧基的保护基团,然后在碱性条件下与ae活性酯反应,制得头孢喹肟。此路线的合成反应式见式iii
上述合成头孢喹肟的路线,其合成途径因起始原料不同而不同。选择不同的起始原料,其合成头孢喹肟的工艺路线、工艺条件和参数如所用的试剂和反应条件等完全不同,其效果也会不同。
合成路线一、合成路线二的共同点都是以7-aca为起始原料,不同点是与ae活性酯和5,6,7,8-四氢喹啉反应顺序不同。
在合成路线一中,7-aca的c7位氨基先与ae活性酯反应,制得头孢噻肟酸,然后在三甲基碘硅烷的作用下,对c4位羧基进行硅烷化保护,再与5,6,7,8-四氢喹啉亲核反应得到产物头孢喹肟。美国专利us4845087就是采用这条路线合成头孢喹肟的。合成过程中使用了大量的三甲基碘硅烷和5,6,7,8-四氢喹啉等试剂,并且要求反应条件无水无氧,反应条件苛刻,反应体系复杂,产生杂质多,所得产品纯度和色级均不符合质量要求。
在合成路线二中,也是以7-aca为起始原料,先对其c4位羧基和c7位氨基进行保护,7-aca的c3位乙酰氧基在三甲基碘硅烷的作用下与5,6,7,8-四氢喹啉反应,制得中间体,然后再和ae活性酯反应得到产物头孢喹肟。美国专利us4667028采用了该条合成路线。在此合成路线中,使用了大大过量的三甲基碘硅烷来保护7-aca结构上的c4位羧基和c7位氨基,极大增加了成本,且反应过程中生成了大量的副产物,增加了后续产品分离提纯的困难,造成产品收率低、质量差。
在合成路线三中,使用gcle(7-苯乙酰胺基-3-氯甲基头孢烷酸对甲氧基苄酯)为起始原料。鉴于gcle结构中3位氯甲基的反应活性要优于7-aca结构中的3位乙酰氧甲基,且c4位羧基和c7位氨基都已被保护,使得c3位进行的亲核取代反应更专一。此路线虽然避免了三甲基碘硅烷使用,但后续要增加c4位和c7位上的保护基团的脱去反应,反应路线延长,工艺操作复杂,使用的化学原料增多,造成副反应多,目标产物收率偏低。
由于合成头孢菌素的原料具有特殊结构,使用一些特殊的试剂,导致合成中存在许多副反应,产生较多副产物,从而造成分离纯化困难,导致头孢喹肟的收率低。这样不仅使生产成本高,而且对环境污染比较严重,因此,寻求头孢喹肟具有工业化价值的新型合成工艺具有重要的意义。
技术实现要素:
为解决合成头孢喹肟技术的种种不足,本发明提供了一种头孢烯类鎓盐化合物,化学名称为7-β-氨基-3-(5,6,7,8-四氢喹啉鎓基)甲基-3-头孢烯-4-羧酸盐。
本发明的第二个目的是提供一种头孢烯类鎓盐化合物的制备方法。
本发明的第三个目的是提供一种以头孢烯类鎓盐化合物合成头孢喹肟的方法。
以本发明提供的头孢烯类鎓盐化合物为中间体制备头孢喹肟,反应不需要昂贵试剂,反应步骤少,无需特殊设备,反应条件温和易控,收率高,可控制较低的生产成本和减少环境污染,适宜工业化生产,并且制得的头孢喹肟纯度高,产品质量稳定。
本发明是通过如下技术方案实现的:
一种头孢烯类鎓盐化合物,化学名称为7-β-氨基-3-(5,6,7,8-四氢喹啉鎓基)甲基-3-头孢烯-4-羧酸盐,其结构式如式i所示。
其中,x为酸根阴离子,所述的酸根阴离子为硫酸根、乙酸根、硝酸根、三氟乙酸根、烷基磺酸根、氯离子、溴离子或碘离子之一;n为1或2;
所述的头孢烯类鎓盐化合物以式i的游离形式或水合物形式或溶剂化物形式存在。
对式i所述化合物进行结构确证,通过核磁共振、质谱和红外等测定确证结构为式i所示的化合物,以下数据是以化合物硫酸鎓盐为例。
核磁共振数据如下:
1hnmr(500mhz,cdcl3),
δ3.70,3.79(d,j=7.5hz,2h,sch2),4.2(abq,j=13.5hz,1hn+ch2),4.57(abq,j=14.0hz,1hn+ch2),5.12(s,3h,c-6,β-lactam),5.15(s,2h,coch2),5.41(dd,j=4.0hz,j=8.5hz,1h,c-7,β-lactam),6.68(m,2h,cooch2),8.48-8.90(m,9h,pyridninum-h)。
13cnmr(500mhz,cdcl3),
δ64.9,164.1,58.7,26.2,121.7,155.0,144.2,142.7,126.1,127.1,140.6,26.5,28.6,23.0,22.7,161.9,49.1。
其质谱数据为:ms[m+]444.50,[m++1]445.50。
其红外吸收峰数据为:
ir(kbr,max,cm-1):3416,3180,2948,2920,1775,1675,1612,1585,1455,1244。
本发明的第二个目的是提供一种头孢烯类鎓盐化合物的制备方法。
本发明头孢烯类鎓盐化合物的制备方法是采用7-aca为起始原料,用n,n-六甲基二硅氮烷或三甲基氯硅烷对c4位羧基和c7位上的氨基进行硅烷化保护,保护后加入三甲基碘硅烷对c3位的乙酰氧基进行碘代,然后加入5,6,7,8-四氢喹啉进行亲核取代反应,最后加入有机溶剂进行析晶,得到的产物和酸根离子成盐,从而完成如式i所示的化合物的制备。
一种头孢烯类鎓盐化合物的制备方法,包括步骤如下:
1)在有机溶剂中,将7-aca用硅烷化试剂进行保护,得到硅烷化保护的7-aca反应液;
2)向硅烷化保护的7-aca反应液中加入三甲基碘硅烷进行碘代反应,得碘代后的反应液,碘代后的反应液不经分离直接进行下一步;
3)在有机胺的催化下,向碘代后的反应液加入5,6,7,8-四氢喹啉进行亲核取代反应,反应结束后,脱去硅烷化保护基团,然后加入溶剂析出固体;
4)析出的固体与酸根离子成盐,加入有机溶剂析晶,即可得到式i所示的化合物。
上述步骤1)、步骤2)反应液都不经分离,直接进行下一步反应。
优选的,步骤1)中,所述有机溶剂为腈类、卤代烷烃类或腈类与卤代烷烃类的混合溶剂。
优选的,所述的腈类选自乙腈;卤代烷烃类选自二氯甲烷、氯仿或四氯化碳中的任意两种或两种以上混合。
优选的,步骤1)中,硅烷化试剂为n,n-六甲基二硅氮烷或三甲基氯硅烷与三乙胺的混合。
优选的,步骤1)中,硅烷化试剂选用n,n-六甲基二硅氮烷时,7-aca与n,n-六甲基二硅氮烷的摩尔比为1:0.5~20,优选的,7-aca与n,n-六甲基二硅氮烷的摩尔比为1:0.5~5。
优选的,步骤1)中,硅烷化试剂选用三甲基氯硅烷与三乙胺的混合时,7-aca:三甲基氯硅烷:三乙胺的摩尔比为1:0.5~10:0.5~10,优选的,7-aca:三甲基氯硅烷:三乙胺的摩尔比为1:0.5~5:0.5~5。
优选的,步骤1)中,7-aca与有机溶剂的质量体积比为:1:(4~20),单位:g/ml,保护反应为在0~45℃反应1-5小时。
优选的,步骤2)中,7-aca与三甲基碘硅烷的摩尔比为1:0.5~20,优选的,7-aca与三甲基碘硅烷的摩尔比为:1:0.5~10,碘代反应为保温0~10℃反应3-6小时。
优选的,步骤3)中,有机胺为低级脂肪胺,优选的有机胺为三乙胺、三丁胺或正丁胺。
优选的,步骤3)中,7-aca:有机胺:5,6,7,8-四氢喹啉的摩尔比为1:0.01~5:0.05~20,优选的,7-aca:有机胺:5,6,7,8-四氢喹啉的摩尔比为1:0.01~2:0.5~10。
优选的,步骤3)中,亲核取代反应为在0~10℃下反应10-20小时。
优选的,步骤3)中,脱去硅烷化保护基团为加入醇类溶剂进行脱去硅烷化保护基团。
优选的,步骤3)中,析出固体加入的溶剂为酮类、烷烃类或醚类中的一种或两种以上混合。
优选的,步骤3)中,酮类溶剂为丙酮、甲乙酮或戊酮;烷烃类溶剂为石油醚、环己烷或庚烷;醚类溶剂为异丙醚或甲基叔丁基醚。
优选的,步骤4)中,成盐反应温度为-50~100℃,优选的,成盐反应温度为-20~50℃。
优选的,步骤4)中,酸根离子为硫酸根、乙酸根、硝酸根、三氟乙酸根、烷基磺酸根、氯离子、溴离子或碘离子之一。
优选的,步骤4)中,析出的固体与酸根离子的摩尔比为1:0.5~50,优选的,析出的固体与酸根离子的摩尔比为1:0.5~20。
上述头孢烯类鎓盐化合物的制备方法化学反应式如式iv所示:
本发明的第三个目的是提供一种以头孢烯类鎓盐化合物合成头孢喹肟的方法。
一种以头孢烯类鎓盐化合物合成头孢喹肟的方法,以式i所示的化合物作为制备头孢喹肟的中间体,此中间体和ae活性酯进行缩合反应,反应结束后进行酸化结晶后即得头孢喹肟。
一种以头孢烯类鎓盐化合物合成头孢喹肟的方法,包括步骤如下:
采用一锅法反应,将式i所示的化合物投入有机溶剂或有机溶剂与水的混合溶剂中,降至一定温度后,向体系中加入ae活性酯和有机胺进行缩合反应,反应完毕,经过萃取分离,向料液中加入硫酸或盐酸进行酸化,然后加入有机溶剂进行析晶,即可得到头孢喹肟。
合成头孢喹肟的方法中,缩合反应反应温度为-100~100℃,优选的,缩合反应温度为-50~50℃;
合成头孢喹肟的方法中,式i所示的化合物、ae活性酯与硫酸/盐酸的摩尔比为1:0.5~10:0.1~50,优选,式i所示的化合物、ae活性酯与硫酸/盐酸的摩尔比为配比1:0.5~5:0.5~2.0。
合成头孢喹肟的方法中,有机溶剂为低级醇类、腈类、卤代烷烃类或它们的混合溶剂。
优选的,低级醇类为甲醇、乙醇、异丙醇、正丙醇或正丁醇;腈类为乙腈;卤代烷烃类为二氯甲烷、氯仿或四氯化碳中的任意两种或两种以上混合。
合成头孢喹肟的方法中,所用的有机胺为三乙胺、三丁胺或正丁胺。
合成头孢喹肟的方法中,析晶有机溶剂为酮类、烷烃类或醚类。
酮类为丙酮、甲乙酮或戊酮;烷烃类为石油醚、环己烷、正己烷或正庚烷;醚类为异丙醚、异丙醚、甲基叔丁基醚中的任意两种或两种以上混合。
以头孢烯类鎓盐化合物合成头孢喹肟的反应化学式如式v所示:
在制备头孢喹肟的过程中,无需分离中间产物,反应可用hplc监控至反应完全。
本发明的有益效果在于开发出制备头孢喹肟的独特新工艺。本发明提供了如式i所示的头孢烯类鎓盐化合物并通过此化合物来制备头孢喹肟。
式i所示的化合物经过结构鉴别和确定,产品纯度高,质量稳定,收率高,是一种合成头孢产品的新型中间体,以该中间体与ae活性酯一锅法反应合成头孢喹肟,减少了反应步骤和使用的溶剂,降低了生产成本,防治了环境污染。
本发明所制得的硫酸头孢喹肟晶型好,流动性好,质量符合国内外药典要求。
本发明制备头孢喹肟方法中,所用原料试剂廉价易得,工艺条件温和易控,反应简单易行,反应收率高,无需特殊设备,适合工业化生产。
附图说明
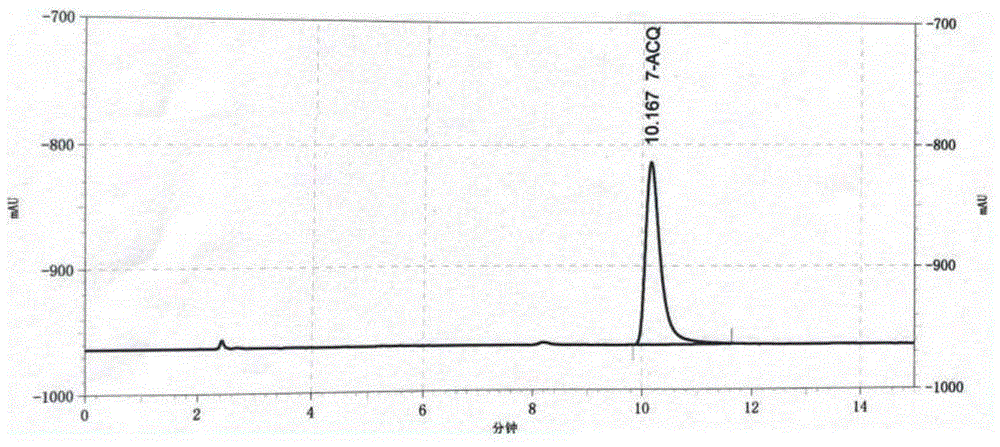
图1为实施例2制备的化合物的一盐酸盐的hplc图谱;
图2为实施例5制备的盐酸头孢喹肟的hplc图谱。
具体实施方式
下面结合实施例对本发明作进一步的说明,但并不能以此作为对本发明保护范围的限制。
实施例1制备式i所示的化合物的二盐酸盐
(1)将27.2g7-aca投入到150ml二氯甲烷中,降温至0~5℃,用时60分钟滴加50ml三乙胺,滴加完毕,然后向料液中滴加27g三甲基氯硅烷,用时90分钟滴加完毕,保温0~5℃反应60分钟,过滤除去固体,用50ml二氯甲烷洗涤,合并滤液;
(2)将合并后的滤液降温至-10~-5℃,加入15ml三乙胺和50g三甲基碘硅烷,保温0~10℃反应3小时。
(3)然后向料液中加入25g5,6,7,8-四氢喹啉,保温反应12小时。然后向料液中滴加异丙醇800ml,逐渐析出固体,搅拌60分钟后过滤,异丙醇洗涤。抽干。
(4)将所得固体加入到100ml水中,加入浓盐酸30ml,固体溶解,然后加入活性炭5g,搅拌脱色30分钟,过滤。将所得滤液滴加到1000ml异丙醇中,析出固体,搅拌30分钟后过滤,适量异丙醇洗涤,抽干,置于真空干燥箱中40℃下干燥,得到式i所示的化合物的二盐酸盐约28g,产品纯度96%,分子结构如下:
实施例2制备式i所示的化合物的一盐酸盐
(1)将27.2g7-aca投入到150ml二氯甲烷中,降温至0~5℃,加入30mln,n-六甲基二硅氮烷和0.1g碘,然后将料液升温至45℃回流反应5小时。
(2)料液降温至0~10℃,加入三乙胺15ml和40g三甲基碘硅烷,保温反应4小时。
(3)然后向料液中加入25g5,6,7,8-四氢喹啉,保温反应15小时。然后向料液中滴加异丙醇800ml,逐渐析出固体,搅拌60分钟后过滤,适量异丙醇洗涤。抽干。
(4)将所得固体加入到100ml水中,加入浓盐酸约10ml,调节料液ph在3.5左右,固体溶解。然后加入活性炭5g,搅拌脱色30分钟,过滤。将所得滤液滴加到800ml丙酮中,析出固体,搅拌30分钟后过滤,适量丙酮洗涤,抽干,置于真空干燥箱中40℃下干燥,得到式i所示的化合物的一盐酸盐约26g,产品纯度96.8%,分子结构如下:
图1为制备的一盐酸盐化合物的hplc图谱。
实施例3式i所示的化合物的对甲基苯磺酸盐的制备
(1)将27.2g7-aca投入到150ml二氯甲烷中,降温至0~5℃,加入30mln,n-六甲基二硅氮烷和0.1g碘。然后将料液升温至45℃回流反应5小时。
(2)料液降温至0~10℃,加入三乙胺15ml和40g三甲基碘硅烷,保温反应4小时。
(3)然后向料液中加入25g5,6,7,8-四氢喹啉,保温反应15小时。然后向料液中滴加正丁烷800ml,逐渐析出固体,搅拌60分钟后过滤,适量正丁烷洗涤。抽干。
(4)将所得固体加入到120ml水中,加入对甲基苯磺酸约20g,调节料液ph在3.0~3.5左右,固体溶解。然后加入活性炭5g,搅拌脱色30分钟,过滤。将所得滤液滴加到1200ml丙酮中,析出固体,搅拌30分钟后过滤,适量丙酮洗涤,抽干,置于真空干燥箱中40℃下干燥,得到式i所示的化合物的对甲基苯磺酸盐约22g,产品纯度95%,分子结构如下:
实施例4硫酸头孢喹肟的制备
将所得的式i所示的化合物硫酸盐20g投入到100ml水和100mldmf的混合液中,降温至5~10℃,加入ae活性酯30g,然后滴加正丁胺约30ml,滴加完毕,料液澄清。保温5℃反应4小时,取样hplc检测至反应合格。
向料液中加入二氯甲烷200ml×3洗涤,保留水相。向所得水相中加入40%硫酸20ml,加入活性炭5g,搅拌脱色30分钟,过滤,然后向滤液中滴加异丙醇1500ml,析出固体。搅拌60分钟,过滤,适量异丙醇洗涤,抽干,40℃下真空干燥至水分≤1.0%,得到白色硫酸头孢喹肟约22g,收率83%,产物纯度98.8%。
图2为制备的硫酸头孢喹肟的hplc图谱。
实施例5盐酸头孢喹肟的制备
将实施例2制得的式i所示的化合物的一盐酸盐15g和ae活性酯20g,投入到200ml二氯甲烷中,降温至0~5℃,用时90分钟滴加三乙胺20ml,滴加完毕,保温反应4小时。取样hplc检测至反应合格。然后向料液中加入纯化水200ml,搅拌15分钟后萃取分层,保留水相。
向所得水相中加入活性炭3g,搅拌脱色30分钟,过滤,30ml水洗涤滤饼。合并滤液,向所得滤液中加入浓盐酸约30ml,搅拌均匀,将料液滴加到1800ml丙酮中,析出固体,搅拌45分钟后过滤,适量丙酮洗涤,抽干,40℃下真空干燥至水分≤1.0%,得到白色盐酸头孢喹肟约18g,收率86%,产物纯度98.5%。
- 还没有人留言评论。精彩留言会获得点赞!