吖啶衍生物多晶型及其制备方法和应用与流程
本发明属于生物医药领域,涉及吖啶衍生物多晶型及其制备方法和应用。
背景技术:
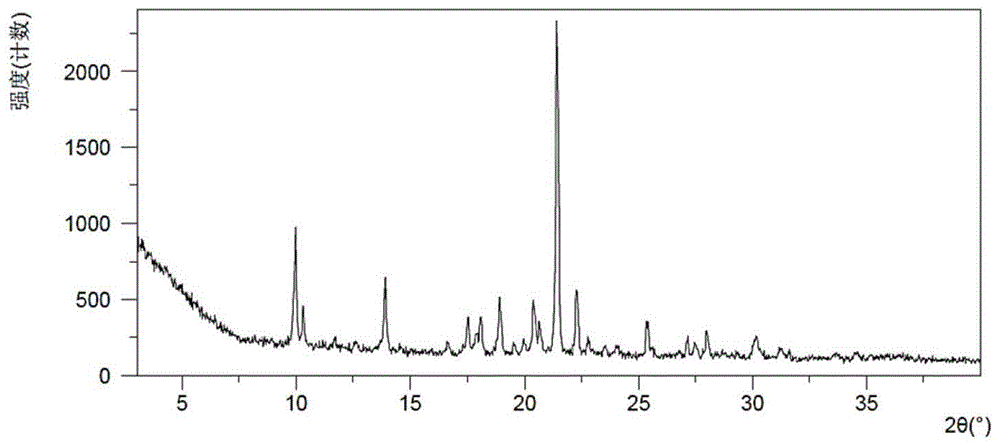
:琥珀八氢氨吖啶为双重的胆碱酯酶抑制剂,能可逆的抑制乙酰胆碱酯酶和丁酰胆碱酯酶,对乙酰胆碱酯酶选择性约是丁酰胆碱酯酶的10倍。经相关试验表明琥珀八氢氨吖啶能穿透血脑屏障,抑制脑内乙酰胆碱酯酶,改善阿尔茨海默病相关症状。琥珀八氢氨吖啶是一种全新的化合物,与现有的乙酰胆碱酯酶抑制剂结构均不同。该化合物的合成是全新的化学反应过程,经查新检索,未见有国内外文献报道,工艺技术属首创;该化合物化学合成工艺路线短,原料易得,反应条件温和,不需要高温、高压,废液可回收使用,易于“三废”处理,符合国家环保政策,具有产业化生产的技术基础。琥珀八氢氨吖啶属于吖啶衍生物,已经通过国家临床前新药审批,药理实验证实,该药对东莨菪碱引起的大小鼠学习记忆的损害有预防作用,对海人藻酸,b-淀粉样蛋白及注射d-半乳糖引起的老年性痴呆有治疗作用;对结扎双侧颈动脉及内注射微小栓子引起的血管性痴呆有治疗作用,药物晶型多态性对临床疗效的影响是目前药学界比较关注的问题。同一种药物在疗效上存在差异,其原因除了因生产工艺不同而产生的质量差异外,另一个可能原因就是药物晶型对生物利用度的影响。药物因晶型不同(晶型自由能差异以及分子间作用力不同)导致其生物利用度不同,进而影响药物在体内的吸收,产生药效差异。有研究表明,造成仿制药与原研药、不同企业生产的同种药物、同一企业同种药物的不同生产批次临床疗效差异的原因,大多数是由于固体药物的晶型物质存在状态变化。如,那格列奈的s晶型与临床使用的h晶型溶解度均明显>b晶型;阿司匹林有晶型ⅰ和晶型ⅱ,相同给药剂量下服用ⅱ型的血药浓度超出ⅰ型达70%。目前,文献对于药物晶型的报道大多集中在药物晶型制备和表征及其多晶型的存在形式上,而对药物不同晶型间生物利用度的差异研究则较少涉及。这提示我们需进一步掌握各种固体药物的晶型种类,发现其优势晶型,摸索引起晶型变化的各种控制条件,保证药物晶型的稳定性,改善药物的溶出速率和生物利用度,从而提高临床疗效和安全性。技术实现要素:本发明提供了一种琥珀八氢氨吖啶晶型l,其x-射线粉末衍射图谱在2θ值为10.3°±0.2°,21.4°±0.2°,13.9°±0.2°的相应位置对应有主要特征衍射峰。进一步,晶型l的x-射线粉末衍射图谱在2θ值为9.9°±0.2°,22.2°±0.2°,18.9°±0.2°的相应位置对应有次要特征衍射峰。更进一步,晶型l的x-射线粉末衍射图谱在2θ值为17.5°±0.2°,25.3°±0.2°,18.1°±0.2°的相应位置对应有再次特征衍射峰。晶型l具有基本上如图1所示的x-射线粉末衍射图。本发明提供了一种琥珀八氢氨吖啶晶型i,其x-射线粉末衍射图谱在2θ值为9.1°±0.2°,10.0°±0.2°,16.9°±0.2°的相应位置对应有主要特征衍射峰。进一步,晶型i的x-射线粉末衍射图谱在2θ值为23.9°±0.2°,12.4°±0.2°,24.5°±0.2°的相应位置对应有次要特征衍射峰。更进一步,晶型i的x-射线粉末衍射图谱在2θ值为19.7°±0.2°,22.3°±0.2°,30.0°±0.2°的相应位置对应有再次特征衍射峰。晶型i具有基本上如图2所示的x-射线粉末衍射图。本发明提供了一种琥珀八氢氨吖啶晶型m,其x-射线粉末衍射图谱在2θ值为9.3±0.2°,25.2±0.2°,22.6±0.2°的相应位置对应有主要特征衍射峰。进一步,晶型m的x-射线粉末衍射图谱在2θ值为16.7±0.2°、23.4±0.2°,17.1±0.2°的相应位置对应有次要特征衍射峰。晶型m具有基本上如图3所示的x-射线粉末衍射图。本发明提供了一种琥珀八氢氨吖啶晶型b,其x-射线粉末衍射图谱在2θ值为9.3°±0.2°,20.7°±0.2°,22.4°±0.2°的相应位置对应有主要特征衍射峰。进一步,晶型b的x-射线粉末衍射图谱在2θ值为28.8°±0.2°,22.9°±0.2°,11.4°±0.2°的相应位置对应有次要特征衍射峰。更进一步,晶型b的x-射线粉末衍射图谱在2θ值为28.0°±0.2°,9.9°±0.2°,37.8°±0.2°的相应位置对应有再次特征衍射峰。晶型b具有基本上如图4所示的x-射线粉末衍射图。本发明提供了一种琥珀八氢氨吖啶晶型k,其x-射线粉末衍射图谱在2θ值为12.4°±0.2°,23.6°±0.2°,16.2°±0.2°的相应位置对应有主要特征衍射峰。晶型k具有基本上如图5所示的x-射线粉末衍射图。本发明还提供了琥珀八氢氨吖啶晶型l的制备方法,所述方法选自以下组:气固扩散、缓慢挥发、缓慢降温、室温悬浮搅拌、50℃悬浮搅拌、50-5℃循环搅拌、气液扩散、高聚物诱导(挥发)。本发明还提供了琥珀八氢氨吖啶晶型i的制备方法,所述方法选自以下组:缓慢挥发、缓慢降温、室温悬浮搅拌、50℃悬浮搅拌、气液扩散、高聚物诱导(挥发)。本发明还提供了琥珀八氢氨吖啶晶型m的制备方法,所述方法是50℃悬浮搅拌。本发明还提供了琥珀八氢氨吖啶晶型b的制备方法,所述方法是反溶剂添加试验或室温悬浮搅拌。本发明还提供了琥珀八氢氨吖啶晶型k的制备方法,所述方法是将前面所述的晶型b在室温下放置晾干后得到的。本发明所述的晶型可以被配制成适于哺乳动物医疗用途的药物组合物。可以采用任何合适的给药途径向患者提供包含有效量晶型l、晶型i、晶型m、晶型b、晶型k中任意一种或多种晶型的药物组合物。药物组合物的剂型可以是药学上可用的任何剂型,包括但不限于,胶囊、片剂、分散液、悬浮液等。药物组合物可以常规地存在于单位剂型中,并通过药学领域中的任何方法制备。本发明的药物组合物包括治疗有效量的晶型、一种或多种惰性的药学上可接受的载体、可选的任何其他的治疗成分、稳定剂等。载体必须是药学上可接受的,这是指载体与药物组合物中的其他成分相容并且不会不利地对其接收者造成损害。此组合物还可包括稀释剂、缓冲剂、粘结剂、崩解剂、增稠剂、润滑剂、防腐剂(包括抗氧化剂)、调味剂、掩味剂、无机盐(例如氯化钠)、抗菌剂(例如苯扎氯铵)、甜味剂、抗静电剂、表面活性剂、脱水山梨醇酯、脂类(例如,磷脂如卵磷脂和其他磷脂酰胆碱、磷脂酰乙醇胺、脂肪酸和脂肪酯、类固醇(例如胆固醇))以及螯合剂(例如edta、锌和其他合适的阳离子)。适用于本发明的药物组合物中的其他药物赋形剂和/或添加剂被列于“remington:thescience&practiceofpharmacy”,第19版,williams&williams,(1995)、“physician’sdeskreference”,第52版,medicaleconomics,montvale,nj(1998)和“handbookofpharmaceuticalexcipients”,第3版,ed.a.h.kibbe,pharmaceuticalpress,2000。本发明的晶型所配制成的药物组合物包括适合口服给药、直肠给药、局部给药、鼻腔给药、眼部给药或肠道外给药(包括腹腔、静脉、皮下或肌肉注射)的那些。药物组合物中晶型含量可以根据各种因素(包括剂型、治疗的病症、目标患者人群和其他考虑)而改变,并且通常容易由本领域的技术人员确定。实际上,该量依赖于具体晶型、治疗的病症的严重程度、患者人群、其他药物成分的稳定性等可以有很大变化。药物组合物通常包含从约0.001wt%至约99wt%之间任何量的晶型,优选约0.01wt%至约5wt%的晶型,更优选约0.01wt%至约2wt%的晶型,该量还依赖于药物组合物中所含的赋形剂/添加剂的相对量。本领域的技术人员采用常规的确定剂量的试验并结合试验数据,可以确定给定条件下的一种药剂的最佳剂量。对于口服给药,通常采用的示例性的每日剂量为约0.001-1000mg/kg体重,更优选约0.001-50mg/kg体重,治疗过程以合适的时间间隔重复。在实施本发明时,最适合的给药途径以及治疗剂量大小依赖于治疗的疾病的性质和严重程度。剂量和剂量频率也可以根据各个患者的年龄、体重和反应而变化。通常,合适的口服剂型可以包括每日总剂量为5-250mg的剂量范围,可以单次给药或平均分成多次给药。优选的剂量范围为10-80mg。通常,合适的肠胃外剂型可以包括每日总剂量为5-200mg的剂量范围,可以单次给药或平均分成多次给药。优选的剂量范围为10-100mg。本发明的药物组合物可作为单一疗法或与另一疗法或其他疗法组合投与。如可经由投与佐剂增强本发明的药物组合物的治疗效果(即,该佐剂本身可能仅有最小的治疗效果,但是当与另一治疗剂组合时,对患者的整体治疗效果将增强)。或者,仅举例而言,可经由投与如本文所述药物组合物一样也具有治疗效果的另一治疗剂,增加患者所经历的疗效。无论经治疗的疾病、失调症或病症,该患者所经历的整体疗效可简单地为该两种治疗剂的加成或该患者可经历协同疗效。由于不同治疗剂之间可能存在不同的物理及化学特性,因此本文所述的药物组合物可以与其他治疗剂经由不同途径投与。例如,如本文所述的药物组合物可经口投与,以产生并保持其良好的血液水平,而其他治疗剂可经静脉內投与。投药模式及投药适当性的确定为熟练的临床医师所熟知。例如,可根据相关领域中已知的确定方案,进行最初投药,且随后基于所观察到的效果,可由熟练的临床医师改变剂量、投药模式及投药时间。本发明还提供了一个套组,所述套组包含前面所述的任意一种或多种晶型、或前面所述的药物组合物。进一步,所述套组还包括教示根据本文所述的各种方法及途径使用该套组的说明指南。所述套组也可包括信息,如科学文献参考资料、包装填入材料、临床试验结果、及/或此等之概要等等,其指示或确定该组合物的活性及/或优点,及/或描述剂量、投药、副作用、药物相互作用、或其他对医护提供者有用的信息。此信息是基于各种研究结果,例如,使用包括活体內模型的实验动物的研究及基于人类临床试验的研究。可将本文所述的套组提供、销售及/或推销给健康提供者,其包括医师、护士、药剂师、处方人员等等。在某些实施方案中,也可将套组直接销售给消费者。在某些实施方案中,本发明提供一种套组,其包括晶型l、晶型i、晶型m、晶型b、或晶型k;还包括双层低密度聚乙烯塑料袋,及hdpe容器。在其他实施方案中,所述套组另外包括箔袋(例如,无水箔袋,如热密封型无水箔袋)。在某些实施方案中,所述套组另外包括干燥剂;在其他实施方案中,干燥剂为不需要及/或不存在。本文所述的晶型及药物组合物可用于诊断及作为研究试剂。例如,本文所述的晶型及药物组合物可单独或与其他化合物组合地用作差异及/或组合分析中的工具,以阐明细胞及组织内所表达的基因之表达模式。作为一非限制性实例,将经一种或多种化合物处理之细胞或组织内之表达模式与未经化合物处理之对照细胞或组织作比较,分析所产生模式的基因表达的差异水平,其与(例如)所检测基因的疾病结合、信号路径、细胞定位、表达水平、大小、结构或功能有关。此等分析可在受刺激或未受刺激的细胞中及在存在或不存在影响表达模式的其他化合物下进行。本发明还提供了前面所述的琥珀八氢氨吖啶晶型、结晶组合物或药物组合物在制备治疗胆碱酯酶过度激活导致的疾病的药物中的应用。进一步,胆碱酯酶过激活导致的疾病包括但不限于,阿尔茨海默病、重症肌无力、肌肉萎缩、脊髓灰白质炎后遗症、儿童脑型麻痹、外伤性感觉运动障碍、多发性神经炎及脊神经根炎、腹气胀和尿潴留、阵发性室上性心动过速、非去极化型肌松药中毒的解救、青光眼、肌松药拮抗、炎症、肾脏疾病、肥胖、脂肪肝、甲状腺功能亢进症、精神分裂症、溶血性贫血、巨幼细胞性贫血。本发明还提供了前面所述的琥珀八氢氨吖啶晶型、结晶组合物或药物组合物在制备治疗胆碱功能减低有关疾病的药物中的应用。进一步,胆碱功能减低有关疾病包括失眠、血管性痴呆、记忆丧失、注意力障碍及其他睡眠障碍、与胆碱耗损相关的认知缺损疾病。除用于人类治疗以外,本文所述的晶型及药物组合物也可用于同伴动物(例如,狗、猫)、珍稀动物及农场动物(例如,马)(其包括哺乳动物、啮齿动物等等)的兽医治疗。需要说明的是,在x-射线衍射光谱中,由结晶化合物得到的衍射谱图对于特定的晶型往往是特征性的,其中谱带(尤其是在低角度)的相对强度可能会因为结晶条件、粒径和其它测定条件的差异而产生的优势取向效果而变化。因此,衍射峰的相对强度对所针对的晶型并非是特征性的,判断是否与已知的晶型相同时,更应该注意的是峰的相对位置而不是它们的相对强度。此外,对任何给定的晶型而言,峰的位置可能存在轻微误差,这在晶体学领域中也是公知的。例如,由于分析样品时温度的变化、样品移动、或仪器的标定等,峰的位置可以移动,2θ值的测定误差有时约为±0.2°,典型地约为±0.1°。因此,在确定每种晶型结构时,应该将此误差考虑在内。在xrd图谱中通常用2θ角或晶面距d表示峰位置,两者之间具有简单的换算关系:d=λ/2sinθ,其中d代表晶面距,λ表入射x射线的波长,θ为衍射角。对于同种化合物的同种晶型,其xrd谱的峰位置在整体上具有相似性,相对强度误差可能较大。还应指出的是,在混合物的鉴定中,由于含量下降等因素会造成部分衍射线的缺失,此时,无需依赖高纯试样中观察到的全部谱带,甚至一条谱带也可能对给定的晶体是特征性的。也就是说,本领域技术人员应理解,x-射线衍射光谱中的衍射峰并不能显示本发明所述的晶型所显示衍射峰的详尽情况。x射线粉末衍射图的2θ值是可以随着机器以及随着样品制备中的变化和批次间变化而轻微变化,所引用的值不视为绝对值。还应理解的是,峰的相对强度可能随取向效应而变,因此本发明所含的xprd迹线中所示的强度是示例性的,并不用于绝对比较。dsc测定当晶体由于其晶体结构发生变化或晶体熔融而吸收或释放热时的转变温度。对于同种化合物的同种晶型,在连续的分析中,热转变温度和熔点误差典型的在约5℃之内,通常在约3℃之内,当我们说一个化合物具有某一给定的dsc峰或熔点时,这是指该dsc峰或熔点±5℃。dsc提供了一种辨别不同晶型的辅助方法。不同的晶体形态可根据其不同的转变温度特征而加以识别。需要指出的是对于混合物而言,其dsc峰或熔点可能会在更大的范围内变动。此外,由于在物质熔化的过程中伴有分解,因此熔化温度与升温速率相关。本文使用的术语“有效量”系指足以治疗或预防特定疾病或病症的至少一种所投与的药剂或化合物之含量。结果可以为减少及/或减轻疾病的征兆、症状、或原因,或任何其他所需的生物系统的改变。例如,针对治疗用途的“有效量”是临床上显著减少疾病所需的包含如本文所披露的化合物之组合物的含量。在任何个別情况下,适当的“有效”量可利用各种技术(如剂量增加研究)来决定。本文使用的术语“基本上相同”是指x射线粉末衍射图谱或差示扫描量热图谱可与那些本文所述者不同,但是落于本领域技术人员所考虑的实验誤差范围内。附图说明图1显示晶型l的x射线粉末衍射图;图2显示晶型i的x射线粉末衍射图;图3显示晶型m的x射线粉末衍射图;图4显示晶型b的x射线粉末衍射图;图5显示晶型k的x射线粉末衍射图;图6显示晶型l的tga/dsc图,其中,a:tga图;b:dsc图;图7显示晶型i的tga/dsc图,其中,a:tga图;b:dsc图;图8显示晶型m的tga/dsc图,其中a:tga图;b:dsc图;图9显示晶型l的1hnmr图;图10显示晶型i的1hnmr图;图11显示晶型i加热前后的xrpd叠图;图12显示晶型m的1hnmr图;图13显示晶型m加热前后的xrpd叠图;图14显示晶型b的1hnmr图;图15显示晶型l稳定性测试前后的xrpd叠图;图16显示晶型l平衡溶解度测试前后的xrpd叠图;图17显示晶型l的dvs图;图18显示晶型l在dvs测试前后的xrpd叠图;图19显示晶型l气固渗透后的xrpd图;图20显示晶型l气固渗透后的tga/dsc图;图21显示晶型l湿样的透射xrpd图;图22显示晶型l的plm图。具体的实施方式下面结合附图和实施例对本发明作进一步详细的说明。以下实施例仅用于说明本发明而不用于限制本发明的范围。实施例中未注明具体条件的实验方法,通常按照常规条件,或按照制造厂商所建议的条件。实施例1琥珀八氢氨吖啶晶型的制备1、晶型l的制备1)气固扩散称取约15mg每份的琥珀八氢氨吖啶(商品名:神尔洋,购自长春华洋高科技有限公司)于3ml小瓶中,另在20ml小瓶中加入约4ml水,将3ml小瓶敞口置于20ml小瓶中后,将20ml小瓶密封。室温下静置9天后收集固体。2)缓慢挥发称取约15mg琥珀八氢氨吖啶至3ml小瓶中,分别加入0.4-3.0ml水/四氢呋喃(1:1),经震荡过滤后取其上清液,用封口膜封住装有澄清溶液的小瓶并在上面扎几个小孔,放置在室温下缓慢挥发。当溶剂完全挥干后,收集所得固体。3)缓慢降温称取约20mg每份的琥珀八氢氨吖啶于5ml小瓶中,加入1.0-3.0ml异丙醇,在50℃下搅拌约2小时后过滤取上清液,将所得上清液放置在生化培养箱中,以0.1℃/分钟的降温速度从50℃降温至-20℃,未析出固体,转移至室温下挥发获得。4)室温悬浮搅拌称量约20mg琥珀八氢氨吖啶至hplc小瓶中,加入0.3ml水/四氢呋喃(1:1)或异丙醇/甲酸乙酯(1:1),得到的浑浊液置于室温下磁力搅拌(~1000转/分)约3天后,离心收集固体。5)50℃悬浮搅拌称量约25mg每份琥珀八氢氨吖啶至hplc小瓶中,分别加入0.4ml甲酸乙酯/乙酸异丙酯(1:1)、水/丙酮(1:9),得到的浑浊液置于50℃下磁力搅拌(~1000转/分)约3天后,离心收集固体。6)50-5℃循环搅拌称量约20mg每份琥珀八氢氨吖啶至hplc小瓶中,分别加入0.4ml甲醇/甲酸乙酯(1:9),得到的悬浊液置于50℃下磁力搅拌2小时后以0.1℃/min的降温速率降到5℃,在5℃下平衡1小时后再以相同速率升温至50℃,如此循环3次后在5℃下搅拌,试验共约3天。离心收集固体。7)气液扩散称取约20mg每份琥珀八氢氨吖啶溶于0.43-2.0ml水中,过滤取得上清液转移至3ml小瓶,另取20ml的小瓶向其中加入约4ml的丙酮或四氢呋喃,将装有清液的3ml小瓶敞口置于20ml小瓶后,密封20ml的小瓶并于室温下静置。当观察到有固体析出时,则分离固体,若16天后无固体析出,则取出3ml小瓶置于室温挥发,收集得到的固体。8)高聚物诱导(挥发)称取约20mg每份样品溶于0.4-3.0ml甲醇/丁酮(1:2),过滤取得上清液转移至装有~1mg混合聚合物的3ml小瓶中,用封口膜封住装有澄清溶液的小瓶并在上面扎几个小孔,放置在室温下缓慢挥发。当溶剂完全挥干后,收集所得固体。2、晶型i的制备1)缓慢挥发称取约15mg琥珀八氢氨吖啶至3ml小瓶中,分别加入0.4-3.0ml甲酸乙酯,经震荡过滤后取其上清液,用封口膜封住装有澄清溶液的小瓶并在上面扎几个小孔,放置在室温下缓慢挥发。当溶剂完全挥干后,收集所得固体。2)缓慢降温称取约20mg每份的琥珀八氢氨吖啶于5ml小瓶中,加入1.0-3.0ml甲酸乙酯、甲醇/甲酸乙酯(1:4)、或异丙醇/甲酸乙酯(1:1),在50℃下搅拌约2小时后过滤取上清液,将所得上清液放置在生化培养箱中,以0.1℃/分钟的降温速度从50℃降温至-20℃,收集析出的固体,未析出固体的琥珀八氢氨吖啶转移至室温下挥发。表中*:缓慢降温后未析出固体,转移至室温下挥发。表1缓慢降温溶剂晶型甲酸乙酯晶型i*甲醇/甲酸乙酯(1:4)晶型i异丙醇/甲酸乙酯(1:1)晶型i*3)室温悬浮搅拌称量约20mg晶型a至hplc小瓶中,加入0.3ml甲酸乙酯或异丙基苯/甲酸乙酯(1:1),得到的浑浊液置于室温下磁力搅拌(~1000转/分)约3天后,离心收集固体。4)50℃悬浮搅拌称量约25mg每份琥珀八氢氨吖啶至hplc小瓶中,分别加入0.4ml甲酸乙酯/乙酸异丙酯(1:1),得到的浑浊液置于50℃下磁力搅拌(~1000转/分)约3天后,离心收集固体。4)气液扩散称取约20mg每份琥珀八氢氨吖啶溶于0.43-2.0ml甲酸乙酯中,过滤取得上清液转移至3ml小瓶,另取20ml的小瓶向其中加入约4ml的异丙基苯或正辛醇,将装有清液的3ml小瓶敞口置于20ml小瓶后,密封20ml的小瓶并于室温下静置。当观察到有固体析出时,则分离固体,若16天后无固体析出,则取出3ml小瓶置于室温挥发,收集得到的固体。5)高聚物诱导(挥发)称取约20mg每份琥珀八氢氨吖啶溶于0.4-3.0ml甲酸乙酯,过滤取得上清液转移至装有~1mg混合聚合物b(聚己酸内酯,聚乙二醇,聚甲基丙烯酸甲酯,海藻酸钠和羟乙基纤维素(等质量混合))的3ml小瓶中,用封口膜封住装有澄清溶液的小瓶并在上面扎几个小孔,放置在室温下缓慢挥发。当溶剂完全挥干后,收集所得固体。3、晶型m的制备50℃悬浮搅拌称量约25mg每份的琥珀八氢氨吖啶至hplc小瓶中,分别加入0.4mlthf,得到的浑浊液置于50℃下磁力搅拌(~1000转/分)约3天后,离心收集固体。4、晶型b的制备1)反溶剂添加试验称取约20mg琥珀八氢氨吖啶加至20ml的小瓶内,用0.2-2.0ml的二甲基亚砜溶解后,向该澄清溶液中加入反溶剂甲基异丁基酮,边滴加边搅拌(~1000转/分),至有固体析出,若加入约10ml反溶剂后仍无固体析出则转移至5℃继续搅拌析出固体,经鉴定为半琥珀酸盐。2)室温悬浮搅拌称量约20mg每份琥珀八氢氨吖啶至hplc小瓶中,分别加入0.3ml二甲基亚砜/环己烷(1:9),得到的浑浊液置于室温下磁力搅拌(~1000转/分)约3天后,离心收集固体。5、晶型k的制备晶型k是将晶型b在室温下放置1天晾干后得到的。由于晶型k为晶型b干燥后得到,推测也为半琥珀酸盐。实施例2琥珀八氢氨吖啶多晶型的表征鉴定1、x射线粉末衍射(xrpd)检测步骤:xrpd图在panalytacalempyreanx射线粉末衍射分析仪上采集,扫描参数如表2所示。表2xrpd测试参数结果:(1)晶型l的x射线粉末衍射结果分别如图1和表3所示。晶型l的x-射线粉末衍射图谱在2θ值为10.3°±0.2°,21.4°±0.2°,13.9°±0.2°的相应位置对应有主要特征衍射峰,在2θ值为9.9°±0.2°,22.2°±0.2°,18.9°±0.2°的相应位置对应有次要特征衍射峰,在2θ值为17.5°±0.2°,25.3°±0.2°,18.1°±0.2°的相应位置对应有再次特征衍射峰。表3晶型l的xrpd衍射峰数据(2)晶型i的x射线粉末衍射结果分别如图2和表4所示。晶型i的x-射线粉末衍射图谱在2θ值为9.1°±0.2°,10.0°±0.2°,16.9°±0.2°的相应位置对应有主要特征衍射峰,在2θ值为23.9°±0.2°,12.4°±0.2°,24.5°±0.2°的相应位置对应有次要特征衍射峰,在2θ值为19.7°±0.2°,22.3°±0.2°,30.0°±0.2°的相应位置对应有再次特征衍射峰。表4晶型i的x射线衍射峰数据晶型m的x射线粉末衍射结果如图3和表5所示,在2θ值为9.3±0.2°,25.2±0.2°,22.6±0.2°的相应位置对应有主要特征衍射峰,为16.7±0.2°、23.4±0.2°,17.1±0.2°的相应位置对应有次要特征衍射峰。表5晶型m的x射线衍射峰数据晶型b的x射线粉末衍射结果分别如图4和表6所示,在2θ值为9.3°±0.2°,20.7°±0.2°,22.4°±0.2°的相应位置对应有主要特征衍射峰,在2θ值为28.8°±0.2°,22.9°±0.2°,11.4°±0.2°的相应位置对应有次要特征衍射峰。表6晶型b的x射线衍射峰数据晶型k的x射线粉末衍射结果分别如图5和表7所示,在2θ值为12.4°±0.2°,23.6°±0.2°,16.2°±0.2°的相应位置对应有主要特征衍射峰。表7晶型k的x射线衍射峰数据2、热重分析(tga)和差示扫描量热(dsc)检测步骤:tga在taq500/5000热重分析仪上采集,dsc在taq200/2000差示扫描量热仪上采集,采集参数如表8所示。表8tga和dsc测试参数结果:晶型l的热重分析结果和差示扫描量热结果如图6所示。晶型l在155.7℃和185.6℃(起始温度)处有两个吸热峰;晶型l加热至170℃失重1.9%。晶型i的热重分析结果和差示扫描量热结果如图7所示。晶型i在13.5%,在126.0℃(起始温度)和164.9℃(峰值温度)处有两个吸热峰;晶型i加热至150℃失重13.5%。晶型m的热重分析结果和差示扫描量热结果如图8所示。晶型m在137.4℃、183.8℃(峰值温度)和201.6℃(起始温度)处有三个吸热峰;晶型m加热至190℃失重11.7%。3、液态核磁氢谱(1hsolutionnmr)检测步骤:液态核磁氢谱在bruker400m核磁共振仪上采集,dmso-d6作为溶剂。结果:(1)晶型l的1hnmr如图9所示,样品中无溶剂乙酸异丙酯或甲酸乙酯残留。根据较小的tga失重和1hnmr结果,推测晶型l为无水晶型。(2)晶型i的1hnmr如图10所示,样品中检测到甲酸出峰,但未检测到甲酸乙酯出峰,推测甲酸乙酯分解产生甲酸,同时甲酸与琥珀八氢氨吖啶的摩尔比为1.0:1(12.6wt%)。将晶型i样品加热至130℃并冷却至室温,xrpd结果(图11)显示加热后样品转变为晶型l。根据1hnmr和加热试验结果推测,晶型i脱去甲酸后转变为无水晶型,表明晶型i为甲酸溶剂合物。(3)晶型m的1hnmr如图12所示,该样品中溶剂四氢呋喃与琥珀八氢氨吖啶的摩尔比约为0.6:1(11.5wt%)。将晶型m样品分别加热至140℃和180℃并冷却至室温后测xrpd,结果如图13所示。晶型m样品加热至140℃后转变为晶型a+m的混合物,加热至180℃后转变为无水晶型a(晶型a记载在与本申请同日申请的专利中)。根据1hnmr和加热试验结果推测,晶型m为四氢呋喃溶剂合物,加热后脱去溶剂四氢呋喃后转变为无水晶型。(4)晶型b的1hnmr如图14所示,样品中琥珀酸与琥珀八氢氨吖啶的摩尔比为0.5:1,因此推测晶型b为半琥珀酸盐。实施例3琥珀八氢氨吖啶晶型的稳定性研究晶型l的稳定性研究:(1)将晶型l在80℃条件下闭口放置24小时后进行物理和化学稳定性评估。(2)将晶型l在25℃/60%相对湿度、40℃/75%相对湿度条件下敞口放置1周后进行物理和化学稳定性评估。通过xrpd和hplc测试样品的物理和化学稳定性。结果:xrpd结果显示(图15)在25℃/60%相对湿度、40℃/75%相对湿度条件下晶型l未发生改变;hplc结果显示,晶型l的化学纯度在三种测试条件下均未发生变化。表明,晶型l在25℃/60%相对湿度和40℃/75%相对湿度条件下具有较好的物理和化学稳定性。实施例4琥珀八氢氨吖啶晶型平衡溶解度研究水中平衡溶解度测定步骤:室温对晶型l样品在水中的24小时平衡溶解度进行了测试。试验中将不同晶型的样品与水混合成悬浊液后(起始浓度约100mg/ml),室温下搅拌24小时(1000rpm),离心后将上清液过滤测溶解度,剩余固体进行xrpd测试。结果:晶型l在h2o中24小时的平衡溶解度为41.9mg/ml,与本申请同日申请的专利中晶型a在h2o中24小时后转变为晶型l,且晶型l的化学纯度未发生变化(图16)。实施例5琥珀八氢氨吖啶多晶型引湿性研究为了评估晶型l在不同湿度条件下的稳定性,在25℃恒温条件下对晶型l样品进行了动态水分吸附(dvs)试验结果。结果:晶型l的dvs结果如图17所示,样品在80%相对湿度时开始吸水至7.6%;xrpd对比结果如图18所示,晶型l在dvs测试前后的晶型不变。由于晶型l在80%相对湿度时开始吸水至7.6%,因此推测晶型l在高湿度下可能转为水合物。为了表征该水合物,将晶型l样品置于水中气固渗透11小时,并对得到的样品进行xrpd和tga/dsc表征。如图19和图20所示,水中气固渗透后样品的xrpd与晶型l参比一致,同时样品加热至170℃失重6.2%,在92.8℃(起始温度)、154.4℃和182.5℃(起始温度)处有三个吸热峰。由此推测高湿度下晶型l会转为水合物状态,但xrpd与无水物状态一致。此外,将从h2o中分离出的晶型l湿样(通过与本申请同日申请的专利中的晶型a样品置于h2o中室温搅拌24小时后得到)用塑料薄膜密封后(防止水分蒸发)测试透射xrpd。图21的xrpd对比图显示,湿样的xrpd与晶型l参比一致,进一步证实高湿度下得到的水合物的xrpd与无水晶型l一致。在dvs测试后晶型不变。作为对比,与本申请同日申请的专利中的晶型a在dvs测试后部分转变为晶型l,推测晶型a在高湿条件下转变为晶型l。实施例6琥珀八氢氨吖啶多晶型间的转化关系混悬竞争实验加以证明由于晶型l存在水合物状态,为了进一步研究与本申请同日申请的专利中的晶型a和晶型l在不同水活度下的稳定性关系,设置了室温下晶型a、l在不同水活度(aw=0~1)的溶剂体系中的混悬竞争试验。具体步骤如下:分别称取等量的晶型a、l样品(各约3mg)至hplc小瓶中,再加入起始样品在不同水活度的甲醇/水中的饱和溶液形成混悬液。在室温下磁力搅拌(~1000转/分)6天后分离固体测试xrpd。结果总结于表9中。结果显示:晶型l在甲醇/水(水活度aw=0~1)溶剂体系中混悬竞争试验,aw≤0.2时晶型l转变为晶型a;aw≥0.8时晶型a转变为晶型l;aw=0.4,0.6时得到晶型a和l的混合物。根据上述结果,室温高水活度时晶型l为更稳定晶型。表9晶型混悬实验溶剂(v/v)反应物甲醇晶型a甲醇/水(94:6,aw~0.2)晶型a甲醇/水(84:16,aw~0.4)晶型a+l甲醇/水(70:30,aw~0.6)晶型a+l甲醇/水(43:57,aw~0.8)晶型l水晶型l实施例7颗粒形貌表征利用偏光显微镜(plm)对晶型l样品进行表征,结果如图22所示。晶型l为不规则颗粒状的晶体,粒径约0~30μm。上述实施例的说明只是用于理解本发明的方法及其核心思想。应当指出,对于本领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以对本发明进行若干改进和修饰,这些改进和修饰也将落入本发明权利要求的保护范围内。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1