一株高产丙二酰辅酶A的基因工程菌及其构建方法和应用与流程
本发明属于基因重组技术领域,具体涉及一株高产丙二酰辅酶a的基因工程菌及其构建方法和应用。
背景技术:
丙二酰辅酶a(malonylcoenzymea)又称丙二酸单酰辅酶a,是一种辅酶a的衍生物,是微生物必须代谢途径中的关键化合物。在大肠杆菌(escherichiacoli)代谢网络中,以葡萄糖为碳源,经过糖酵解途径生成乙酰辅酶a,大部分乙酰辅酶a经过柠檬酸循环代谢为二氧化碳和水,少部分乙酰辅酶a通过脂肪酸代谢途径被乙酰辅酶a羧化酶转化为丙二酰辅酶a,广泛参与到脂质、碳水化合物和氨基酸等多种重要物质的代谢活动中。同时,丙二酰辅酶a还是重要的信号分子,在许多生物体内参与脂质生物合成的转录调控和脂肪酸生物合成的转录调控。
丙二酰辅酶a在动植物体内均广泛参与多种代谢产物的合成。在脂肪酸合成中丙二酰辅酶a为脂肪酸提供二碳单位,将脂肪酸碳链延伸两个碳单位。枯草芽孢杆菌(bacillussubtilis)中存在丙二酰辅酶a-fapr的调控系统,当胞内丙二酰辅酶a浓度充足时会解开fapr对脂肪酸合成基因的抑制,促进脂肪酸合成基因表达。同时丙二酰辅酶a参与了动物体内脂肪的沉积活动,乙酰辅酶a和丙二酰辅酶a在脂肪酸合酶的催化下转变为甘油三酯,丙二酰辅酶a作为脂肪代谢的重要中间产物,在脂肪细胞的代谢过程中也起着重要的调控作用。丙二酰辅酶a还参与了植物体内黄酮类化合物、多酮类化合物的合成,利用相关途径基因的组装可以在微生物体内实现这些高值化合物的微生物法合成,受限于微生物胞内较低的丙二酰辅酶a浓度,丙二酰辅酶a成为微生物法合成姜黄素、柚皮素、白藜芦醇等高价值化合物的关键限速前体物质,其中黄酮类化合物因具有极高的药理功能而受到广泛关注。
为了提高胞内丙二酰辅酶a的浓度,突破黄酮类化合物微生物法合成瓶颈,前人已经做了许多尝试。大肠杆菌通过乙酰辅酶a羧化酶将胞内乙酰辅酶a转化为丙二酰辅酶,大肠杆菌的乙酰辅酶a羧化酶包括生物素羧化酶亚基、生物素羧基载体蛋白亚基(bccp)、羧基转移酶α和β亚基共四个亚基,lu等通过过表达大肠杆菌自身的乙酰辅酶a羧化酶,提高了胞内乙酰辅酶a转化效率,提高了胞内丙二酰辅酶a的含量。但是有研究表明大肠杆菌的生物素羧基载体蛋白亚基会对胞内乙酰辅酶a羧化酶形成负反馈调节,降低胞内乙酰辅酶a羧化酶含量。因此miyahisa等通过过表达谷氨酸棒状杆菌(corynebacteriumglutamicum)由accbc和dtsr1基因编码的两个乙酰辅酶a羧化酶亚基大幅提高胞内丙二酰辅酶a的浓度,使柚皮素和生松素产量达到60mg/l。
胞内乙酰辅酶a参与柠檬酸循环,也是重要的中间代谢产物,过表达乙酰辅酶a羧化酶必然会影响到胞内乙酰辅酶a的供给,因此leonard等通过过表达乙酰辅酶a合成酶将大肠杆菌代谢的副产物乙酸转化为乙酰辅酶a,为丙二酰辅酶a合成提供充足的前体物质。乙酸在体系中部分电离,未电离的乙酸进入细胞会减弱质子推动力,影响atp的生成进而影响菌体生长和重组蛋白的表达,这一途径在降低了环境中乙酸的浓度的同时又保证了胞内乙酰辅酶a的供给。在大肠杆菌代谢网络中,乙酰辅酶a也会流向一些非必需的副产物,导致胞内乙酰辅酶a含量的降低。这些研究均一定程度上提高了微生物胞内的丙二酰辅酶a的含量,但是对于途径的相关基因缺乏深入的挖掘,对于多种途径缺少有效的整合。
技术实现要素:
发明目的:针对现有技术存在的问题,本发明提供一株高效积累丙二酰辅酶a的基因工程菌,本发明产丙二酰辅酶a的基因工程菌为经过底盘微生物改造的产丙二酰辅酶a的基因工程菌,通过对工程菌内部乙酰辅酶a流出途径进行抑制,并将丙二酰辅酶a合成途径构建到不同的表达载体转入工程菌,实现丙二酰辅酶a的高效积累。本发明的工程菌可以以大肠杆菌的代谢副产物乙酸为底物,高效合成黄酮类化合物的前体物质丙二酰辅酶a,并利用该菌突破黄酮类化合物合成途径的瓶颈,提高微生物法合成黄酮类化合物的效率。
本发明还提供一株高产丙二酰辅酶a的基因工程菌的构建方法和应用。
技术方案:为了实现上述目的,如本发明所述一株高产丙二酰辅酶a的基因工程菌,所述基因工程菌由敲除大肠杆菌基因组中ldha、pta、frda、poxb、adhe五个基因,再导入丙二酰辅酶a合成途径基因构建而成,所述丙二酰辅酶a合成途径基因包括大肠杆菌的乙酰辅酶a合成酶基因acs,肠沙门氏菌的乙酰辅酶a羧化酶基因基因seaccacd和谷氨酸棒状杆菌的生物素连接酶基因cgbira,所述肠沙门氏菌的乙酰辅酶a羧化酶基因seaccacd包括三个亚基seacca、seaccc、seaccd。
其中,所述ldha(geneid:946315)、pta(geneid:946778)、frda(geneid:948667)、poxb(geneid:946132)、adhe(geneid:945837)五个基因分别为流向乳酸的竞争代谢途径相关基因ldha,流向乙酰磷酸的竞争代谢途径相关基因pta,流向琥珀酸的竞争代谢途径相关基因frda,流向乙酸的竞争代谢途径相关基因poxb,流向乙醇的竞争代谢途径相关基因adhe。
其中,所述大肠杆菌的乙酰辅酶a合成酶基因acs转录翻译合成乙酰辅酶a合成酶将乙酸转化为乙酰辅酶a;肠沙门氏菌的乙酰辅酶a羧化酶基因seaccacd转录翻译合成乙酰辅酶a羧化酶将乙酰辅酶a转化为丙二酰辅酶a;谷氨酸棒状杆菌的生物素连接酶基因cgbira转录翻译合成生物素连接酶辅助提高乙酰辅酶a羧化酶的活力。
本发明所述产丙二酰辅酶a的基因工程菌为经过底盘微生物改造的产丙二酰辅酶a的基因工程菌,所述底盘微生物改造包括对ldha、pta、adhe、poxb、frda基因进行了敲除;同时在工程菌内部构建了一个全新的丙二酰辅酶a的合成途径和全新的丙二酰辅酶a合成途径基因,所述丙二酰辅酶a合成基因包括大肠杆菌的乙酰辅酶a合成酶基因acs,肠沙门氏菌的乙酰辅酶a羧化酶亚基seacca、seaccc、seaccd,谷氨酸棒状杆菌的生物素连接酶基因cgbira。
本发明中ldha、pta、adhe、poxb、frda基因为乙酰辅酶a流向乳酸、乙酰磷酸、乙醇、乙酸和琥珀酸的基因,敲除后抑制了乙酰辅酶a的流出,提高了胞内乙酰辅酶a的含量,乙酰辅酶a作为丙二酰辅酶a的直接前体物质,可以间接提高胞内丙二酰辅酶a的含量;大肠杆菌的乙酰辅酶a合成酶acs可以将代谢副产物乙酸转化为乙酰辅酶a,进一步提高胞内乙酰辅酶a的含量,肠沙门氏菌的乙酰辅酶a羧化酶seaccacd可以将乙酰辅酶a转化为丙二酰辅酶a,提高胞内丙二酰辅酶a的含量。谷氨酸棒状杆菌的生物素连接酶cgbira可以协助seaccacd发挥活力,进一步提高胞内丙二酰辅酶a的含量。丙二酰辅酶a含量从0.085nmol/mgdcw提升至0.812nmol/mgdcw。
本发明所述的丙二酰辅酶a合成途径,所述途径包括由乙酰辅酶a合成酶将微生物的代谢副产物乙酸合成乙酰辅酶a,提高胞内乙酰辅酶a的含量,然后利用乙酰辅酶a羧化酶将乙酰辅酶a转化为丙二酰辅酶a,同时共表达生物素连接酶进一步提高乙酰辅酶a羧化酶的活力,将乙酰辅酶a高效转化为丙二酰辅酶a。
本发明的表达载体,包含所述的大肠杆菌的乙酰辅酶a合成酶基因acs,肠沙门氏菌的乙酰辅酶a羧化酶基因seaccacd和谷氨酸棒状杆菌的生物素连接酶基因cgbira。
进一步地,所述的表达载体包含所述的丙二酰辅酶a合成途径,所述基因工程菌包含丙二酰辅酶a合成途径的表达载体包括由acs、seaccacd、cgbira形成的载体o-acs-cgbira和a-seaccacd。其中载体o-acs-cgbira包含两个基因acs和cgbira;载体a-seaccacd包含基因seaccacd,或是形成的质粒pcola-acs-cgbira和pacyc-seacca-seaccc-sseaccd。
更进一步地,所述的表达载体将大肠杆菌的乙酰辅酶a合成酶基因acs,肠沙门氏菌的乙酰辅酶a羧化酶基因seaccacd和谷氨酸棒状杆菌的生物素连接酶基因cgbira构建到表达载体上便于在微生物体内复制以及表达。
本发明丙二酰辅酶a合成途径主要是将表达载体o-acs-cgbira和a-seaccacd一起导入经过底盘改造的基因工程菌内。
本发明所述的一株高产丙二酰辅酶a的基因工程菌的构建方法,包括如下步骤:
(1)运用crispr/cas9方法分别敲除大肠杆菌基因组中ldha、pta、frda、poxb、adhe五个基因;对敲除五个基因的基因序列设计引物进行扩增、酶切、酶连、转化得到基因敲除改造后的菌株;
(2)以大肠杆菌的基因组为模板设计引物,扩增得到大肠杆菌中编码乙酰辅酶a合成酶的acs基因,将其导入步骤(1)基因敲除改造后的菌株中得到含有大肠杆菌中编码乙酰辅酶a合成酶的acs基因的菌株;
(3)以肠沙门氏菌基因组基因组为模板设计引物,扩增得到肠沙门氏菌的乙酰辅酶a羧化酶基因,将其导入步骤(2)含有大肠杆菌中编码乙酰辅酶a合成酶的acs基因的菌株中得到含有大肠杆菌中编码乙酰辅酶a合成酶的acs基因和肠沙门氏菌的乙酰辅酶a羧化酶基因的菌株;
(4)以谷氨酸棒状杆菌基因组为模板设计引物,扩增得到谷氨酸棒状杆菌的生物素连接酶基因cgbira,将其导入步骤(3)含有大肠杆菌中编码乙酰辅酶a合成酶的acs基因和肠沙门氏菌的乙酰辅酶a羧化酶基因的菌株中得到高产丙二酰辅酶a的基因工程菌。
上述方法为本发明高产丙二酰辅酶a的基因工程菌构建示意流程方法,在实际构建中可以将三个基因同时导入基因敲除改造后的菌株;或者分开导入基因敲除改造后的菌株;或者是将如谷氨酸棒状杆菌的生物素连接酶基因cgbira克隆到含有大肠杆菌中编码乙酰辅酶a合成酶acs基因的载体上,与含肠沙门氏菌的乙酰辅酶a羧化酶三个亚基基因的载体转入步骤(1)经过基因敲除的基因工程菌中共表达得到高产丙二酰辅酶a的基因工程菌。
本发明所述的高产丙二酰辅酶a的基因工程菌在高效积累丙二酰辅酶a中的应用。其中,胞内丙二酰辅酶a含量从0.085nmol/mgdcw提升至0.812nmol/mgdcw。
本发明所述的高产丙二酰辅酶a的基因工程菌在提高黄酮类化合物微生物法合成效率中的应用,将柚皮素途径基因导入高产丙二酰辅酶a的基因工程菌中,柚皮素微生物法合成的产量从16mg/l提升至252.31mg/l。即本发明的基因工程菌可以利用胞内较高浓度的丙二酰辅酶a,提高微生物法合成黄酮类化合物骨架前体物质柚皮素的产量,将柚皮素的产量从16mg/l提升至252.31mg/l。
本发明针对微生物胞内丙二酰辅酶a较低,无法满足黄酮类化合物合成所需的缺点,首先对发酵菌株进行底盘改造,敲除5个乙酰辅酶a流出途径基因,抑制乙酰辅酶a的消耗,提高胞内乙酰辅酶a的含量,再设计利用乙酰辅酶a合成酶将微生物的代谢副产物乙酸合成乙酰辅酶a,进一步提高胞内乙酰辅酶a的含量,然后利用乙酰辅酶a羧化酶将乙酰辅酶a转化为丙二酰辅酶a,同时共表达生物素连接酶进一步提高乙酰辅酶a羧化酶的活力,将乙酰辅酶a高效转化为丙二酰辅酶a。本发明首次在基因工程菌中组合应用一系列策略,综合提高胞内丙二酰辅酶a的含量,同时首次优选大肠杆菌的乙酰辅酶a合成酶有最高的乙酰辅酶a合成活力,同时肠沙门氏菌的三个乙酰辅酶a羧化酶亚基在谷氨酸棒状杆菌的生物素连接酶协助下可以有很高的丙二酰辅酶a合成活力,胞内丙二酰辅酶a含量从起始的0.085nmol/mgdcw提升至0.812nmol/mgdcw。
总体而言本发明主要体现在以下5个方面:
1、本发明的第一个方面是提供一个可以将乙酸合成丙二酰辅酶a的全新途径以及全新的丙二酰辅酶a合成途径基因。
合成途径包括由乙酰辅酶a合成酶将微生物的代谢副产物乙酸合成乙酰辅酶a,提高胞内乙酰辅酶a的含量,然后利用乙酰辅酶a羧化酶将乙酰辅酶a转化为丙二酰辅酶a,同时共表达生物素连接酶进一步提高乙酰辅酶a羧化酶的活力,将乙酰辅酶a高效转化为丙二酰辅酶a。
丙二酰辅酶a合成途径基因包括大肠杆菌自身的乙酰辅酶a合成酶,肠沙门氏菌的乙酰辅酶a羧化酶a亚基、肠沙门氏菌的乙酰辅酶a羧化酶c亚基,肠沙门氏菌的乙酰辅酶a羧化酶d亚基,谷氨酸棒状杆菌的生物素连接酶,。
2、本发明的第二个方面是提供一株高效积累丙二酰辅酶a的基因工程菌,可以以乙酸为底物高效合成丙二酰辅酶a,提高胞内丙二酰辅酶a的含量。
3、本发明第三个方面是提供一种本发明第一个方面所述的高效积累丙二酰辅酶a的底座细胞(即基因工程菌)的构建方法,包括以下方面:(1)大肠杆菌胞内乙酰辅酶a代谢流的调节(即基因敲除);(2)过表达乙酰辅酶a合成酶;(3)过表达乙酰辅酶a羧化酶;(4)过表达生物素连接酶。
4、本发明的第四个方面是提供一种表达载体,将高效积累丙二酰辅酶a相关基因构建到表达载体上便于在微生物体内复制以及表达。
5、本发明的第五个方面是本发明构建的能高效积累丙二酰辅酶a的基因工程菌的应用,将大肠杆菌的代谢副产物乙酸转化为丙二酰辅酶a,提高了工程菌胞内丙二酰辅酶a的含量,可以用于黄酮类化合物及多酮类化合物的微生物法合成,能有效突破途径的瓶颈步骤,提高合成效率。
有益效果:与现有技术相比,本发明具有如下优点:
本发明对于途径的相关基因进行深入的挖掘,对于多种提高胞内丙二酰辅酶a含量的途径进行有效的整合。本发明构建了一株全新的高产丙二酰辅酶a的基因工程菌和将乙酸转化为丙二酰辅酶a的全新途径。本发明对基因工程菌进行底盘改造,同时敲除5个乙酰辅酶a流出途径基因,胞内丙二酰辅酶a含量从起始的0.085nmol/mgdcw提高至0.190nmol/mgdcw;筛选出最高效率的乙酰辅酶a合成酶,在底盘改造过的基因工程菌中表达,将胞内丙二酰辅酶a浓度0.085nmol/mg提高到0.406nmol/mgdcw;在优选的乙酰辅酶a合成酶基础上再筛选出最高效率的乙酰辅酶a羧化酶和生物素连接酶,将胞内的丙二酰辅酶a含量从起始的0.085nmol/mgdcw提升至0.812nmol/mgdcw;利用乙酰辅酶a合成酶将代谢副产物乙酸转化为乙酰辅酶a,提高胞内乙酰辅酶a浓度的同时降低了乙酸含量,避免乙酸的积累对基因工程菌造成毒性。以高产丙二酰辅酶a的基因工程菌作为底座细胞,将柚皮素合成途径转入基因工程菌中,柚皮素的产量从16mg/l提升至252.31mg/l。
本发明的工程菌可以以大肠杆菌的代谢副产物乙酸为底物,高效合成黄酮类化合物的前体物质丙二酰辅酶a,并利用该菌突破黄酮类化合物合成途径的瓶颈,提高微生物法合成黄酮类化合物的效率。
附图说明
图1乙酰辅酶a代谢途径改造对胞内乙酰辅酶a和丙二酰辅酶a含量影响示意图;
图2过表达不同来源的乙酰辅酶a合成酶对胞内乙酰辅酶a和丙二酰辅酶a含量影响示意图;
图3过表达不同来源的乙酰辅酶a羧化酶对胞内丙二酰辅酶a含量影响示意图;
图4过表达不同来源的生物素连接酶对胞内丙二酰辅酶a浓度的影响示意图;
图5柚皮素高效液相色谱图。
具体实施方式
以下结合附图和实施例作进一步说明。
本发明中所有原料、试剂、载体、菌株、限制性内切酶等如无特殊说明,均市售可得。本发明中基因编号(geneid)均为ncbi上编号。
实施例1
敲除5个乙酰辅酶a流出途径基因的底座细胞构建
菌株和质粒
大肠杆菌(escherichiacolijm109)用于质粒的复制,大肠杆菌escherichiacolibl21(de3)用于途径质粒的表达及菌株发酵,菌株购置于novagen公司(达姆施塔特,德国)。质粒pcdfduet-1、petduet-1、pcoladuet-1、pacycduet-1均购自于novagen公司(达姆施塔特,德国)。
酶及其他试剂
各种抗生素包括氨苄青霉素和氯霉素均购自于上海生工有限公司。各种化学试剂均为分析纯,购自于上海生工有限公司。各种限制性内切酶和dna连接酶购自于thermo公司。1kbdnaladder、质粒小量提取试剂盒、胶回收试剂盒、细菌基因组提取试剂盒购自于上海生工有限公司。
采用上海生工有限公司质粒小提试剂盒进行质粒提取操作。采用上海生工有限公司基因组提取试剂盒进行基因组提取操作。
质粒的酶切和连接按照thermo公司的限制性内切酶和连接酶试剂盒使用说明书进行操作。一步法克隆按照诺维赞公司的onestepkit试剂盒使用说明书进行操作。
同时本发明实施例中pcr扩增,检测,酶连,转化,表达等方法均为常规的分子生物学方法。
主要仪器设备(见表1)
表1使用到的仪器
培养基的配制
lb(luriabroth)液体培养基:10g/l胰蛋白胨,5g/l酵母提取物,10g/lnacl,ph7.0;121℃高压蒸汽灭菌20min。
lb固体培养基:在lb(luriabroth)液体培养基基础上添加2%琼脂粉;121℃高压蒸汽灭菌20min。
发酵培养基:
10×mops母液:(1)77.76gmops(potassiummorpholinopropanesulfonate)加300ml去离子水溶解后用koh调ph至7.4,用去离子水定容至400ml,用0.45μl无菌水系滤膜过滤;(2)7.1668gtricine(n-tris(hydroxymethyl)-methylglycine)加30ml去离子水溶解后用koh调ph在7.4;(3)0.0278gfeso4·7h2o;5.08156gnh4cl;0.481gk2so4;29.22gnacl加入400ml去离子水溶解,121℃高压下蒸汽灭菌20min;(4)微量元素:3.708×10-2g(nh4)6(mo7)24·4h2o,2.4732×10-1gh3bo2,7.1379×10-2gcocl2·6h2o,2.497×10-2gcuso4·5h2o,1.583×10-1gmncl2·3h2o,2.8756×10-2gznso4·7h2o,5.549×10-1gcacl2,加10ml去离子水溶解后过0.45μl滤膜除菌。
400ml组分(1);40ml组分(2);400ml组分(3);1μl组分(4);去离子水定容至1l。
mops培养基:10×mops母液100ml,0.296gk2hpo4·3h2o,5g葡萄糖,4g氯化铵,无菌蒸馏水900ml。121℃高压蒸汽灭菌20min。
crispr/cas9技术敲除改造乙酰辅酶a代谢途径
运用crispr/cas9技术分别敲除大肠杆菌e.colibl21(de3)基因组中ldha、pta、frda、poxb、adhe五个基因,crispr/cas9敲除基因使用到的引物及质粒见表2。
具体操作步骤以敲除基因ldha为例:
以p-target全质粒(takara)为模板,通过引物pf_ldha(knock)(seqidno.1)和pr_ptarget(seqidno.6)进行全质粒pcr引入突变,取代原来sgrna的20bp的n20序列。向pcr体系中加入1μgdpnⅰ,5μlgreenbuffer,37℃反应30min,消化甲基化的模板质粒dna。对dpnⅰ消化后的pcr体系进行1%agrose凝胶电泳,切胶回收目的dna片段,bluntingkination反应磷酸化后,16℃条件下连接16h。将连接液转化至大肠杆菌(escherichiacolijm109)感受态,待转化子长出后挑取菌落测序验证,提取质粒,获得改造过的质粒p-target(ldha)。
对待敲除基因序列的上游500bp序列和下游500bp序列分别设计引物pf_ldha(上游)(bamhi)(seqidno.2)、pr_ldha(上游)(seqidno.3)和pf_ldha(下游)(seqidno.4)、pr_ldha(下游)(xhoi)(seqidno.5)。大肠杆菌(e.colibl21(de3))基因组中待敲除基因序列上游500bp片段和下游500bp片段分别通过引物pf_ldha(上游)(bamhi)、pr_ldha(上游)和pf_ldha(下游)、pr_ldha(下游)(xhoi)由pcr扩增得到。将得到的pcr扩增片段分别进行胶回收,以胶回收纯化后的上游500bp片段和下游500bp片段为模板,通过引物pf_ldha(上游)(bamhi)和pr_ldha(下游)(xhoi)进行融合pcr得到包含待敲除基因序列的上游500bp和下游500bp的融合片段。将该融合片段连入t载体pmdtm19(simple)(takara)中得到t-融合(ldha)质粒。
将pcas质粒(takara)转化到待改造菌株bl21(de3)中。接种bl21(pcas)于5mllb培养基中,向lb培养基中加入5μlkana抗性溶液(终浓度为50μg/ml),过夜震荡培养(30℃,200rpm,12h)。以1%体积比接种量接种于含有50μg/ml的kana抗性溶液的50mllb培养基中,震荡培养(30℃,200rpm)至od600=0.2时加入500μl1mol/l阿拉伯糖(终浓度为10mmol/l)。震荡培养(30℃,200rpm)至od600=0.6,将菌液转移至50mlep管中冰上冷却15min。离心(2500g,10min,4℃)收集菌体,用25ml冷却的无菌水洗涤菌体2次,再用25ml冷却的10%甘油洗涤菌体2次。加入400μl预冷的10%甘油重悬菌体,50μg每管分装后-80℃保存。
通过bamhi/xhoi双酶切获得待敲除基因上游500bp和下游500bp的融合片段,与此同时提取ptarget(ldha)质粒。从-80°冰箱中取出bl21(pcas)感受态细胞冰浴融化,将p-target(ldha)质粒、t-融合(ldha)酶切胶回收片段同时加入bl21(pcas)感受态细胞中,轻柔的吸打混匀后冰浴10min,预冷电击杯,将感受态细胞转移到预冷的2mm点击杯中2.5kv点击,迅速加入1mllb培养基后转移到1.5mlep管中摇床培养(30℃,200rpm,2h)。将菌液涂布到50μg/mlkana抗性、100μg/ml观霉素平板上,倒置培养(30℃,12-16h)培养,挑选长出来的单克隆,送上海生工测序,确定成功敲除基因的阳性克隆。获得敲除大肠杆菌(e.colibl21(de3))基因组中乙酰辅酶a流向乳酸的基因ldha的菌株bl21δldha。其他四个基因pta、adhe、poxb、frda的敲除参照上面方法,各个基因敲除的引物见表2。
根据上述方法,分别敲除大肠杆菌(e.colibl21(de3))基因组5个基因得到:bl21δldha、bl21δpta、bl21δfrda、bl21δpoxb、bl21δadhe五个菌株,以及在大肠杆菌(e.colibl21(de3))基因组连续敲除大肠杆菌(e.colibl21(de3))基因组5个基因得到:bl21δldhaδpta、bl21δdhaδptaδfrda、bl21δldhaδptaδfrdaδpoxb、bl21δldhaδptaδfrdaδpoxbδadhe四个菌株。对基因敲除改造后的菌株进行发酵,如图1所示,最终连续敲除5个基因的菌株bl21δldhaδptaδfrdaδpoxbδadhe的胞内乙酰辅酶a浓度提升至1.436nmol/mgdcw,丙二酰辅酶a浓度提升至0.190nmol/mgdcw。,说明基因敲除的菌株bl21δldhaδptaδfrdaδpoxbδadhe,可以提高一定程度胞内丙二酰辅酶a含量。
表2crispr/cas9敲除基因使用到的引物及质粒
本实施例1中改造的大肠杆菌的制备,发酵,以及乙酰辅酶a含量检测、乙酰辅酶a含量检测如下。
大肠杆菌感受态制备:
本实施例所选用的转基因受体为基因敲除改造后大肠杆菌(escherichiacolibl21δldhaδptaδfrdaδpoxbδadhe),该菌株为噬菌体de3融源型,挑取单菌落于5mllb培养基中,摇床培养(37℃,200rmp)过夜(约12h)。取500μl过夜培养物转接于50ml新鲜lb培养基中,摇床培养(37℃,200rmp)至吸光度在600nm处达到0.35-0.6。将0.1mol/lcacl2溶液置于冰上预冷。吸取25ml菌液至50mlep管中,置于冰上冷却10min。离心(3000g,5min,4℃),弃上清,加入1.666ml预冷0.1mol/lcacl2溶液,用移液枪轻轻上下吸动打匀,使细胞重新悬浮,冰上放置20min。离心(3000g,5min,4℃),弃上清,加入1ml预冷混合溶液(0.1mol/lcacl2混合10%甘油),吸打混匀使细胞重新悬浮。每管100μl细胞悬浮液分装于1.5mlep管中,于-70℃冷冻贮存备用。
质粒转化入基因敲除改造后大肠杆菌:待感受态细胞融化后,向100μl感受态细胞中转入1μl质粒,缓慢轻弹混匀。冰上放置30min后42℃水浴锅中热激90s,冰上放置10min。向感受态细胞ep管中加入1ml新鲜lb培养基,放入摇床培养(37℃,200rpm,1h)。将大肠杆菌全部加入质粒相应的抗性平板中,充分涂布均匀。转化平板放入37℃培养箱倒置培养12-24h。(实施例2-4)
发酵:
工程菌首先在lb培养基中摇床培养(37℃,200rpm)过夜,然后以初始od600为0.1的接种量重新转接到新鲜的mops发酵培养基中,同时向发酵体系内加入终浓度为5g/l的碳源葡萄糖,摇床培养(37℃,200rpm)至od600为0.5。此时以1mmol/l的iptg(isopropyl-β-d-thiogalactopyranoside)终浓度进行诱导,并进行终浓度为5g/l葡萄糖的补料、终浓度为1g/l的底物乙酸钠和终浓度为2.5g/l底物酪氨酸的补料后摇床培养(30℃,200rpm)发酵52h。
乙酰辅酶a含量检测
取发酵液1ml,13000rpm,4℃离心10min,弃上清。用1ml6%的高氯酸重悬裂解细胞,加入0.3ml3mol/l的碳酸钾中和,再13000rpm,4℃离心10min,收集上清作液相-质谱联用仪和agilentzorbaxeclipseplus反向c18色谱柱(4.6mm×250mm,5μm)分析。为了量化细胞干重,取相同的细胞培养物1ml,过0.45μm纤维素滤膜。用蒸馏水洗涤纤维素滤膜,在烘箱中烘干至恒重,细胞干重等于带有细胞残留物膜和空膜的差值。测定胞内乙酰辅酶a所用的lc-ms仪器和色谱柱和测定柚皮素时所用的相同。其中液相条件为:流速为0.3ml/min;流动相a为15mmol/l甲酸铵,b为溶有10%的10mmol/l醋酸铵的甲醇溶液;色谱条件为:在0-10min,2%-60%b,10-20min,60%-76%b;20-25min,76%-2%b。ms/ms系统在负离子模式下运行,检测电压:1.60kv,雾化气(n2)流:1.5l/min,干燥气体流:200kpa,离子积累时间:30ms,ms1的m/z扫描范围:100-1000,ms2的m/z扫描范围:100-500。
丙二酰辅酶a含量测定
取发酵液10ml,在13000rpm,4℃离心10min,弃上清。将得到的细胞重悬在2ml的6%的高氯酸中进行细胞破碎。再加入0.6ml的3mol/l碳酸钾,震荡中和里面的酸。再13000rpm,4℃离心10min,收集上清液进行丙二酰辅酶a浓度的测定。为了测定细胞干重,取2ml相同的细胞液,过0.45μm的纤维素滤膜,将得到的菌体用蒸馏水进行洗涤并在烘箱中烘干至恒重。细胞干重等于空膜和带有细胞残留物膜的差值。
实施例2
过表达不同来源的乙酰辅酶a合成酶
大肠杆菌(escherichiacolijm109)用于质粒的复制,大肠杆菌escherichiacolibl21(de3)用于途径质粒的表达及菌株发酵,菌株购置于novagen公司(达姆施塔特,德国)。质粒pcdfduet-1、petduet-1、pcoladuet-1、pacycduet-1均购自于novagen公司(达姆施塔特,德国)。酿酒酵母atcc9080购自上海钦诚生物科技有限公司、解脂耶氏酵母atcc9773购自上海柯维化学技术有限公司、肠沙门氏菌atcc10723购自北京百欧博伟生物技术有限公司和谷氨酸棒状杆菌atcc13032购自北京强欣博瑞生物技术有限公司,铜绿假单胞菌atcc27853购自上海恒斐生物科技有限公司。
为了得到大肠杆菌中编码乙酰辅酶a合成酶的acs基因(geneid:948572),以大肠杆菌(e.colibl21(de3))的基因组为模板,通过引物pf_acs(ndei)(seqidno.7)和pr_acs(kpni)(seqidno.8)扩增,将得到的acs基因连入t载体pmd19t(simple)(takara)中得到t-acs。通过ndei/kpni双酶切,将acs基因从t-acs克隆到pcoladuet-1的ndei/kpni位点得到pcola-acs质粒;为了得到肠沙门氏菌中编码乙酰辅酶a合成酶的seacs基因(geneid:1250688),以肠沙门氏菌(salmonellaenterica)基因组为模板,通过引物pf_seacs(ndei))和pr_seacs(xhoi)扩增,将得到的野生型seacs基因连入t载体pmd19t(simple)(takara)中得到t-seacs质粒。通过ndei/kpni双酶切,将seacs基因从t-seacs中克隆到petduet-1,得到pcola-seacs质粒。
为了得到酿酒酵母(saccharomycescerevisiae)基因组中编码乙酰辅酶a合成酶的基因scacs1(geneid:851245)和scacs2(geneid:850846),以酿酒酵母基因组为模版,通过引物pf_scacs1(ndei)、pr_scacs1(xhoi)和pf_scacs2(ndei)、pr_scacs2(xhoi)扩增,将得到的基因连入t载体pmd19t(simple)(takara)中得到t-scacs1和t-scacs2。通过ndei/kpni双酶切,将基因从t-scacs1和t-scacs2克隆到pcoladuet-1的ndei/kpni位点得到pcola-scacs1和pcola-scacs2;为了得到解脂耶氏酵母(yarrowialipolytica)基因组中编码乙酰辅酶a合成酶的基因ylacs(guoh,madzakc,dug,etal.effectsofpyruvatedehydrogenasesubunitsoverexpressionontheα-ketoglutarateproductioninyarrowialipolyticawsh-z06[j].applmicrobiolbiotechnol,2014,98(16):7003-7012),通过引物pf_ylacs(ndei)和pr_ylacs(xhoi)扩增,以解脂耶氏酵母基因组为模板,将得到的基因连入t载体pmd19t(simple)(takara)中得到t-ylacs。通过ndei/kpni双酶切,将基因从t-ylacs克隆到pcoladuet-1的ndei/kpni位点得到pcola-ylacs。
将上述不同来源的含乙酰辅酶a合成酶基因的质粒转化到实施例1进行底盘改造的菌株bl21δldhaδptaδfrdaδpoxbδadhe中进行表达发酵,并取发酵液测定丙二酰辅酶a含量,比较不同来源的乙酰辅酶a合成酶对于提高胞内丙二酰辅酶a含量的影响(参考实施例1中的发酵和检测方法),发现大肠杆菌自身的乙酰辅酶a合成酶活力最高,如图2所示,胞内乙酰辅酶a浓度为5.216nmol/mgdcw,丙二酰辅酶a浓度为0.406nmol/mgdcw,说明导入乙酰辅酶a合成酶可以有效提高胞内丙二酰辅酶a含量;说明导入大肠杆菌乙酰辅酶a合成酶基因acs可以提高胞内丙二酰辅酶a浓度。
同时本发明还构建从丙二酸到丙二酰辅酶a的途径,通过过量表达来自三叶草根瘤菌的丙二酸单酰辅酶a合成酶基因matb和丙二酸载体蛋白基因matc,将胞外的丙二酸转移至胞内并转化为丙二酰辅酶a。利用来自三叶草根瘤菌的matb和matc基因构建从丙二酸到丙二酰辅酶a的通路,实现丙二酸从胞外到胞内转运以及向丙二酰辅酶a的转化,在实施例2相同的条件下将胞内丙二酰辅酶a浓度从0.085nmol/mgdcw提升到了0.174nmol/mgdcw。由于丙二酸途径的低效性和丙二酸的价格无法应用于工业生产,因此进一步探索乙酸途径对丙二酰辅酶a的影响。
实施例3
过表达不同来源的乙酰辅酶a羧化酶
为了得到来自谷氨酸棒状杆菌(corynebacteriumglutamicum)的乙酰辅酶a羧化酶dtsr1(geneid:1018707)和accbc(geneid:1019303),利用引物pf_accbc(bamhi)/pr_accbc(hindiii)和pf_dtsr1(ndei)/pr_dtsr1(kpni),以谷氨酸棒状杆菌基因组为模板扩增,将得到的基因连入t载体pmd19t(simple)(takara)中得到t-dtsr1和t-accbc,通过ndei/kpni双酶切将t-dtsr1克隆到pacycduet-1的ndei/kpni处,得到pacyc-dtsr1。通过bamhi/hindiii双酶切将基因从t-accbc克隆到pacyc-dtsr1的bamhi/hindiii位点处,得到质粒pacyc-dtsr1-accbc。
为了得到来自铜绿假单胞菌(pseudomonasaeruginosa)的乙酰辅酶a羧化酶基因paacc(geneid:880489),利用引物pf_paacc(ndei)和pr_paacc(bglii),以铜绿假单胞菌基因组为模板扩增,将得到的基因连入t载体pmd19t(simple)(takara)中得到t-paacc,通过ndei/bglii双酶切将基因从t-paacc克隆到pacycduet-1的ndei/bglii位点处,得到质粒pacyc-paacc。
为了得到来自肠沙门氏菌(salmonellaenterica)的乙酰辅酶a羧化酶的不同亚基seacca(geneid:1251750)、seaccb(geneid:1254902)、seaccc(geneid:1254903)、seaccd(geneid:1253888),利用引物pf_seacca(ncoi)/pr_seacca(bamhi)(seqidno.9-10)、pf_seaccb(ncoi)/pr_seaccb(bamhi)(seqidno.11-12)、pf_seaccc(ncoi)/pr_seaccc(bamhi)(seqidno.13-14)和pf_seaccd(ncoi)/pr_seaccd(bamhi)(seqidno.15-16),以肠沙门氏菌基因组为模板扩增,将得到的基因连入t载体pmd19t(simple)(takara)中得到t-seacca、t-seaccb、t-seaccc、t-seaccd,通过ncoi/bamhi双酶切将基因从t载体克隆到pacycduet-1的ncoi/bamhi处,得到质粒pacyc-seacca、pacyc-seaccb、pacyc-seaccc、pacyc-seaccd,为了将seaccb连入pacyc-seacca,进行一步法克隆。1、载体线性化:对pacyc-seacca载体进行bamhi/ecori双酶切线性化,胶回收。2、pcr扩增:设计引物pf_t7seaccb(bamhi)/pr_t7seaccb(ecori)(seqidno.17-18)扩增,在引物的5’端和3’最末端加入15bp载体的同源序列(同源序列是表达载体酶切位点两端的序列,便于基因片段进行同源重组)。3、插入片段的pcr扩增:用设计的引物对连入表达载体的seaccb进行扩增,扩增出t7启动子和seaccb基因,胶回收,pcr扩增体系见表3。
表3pcr扩增体系
4、重组反应体系配置
在冰水浴中配置重组反应体系。移液枪轻柔吹打混匀,37℃金属浴反应30min,反应完立即置于冰水浴中5min,-20℃冰箱保存,重组反应体系见表4。
表4重组反应体系
5、转化
取10μl重组液加入jm109感受态,转化。挑取单菌落进行菌落pcr验证,验证后提取质粒应该得到pacyc-seacca-seaccb。
设计引物pf_t7seaccc(ecori)/pr_t7seaccc(hindiii)(seqidno.19-20)以及pf_t7seaccd(hindiii)/pr_t7seaccd(ndei)(seqidno.21-22)扩增,按照上述方法依次将seaccc和seaccd连接到载体上,最终得到pacyc-seacca-seaccb-seaccc-seaccd和pacyc-seacca-seaccc-sseaccd。
此外,采用上述相同的一步法克隆依次将不同的亚基构建至表达载体得到质粒pacyc-seacca-seaccc、pacyc-seacca-seaccdpacyc-seaccc-seaccd、pacyc-seaccb-seaccc、pacyc-seaccb-seaccd、pacyc-seaccb-seaccc-seaccd、pacyc-seacca-seaccb-seaccc等。
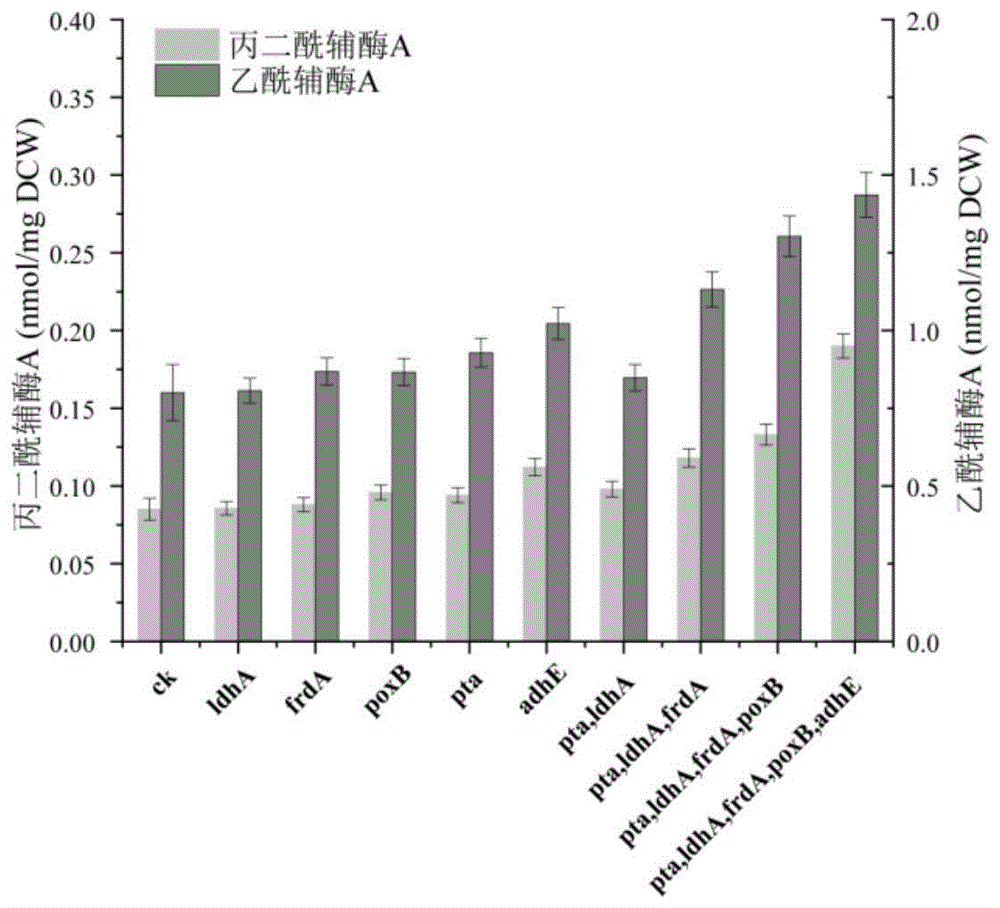
将上述不同来源的含乙酰辅酶a羧化酶基因的质粒转化实施例2中含大肠杆菌乙酰辅酶a合成酶acs基因的底盘改造的菌株bl21δldhaδptaδfrdaδpoxbδadhe进行表达发酵,并取发酵液测定丙二酰辅酶a含量,比较不同来源的乙酰辅酶a合成酶(参考实施例1中的发酵和检测方法),发现来自谷氨酸棒状杆菌的乙酰辅酶a羧化酶有最高的丙二酰辅酶a转化活力,如图3所示,胞内丙二酰辅酶a浓度达到0.776nmol/mgdcw,同时肠沙门氏菌(salmonellaenterica)的乙酰辅酶a羧化酶基因seaccacd也能显著提高丙二酰辅酶a转化活力,说明在表达乙酰辅酶a合成酶的基础上过表达乙酰辅酶a羧化酶可以1进一步提高微生物胞内丙二酰辅酶a的含量。
进一步分析结果发现,肠沙门氏菌乙酰辅酶a羧化酶结果比较特殊,表达肠沙门氏菌的seacca基因对胞内丙二酰辅酶a浓度有明显提升而同时表达seacca和seaccb时胞内丙二酰辅酶a浓度却下降了,而表达编码大肠杆菌自身的乙酰辅酶a羧化酶的bccp亚基的基因accb会降低胞内乙酰辅酶a羧化酶的浓度,因此推测是seaccb基因编码的亚基对胞内乙酰辅酶a羧化酶起到负调控,降低了胞内乙酰辅酶a羧化酶浓度。所以本发明中只表达肠沙门氏菌的seacca、seaccc和seaccd基因,进行发酵并测定胞内丙二酰辅酶a浓度,发现同时过表达seacca、seaccc或者seacca、seaccc、seaccd时胞内丙二酰辅酶a浓度较和seaccb同时过表达时高,分别为0.664nmol/mgdcw和0.774nmol/mgdcw。和柚皮素途径基因共表达,柚皮素产量分别为171.2mg/l和190.25mg/l。
实施例4
过表达不同来源的生物素连接酶
为了得到来自谷氨酸棒状杆菌的生物素连接酶基因cgbira(geneid:1018708)、肠沙门氏菌的生物素连接酶基因sebira(geneid:1255664)、铜绿假单胞菌的生物素连接酶基因pabira(geneid:881668),利用引物pf_cgbira(ncoi)/pr_cgbira(bamhi)(seqidno.23-24)、pf_sebira(ncoi)/pr_sebira(bamhi)和pf_pabira(ncoi)/pr_pabira(bamhi),以谷氨酸棒状杆菌、肠沙门氏菌、铜绿假单胞菌的基因组为模板扩增,将得到的生物素连接酶基因连入t载体pmd19t(simple)(takara)中得到t-cgbira,t-sebira,t-pabira,将cgbira,sebira,pabira通过ncoi/bamhi双酶切将基因从t-cgbira,t-sebira,t-pabira克隆到pcola-acs的ncoi/bamhi位点(实施例2),得到质粒pcola-acs-cgbira,pcola-acs-sebira,pcola-acs-pabira。
将pcola-acs-cgbira,pcola-acs-sebira,pcola-acs-pabira分别转入经过底盘改造的基因工程菌bl21δldhaδptaδfrdaδpoxbδadhe中,和实施例3得到的不同的乙酰辅酶a羧化酶基因质粒共表达,进行表达发酵并取发酵液测定丙二酰辅酶a含量。比较生物素连接酶和不同乙酰辅酶a羧化酶的组合,发现肠沙门氏菌的乙酰辅酶a羧化酶在谷氨酸棒状杆菌生物素连接酶的协助下可以发挥最大活力(参考实施例1中的发酵和检测方法),如图4所示,将胞内丙二酰辅酶a浓度提高至0.812nmol/mgdcw,与没有生物素连接酶协助的状态相比提高了0.046nmol/mgdcw。
综上,实施例1-4敲除大肠杆菌基因组中ldha、pta、frda、poxb、adhe五个基因,再导入丙大肠杆菌的乙酰辅酶a合成酶基因acs,肠沙门氏菌的乙酰辅酶a羧化酶基因seaccacd(含肠沙门氏菌的乙酰辅酶a羧化酶的3个亚基seacca、seaccc、seaccd)和谷氨酸棒状杆菌的生物素连接酶基因cgbira构建而成高产丙二酰辅酶a的基因工程菌(实施例4),胞内丙二酰辅酶a浓度从0.085nmol/mgdcw提升提高至0.812nmol/mgdcw,从而实现丙二酰辅酶a的高产。
实施例2-4中所用引物如表5所示。
表5本发明实施例2-4使用到的引物
实施例5
改造后高产丙二酰辅酶a工程菌提高柚皮素微生物法合成产量
选择来自深红酵母的酪氨酸解氨酶(tal),来自香芹菜的4-香豆酰辅酶a连接酶(4cl),来自矮牵牛的查尔酮合成酶(chs)和来自紫苜蓿的查尔酮异构酶(chi)作为柚皮素合成途径基因(吴俊俊.合成生物技术改造大肠杆菌生产黄酮骨架物质[d].无锡:江南大学,2015.),相关基因由上海生工合成,并构建至pcdfduet-1和petduet-1质粒上。
参考多模块代谢工程改造手段,调节tal和4cl基因的表达强度,用trc启动子替换表达载体上原有的t7启动子,表达质粒pcdf-trc-tal-trc-4cl和pet-chs-chi。将pcdf-trc-tal-trc-4cl和pet-chs-chi这两个质粒同时转入bl21(de3)感受态进行共表达,发酵并取样分析。通过高效液相色谱(hplc)检测发酵表达的目标产物柚皮素,使用安捷伦1100色谱仪和反相zorbaxsb-c18色谱柱(4.6×150mm),色谱柱的温度控制在25℃。黄酮骨架物质通过乙腈/水的梯度洗脱进行分离:在0-10min,色谱条件为10-40%的乙腈(体积/体积);在10-15min,色谱条件控制为40-60%的乙腈(体积/体积);在15-17min,色谱条件控制在60-10%的乙腈。产物的检测波长为290nm。
如图5所示,发酵液在15.85min有和柚皮素标品一致的检出峰,对比紫外光谱图,在288.7nm处有和柚皮素标准品一致的最大吸收波长。确定柚皮素合成途径构建成功并且成功表达出目标产物柚皮素,柚皮素产量为15.776mg/l,以此产量为标准,验证改造后的高产丙二酰辅酶a的工程菌对柚皮素产量的影响。
将柚皮素途径基因转入改造后的高产丙二酰辅酶a工程菌(实施例4),进行发酵,改造后的工程菌首先在lb培养基中摇床培养(37℃,200rpm)过夜,然后以初始od600为0.1的接种量重新转接到新鲜的mops发酵培养基中,同时向发酵体系内加入终浓度5g/l的碳源葡萄糖,摇床培养(37℃,200rpm)至od600为0.5。此时以1mmol/l的iptg(isopropyl-β-d-thiogalactopyranoside)终浓度进行诱导,并进行终浓度5g/l底物葡萄糖的补料和过量底物酪氨酸的补料后摇床培养(30℃,200rpm)发酵,并且取发酵53h发酵液进行hplc分析(液相条件同上),柚皮素产量为252.31mg/l,与改造前相比提高了1499.32%,证明提高胞内丙二酰辅酶a的供给能够有效提高黄酮类化合物微生物法合成的效率。
序列表
<110>南京农业大学
<120>一株高产丙二酰辅酶a的基因工程菌及其构建方法和应用
<160>24
<170>siposequencelisting1.0
<210>1
<211>45
<212>dna
<213>人工序列(artificialsequence)
<400>1
ttttcagctcttccagcaccgttttagagctagaaatagcaagtt45
<210>2
<211>31
<212>dna
<213>人工序列(artificialsequence)
<400>2
ggatcccaagcagaatcaagttctaccatgc31
<210>3
<211>50
<212>dna
<213>人工序列(artificialsequence)
<400>3
taaagccggtcagaccttccagagaaagactttctccagtgatgttgaat50
<210>4
<211>51
<212>dna
<213>人工序列(artificialsequence)
<400>4
attcaacatcactggagaaagtctttctctggaaggtctgaccggctttac51
<210>5
<211>32
<212>dna
<213>人工序列(artificialsequence)
<400>5
ctcgagaggaatgcctggtgcccggtaaacag32
<210>6
<211>25
<212>dna
<213>人工序列(artificialsequence)
<400>6
actagtattatacctaggactgagc25
<210>7
<211>27
<212>dna
<213>人工序列(artificialsequence)
<400>7
catatgagccaaattcacaaacacacc27
<210>8
<211>27
<212>dna
<213>人工序列(artificialsequence)
<400>8
ctcgagttacgatggcatcgcgatagc27
<210>9
<211>31
<212>dna
<213>人工序列(artificialsequence)
<400>9
ccatgggcatgagtctgaatttccttgattt31
<210>10
<211>27
<212>dna
<213>人工序列(artificialsequence)
<400>10
ggatccttaggcgtaaccgtagctcat27
<210>11
<211>29
<212>dna
<213>人工序列(artificialsequence)
<400>11
ccatggatattcgtaagattaaaaaactg29
<210>12
<211>30
<212>dna
<213>人工序列(artificialsequence)
<400>12
ggatccttactcgatgacgaccagtggctc30
<210>13
<211>30
<212>dna
<213>人工序列(artificialsequence)
<400>13
ccatgggcatgttggataaaattgtcatcg30
<210>14
<211>27
<212>dna
<213>人工序列(artificialsequence)
<400>14
ggatccttacttttcctgaagaccgag27
<210>15
<211>35
<212>dna
<213>人工序列(artificialsequence)
<400>15
ccatgggcatgagctggattgaacgaattaaaagc35
<210>16
<211>29
<212>dna
<213>人工序列(artificialsequence)
<400>16
ggatcctcaggcctcagactcctgatccg29
<210>17
<211>45
<212>dna
<213>人工序列(artificialsequence)
<400>17
tgagctacggttacgcctaaggatccggatctcgacgctctccct45
<210>18
<211>53
<212>dna
<213>人工序列(artificialsequence)
<400>18
acctgcaggcgcgccgagctcgaattcttactcgatgacgaccagtggctcgt53
<210>19
<211>54
<212>dna
<213>人工序列(artificialsequence)
<400>19
cgagccactggtcgtcatcgagtaagaattcggatctcgacgctctcccttatg54
<210>20
<211>58
<212>dna
<213>人工序列(artificialsequence)
<400>20
ttcgacttaagcattatgcggccgcaagcttttacttttcctgaagaccgagtttttt58
<210>21
<211>55
<212>dna
<213>人工序列(artificialsequence)
<400>21
aaaactcggtcttcaggaaaagtaaaagcttggatctcgacgctctcccttatgc55
<210>22
<211>56
<212>dna
<213>人工序列(artificialsequence)
<400>22
cggccgatatccaattgagatctgccatatgtcaggcctcagactcctgatccggc56
<210>23
<211>32
<212>dna
<213>人工序列(artificialsequence)
<400>23
ccatgggcatgaacgttgacatttcacgatcc32
<210>24
<211>27
<212>dna
<213>人工序列(artificialsequence)
<400>24
ggatccttactgcaggcgaaggtgcgt27
- 还没有人留言评论。精彩留言会获得点赞!
- 一种生物素的酶联免疫检测试剂盒的制作方法
- 药用植物大戟3-羟基-3-甲基戊二酰辅酶a还原酶蛋白编码序列的制作方法
- 用于制备基于芳烃-双(二羧酰亚胺)的半导体材料的方法和用于制备它们的相关中间体的制作方法
- 新的乙酰辅酶a羧化酶(acc)抑制剂及其在糖尿病、肥胖症和代谢综合征中的应用的制作方法
- 新的乙酰辅酶a羧化酶(acc)抑制剂及其在糖尿病、肥胖症和代谢综合征中的应用的制作方法
- 乐昌含笑3-羟基-3-甲基戊二酰辅酶a还原酶蛋白编码序列的制作方法
- N1/n2-内酰胺乙酰辅酶a羧化酶抑制剂的制作方法
- 用作硬脂酰-辅酶a去饱和酶抑制剂的1
- 乙酰辅酶a羧化酶剪接变体及其用途的制作方法
- 新的乙酰辅酶a羧化酶(acc)抑制剂及其在糖尿病、肥胖症和代谢综合征中的应用的制作方法