用于制造3D聚合结构的双固化方法和系统与流程
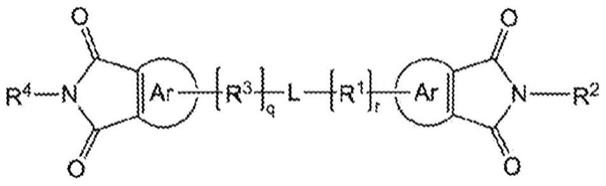
用于制造3d聚合结构的双固化方法和系统
技术领域
1.本发明总体上涉及三维(3d)物体的制造,更具体而言涉及使用一“双固化”系统制造3d结构。
背景技术:
2.3d打印通常涉及通过增材制造(am)工艺生产3d物体。增材制造最初是在20世纪80年代初期开发的,利用紫外可固化液体树脂形成热固性聚合物。固体结构按层构建,每一层对应于结构的横截面切片,并通过液体树脂的沉积和光固化形成。几年后开发了立体光刻增材(sla)制造工艺,在该工艺中,将要形成的物体的横截面图案创建为数字数据,然后根据该图案形成该物体。此后3d打印领域开发了很多技术,对基础增材制造工艺进行了诸多改进和改善:
3.速度和精度大大地提高了,从而能够以非凡的精度制造极小或极其复杂的结构;
4.增材制造目前在诸多应用领域(从血管和器官的“生物打印”到集成电路制造)都实现了大规模商业化;
5.原型制造可通过sla“快速原型技术(rapid prototyping)”快捷、低成本地实施,这是一项省时且节约成本的商业优势;及3d打印材料和设备的成本已降低到个人、小企业及大型组织都可使用该技术的程度。
6.然而,仍需要特别是对被制造物体的机械性能和表面光洁度作出改进。
技术实现要素:
7.因此,本发明涉及解决本领域上述需求的方法和组合物。
8.在一个实施例中,本发明提供用于形成固体聚合结构的双固化方法,该方法包括:
9.(a)将一封端的且酰亚胺封端的预聚物与至少一种可光聚合烯键式单体、至少一种光引发剂、及二胺结合,形成可固化树脂组合物;
10.(b)在能有效使所述至少一种烯键式单体聚合并在一支架内提供一聚烯烃的条件下辐照所述树脂组合物,所述支架包括所述预聚物和所述聚烯烃,其中,所述二胺被物理地俘获在所述支架内;及
11.(c)在能有效使所述预聚物与所述二胺之间发生一转酰亚胺化反应的温度下对所述受辐照的组合物进行热处理,同时释放所述预聚物的端基,提供所述固体聚合结构。
12.应当理解,在3d打印领域实施前述方法时,所述固体聚合结构对应于一3d可打印模型中所包含的预定形状和尺寸的3d物体,例如,可使用计算机辅助设计(cad)软件包、3d扫描仪或基于二维数字图像工作的摄影测量软件产生。
13.在前述实施例的一个方面中,在在步骤(b)中对所述树脂进行辐照之前,将步骤(a)中产生的所述可固化树脂组合物添加至一构建区域,该构建区域在尺寸上对应于所述物体的预定形状和尺寸。
14.在另一实施例中,提供一种用于形成3d物体的层的方法,其例如在增材制造工艺
的背景下实施。该方法包括:
15.(a)将一封端的且酰亚胺封端预聚物与至少一种可光聚合烯键式单体、至少一种光引发剂及二胺组合,形成一可固化树脂组合物;
16.(b)通过涂布、沉积或其他方式在一基底上作为层提供所述可固化树脂组合物;及
17.(c)在能有效使所述烯键式单体聚合并在一支架层内提供一聚烯烃的条件下辐照所述层,所述支架层包括所述预聚物和所述聚烯烃,其中,所述二胺被物理地俘获在所述支架层内。
18.在另一实施例中,本发明提供一种改进的用于形成3d物体的方法,其使用增材制造工艺,该工艺包括在一基底上以与一3d数字图像对应的尺寸以受计算机控制的方式连续形成各层,该改进包括通过以下步骤形成所述各层:
19.(a)在一基底上提供一初始可固化层,其中,该层包括通过结合封端的且酰亚胺末端预聚物、可光聚合烯键式单体、至少一种光引发剂及二胺制备的可固化树脂组合物;
20.(b)在能有效使所述烯键式单体聚合并在第一支架层内提供聚烯烃的条件下辐照所述初始层,其中,该第一支架层包括所述预聚物和所述聚烯烃,所述二胺被物理地俘获在该支架层内;
21.(c)重复步骤(a),在所述第一支架层上提供一额外的层;
22.(d)在能有效使所述烯键式单体聚合并提供一额外的支架层的条件下辐照所述额外的层;
23.(e)重复步骤(c)和(d)直至完全形成所述3d物体;及
24.(f)在能有效使所述预聚物与所述二胺之间发生一转酰亚胺化反应的温度下对所述3d物体进行热处理。
25.在任一前述实施例的一个方面中,所述预聚物具有如下式(i)的结构:
26.(i)
27.其中,
28.l包括未经取代的、经取代的、含杂原子的、或经取代且含杂原子的低聚亚烃基部分;
29.ar为芳基;
30.r1和r3可以相同,也可以不同,且为非低聚连接基团;
31.q和r可以相同,也可以不同,且为0或1;及
32.r2和r4为酰亚胺封端的基团,其可在转酰亚胺化反应中去除。
33.在一相关方面中,ar为苯基,使得所述预聚物具有如下式(ii)的结构:
34.(ii)
35.在另一相关方面中,所述预聚物的重均分子量为约500至约5000。
36.在任一前述实施例的另一方面中,所述可光聚合烯键式单体用作活性稀释剂。
37.在一相关方面中,所述可光聚合烯烃单体为丙烯酸酯或甲基丙烯酸酯单体。
38.在另一相关方面中,所述可光聚合烯键式单体具有如下式(xiv)的结构:
39.(xiv)
40.其中,
41.r5为h或ch3,r6为c1‑
c
36
烃基、经取代的c1‑
c
36
烃基、含杂原子的c1‑
c
36
烃基、或经取代且含杂原子的c1‑
c
36
烃基。
42.在任一前述实施例的另一方面中,所述二胺用作扩链剂,具有如下式(xv)的结构:
43.(xv)h2n
‑
l1‑
nh244.其中,l1为c2‑
c
14
亚烃基、经取代的c2‑
c
14
亚烃基、含杂原子的c2‑
c
14
亚烃基、或经取代且含杂原子的c2‑
c
14
亚烃基。
45.在另一实施例中,本发明提供一种作为新的物质组合物的可固化树脂组合物,其包括封端的端酰亚胺预聚物、至少一种可光聚合烯键式单体、至少一种光引发剂及二胺。
46.在另一实施例中,本发明提供一种用于合成所述封端的且酰亚胺末端预聚物的方法,该合成方法包括:
47.(a)在能有效产生反应产物端邻苯二甲酰亚胺低聚物的条件下,将二邻苯二甲酸酐(diphthalic anhydride)与一端氨基低聚物以至少约2:1的摩尔比结合;及
48.(b)通过将一氨基取代的环式反应物与所述端邻苯二甲酰亚胺低聚物以至少约2:1的摩尔比混合,在高温下反应至少约12h,对所述端邻苯二甲酰亚胺低聚物进行封端。
49.在另一实施例中,本发明提供一种用于形成固体聚合结构的双固化方法,该方法包括:
50.(a)通过如下步骤合成一封端的端酰亚胺预聚物:(i)在能有效产生反应产物端邻苯二甲酰亚胺低聚物的条件下,将二邻苯二甲酸酐(diphthalic anhydride)与一端氨基低聚物以至少约2:1的摩尔比结合;及(ii)通过将一氨基取代的环式反应物与所述端邻苯二甲酰亚胺低聚物以至少约2:1的摩尔比混合,在高温下反应至少约12h,对所述端邻苯二甲酰亚胺低聚物进行封端;
51.(b)将所述预聚物与至少一种可光聚合烯键式单体、至少一种光引发剂、及二胺结合,形成一可固化树脂组合物;
52.(c)在能有效使所述至少一种烯键式单体聚合并在一支架内提供一聚烯烃的条件
下辐照所述树脂组合物,所述支架包括所述预聚物和所述聚烯烃,其中,所述二胺被物理地俘获在所述支架内;及
53.(d)在能有效使所述预聚物与所述二胺之间发生一转酰亚胺化反应的温度下对所述受辐照的组合物进行热处理,由此释放所述预聚物的端基,提供所述固体聚合结构。
54.在还一实施例中,提供一种用于在增材制造工艺中形成一3d物体的层的方法,包括:
55.(a)通过如下步骤合成一封端的且酰亚胺末端预聚物:(i)在能有效产生反应产物端邻苯二甲酰亚胺低聚物的条件下,将二邻苯二甲酸酐(diphthalic anhydride)与一端氨基低聚物以至少约2:1的摩尔比结合;及(ii)通过将一氨基取代的环式反应物与所述端邻苯二甲酰亚胺低聚物以至少约2:1的摩尔比混合,在高温下反应至少约12h,对所述端邻苯二甲酰亚胺低聚物进行封端;
56.(b)将所述预聚物与至少一种可光聚合烯键式单体、至少一种光引发剂、及二胺结合,形成一可固化树脂组合物;
57.(c)在一基底上作为层提供所述可固化树脂组合物;及
58.(d)在能有效使所述烯键式单体聚合并在一支架层内提供一聚烯烃的条件下辐照所述层,所述支架层包括所述预聚物和所述聚烯烃,其中,所述二胺被物理地俘获在所述支架层内。
59.在另一实施例中,本发明提供一种通过用波长可有效固化一可光聚合烯键式单体的光化辐射照射一可固化树脂组合物制备的光固化组合物,其中,该可固化树脂组合物包括封端的且酰亚胺末端预聚物、至少一种所述可光聚合烯键式单体、至少一种光引发剂及二胺。
60.在再一实施例中,本发明提供一种通过如下步骤制备的固体物质组合物:(a)用波长能有效固化一可光聚合烯键式单体的光化辐射照射一可固化树脂组合物,由此提供一光固化的组合物,该树脂组合物包括封端的端酰亚胺预聚物、至少一种所述可光聚合烯键式单体、至少一种光引发剂及一二胺;及
61.(b)通过加热在促进所述封端的端酰亚胺预聚物与所述二胺之间的转酰亚胺化反应的条件下对所述光固化的组合物进行热处理。
具体实施方式
62.1.命名及概述
63.除非另外定义,否则本文使用的所有技术和科学术语都具有本发明所属技术领域中的普通技术人员通常所理解的含义。下面将对对描述本发明特别重要的特定术语进行限定。
64.在本说明书及权利要求书中,单数形式的“一”和“该”包括复数指代,除非上下文中另有明确说明。因此,例如,“一预聚合物”不仅指单一预聚合物,而且指两种或更多种不同预聚合物的组合,“一二胺”指单一二胺或二胺的组合,等等。
65.本文中使用的短语“具有式”或“具有结构”不用于进行限制,而是以与术语“包括”通常使用的方式相同的方式进行使用。
66.本文中使用的术语“烷基”是指含有1至约24个碳原子的带支链或不带支链的饱和
烃基,例如,甲基、乙基、正丙基、异丙基、正丁基、异丁基、叔丁基、辛基、癸基等,还指环烷基,例如,环戊基、环己基等。通常(但也不一定),本文中的烷基含有1至约18个碳原子,优选1至约12个碳原子。术语“低级烷基”是指含有1
‑
6个碳原子的烷基。优选的低级烷基取代基含有1
‑
3个碳原子,特别优选的此类取代基含有1或2个碳原子(即,甲基和乙基)。“经取代的烷基”是指被一个或多个取代基取代的烷基,“含杂原子的烷基”和“杂烷基”是指至少一个碳原子被杂原子(如,o、s或n)取代的烷基。如果没有另外说明,则术语“烷基”和“低级烷基”分别包括直链、带支链、环状、未经取代、经取代、和/或含杂原子的烷基或低级烷基。
67.本文中使用的术语“亚烷基”是指含有1至约24个碳原子的双官能直链、带支链、或环状的饱和烃基,例如,亚甲基、亚乙基、正亚丙基(n
‑
propylene)、正亚丁基(n
‑
butylene)、正亚己基(n
‑
hexylene)、亚癸基(decylene)、亚十四烷基(tetradecylene)、亚十六烷基(hexadecylene)等。优选的亚烷基含有1至约12个碳原子,术语“低级亚烷基”指含有1
‑
6个碳原子、优选1
‑
4个碳原子的亚烷基。术语“经取代的亚烷基”指被一个或多个取代基取代的亚烷基,即,一氢原子被一非氢原子取代基取代;术语“含杂原子的亚烷基”和“杂亚烷基”指至少一个碳原子被杂原子取代的亚烷基。如果没有另外说明,术语“亚烷基”和“低级亚烷基”分别包括直链、带支链、环状、未经取代、经取代、和/或含杂原子的亚烷基和低级亚烷基。本文中也设想低聚和聚合“亚烷基”,例如,经取代或未经取代的任选的含杂原子的聚(亚乙基)(聚乙烯)。
68.本文中使用的术语“烯基”是指2至约24个碳原子的含有至少一个双键的直链、带支链或环状烃基,例如,乙烯基、正丙烯基、异丙烯基、正丁烯基、异丁烯基、辛烯基、癸烯基、十四烯基、十六烯基、十八烯基、廿四烯基等。通常(但也不一定),本文中的烯基含有2至约18个碳原子,优选2至12个碳原子。术语“低级烯基”是指含有2
‑
6个碳原子的烯基,“环烯基”是指环状的烯基,优选具有5
‑
8个碳原子。术语“经取代的烯基”是指被一个或多个取代基取代的烯基,术语“含杂原子的烯基”和“杂烯基”是指至少一个碳原子被杂原子取代的烯基。如果没有另外说明,则术语“烯基”和“低级烯基”分别包括直链、带支链、环状、未经取代、经取代、和/或含杂原子的烯基和低级烯基。
69.本文中使用的术语“亚烯基”是指含有2至约24个碳原子的双官能直链、带支链或环状烃键,例如,亚乙烯基、正亚丙烯基、异亚丙烯基、正亚丁烯基、异亚丁烯基、亚辛烯基、亚癸烯基、亚十四烯基、亚十六烯基、亚十八烯基、亚廿四烯基等。优选的亚烯基键含有2至约12个碳原子,术语“低级亚烯基”是指含有2
‑
6个碳原子的亚烯基键,优选2
‑
4个碳原子。术语“经取代的亚烯基”是指被一个或多个取代基取代的亚烯基键,即,一氢原子被一非氢取代基取代;术语“含杂原子的亚烯基”和“杂亚烯基”是指至少一个碳原子被杂原子取代的亚烯基键。如果没有另外说明,则术语“亚烯基”和“低级亚烯基”分别包括直链、带支链、环状、未经取代、经取代、和/或含杂原子的亚烯基和低级亚烯基。本文中也设想低聚和聚合“亚烯基键”,例如,经取代或未经取代的任选的含杂原子的聚(亚乙烯基)连接基团,其可形成低聚物或聚合物的桥接两个端基的主体。
70.除非另外说明,否则本文中使用的术语“芳基”是指含有单个芳环、或多个稠合在一起、直接连接或间接连接(使得不同芳环结合至一共同的基团,例如,亚甲基或亚乙基部分)的芳环的芳香族取代基。优选芳基含有5至24个碳原子,特别优选芳基含有5至14个碳原子。示例性芳基含有一个芳环或两个稠合或连接的芳环,例如,苯基、萘基、联苯基、二苯醚、
二苯胺、二苯甲酮等。“经取代的芳基”是指被一个或多个取代基取代的芳基部分,术语“含杂原子的芳基”和“杂芳基”是指至少一个碳原子被杂原子取代的芳基取代基,下文中将对此作进一步详述。如果没有另外说明,则术语“芳基”包括未经取代、经取代、和/或含杂原子的芳香族取代基。
71.术语“亚芳基”是指含有1至3个稠合或连接的、未经取代或被一个或多个取代基取代的芳环的二价芳香族基团。除非另外说明,否则术语“亚芳基”包括经取代的亚芳基和/或含杂原子的亚芳基。
72.术语“烷芳基”是指具有烷基取代基的芳基,术语“芳烷基”是指具有芳基取代基的烷基,其中,“芳基”和“烷基”如上文所定义。优选芳烷基含有6至24个碳原子,特别优选芳烷基含有6至16个碳原子。芳烷基的示例包括,但不限于,苄基、2
‑
苯基
‑
乙基、3
‑
苯基
‑
丙基、4
‑
苯基
‑
丁基、5
‑
苯基
‑
戊基、4
‑
苯基环己基、4
‑
苄基环己基、4
‑
苯基环己基甲基、4
‑
苄基环己基甲基等。烷芳基包括,例如,对甲苯基、2,4
‑
二甲苯基、对环己基苯基、2,7
‑
二甲基萘基、7
‑
环辛基萘基、3
‑
乙基
‑
环戊
‑
1,4
‑
二烯等。术语“烷芳氧基”和“芳烷氧基”是指式
‑
or取代基,其中,r为如上定义的烷芳基或芳烷基。
73.术语“酰基”是指具有式
‑
(co)
‑
烷基、
‑
(co)
‑
芳基或
‑
(co)
‑
芳烷基的取代基,术语“酰氧基”是指具有式
‑
o(co)
‑
烷基、
‑
o(co)
‑
芳基或
‑
o(co)
‑
芳烷基的取代基,其中,“烷基”、“芳基”和“芳烷基”如上所定义。
74.术语“环(状)的”是指脂环族或芳香族取代基,其可以被取代,也可以不被取代,和/或含有杂原子;可以是单环的、二环的或多环的。
75.术语“脂环族”按常规意义使用,指脂肪族环部分,与芳香族环部分不同;可以是单环的、二环的或多环的。脂环族化合物或取代基可含有杂原子和/或被取代,但通常未经取代且不含杂原子,即为碳环形的。
[0076]“含杂原子的烷基”(也被称为“杂烷基”)或“含杂原子的芳基”(也被称为“杂芳基”)中的术语“含杂原子的”是指一个或多个碳原子被碳以外的原子取代的分子、连接基、或取代基,除碳以外的原子例如是氮、氧、硫、磷或硅,通常是氮、氧或硫,优选氮或氧。类似地,术语“杂烷基”是指含杂原子的烷基取代基,术语“杂环”是指含杂原子的环式取代基,术语“杂芳基”和“芳香杂环”分别指含杂原子的“芳基”和“芳香族”取代基,等等。杂烷基的示例包括烷氧基芳基(alkoxyaryl)、烷基硫烷基(alkylsulfanyl)取代的烷基、n
‑
烷基化氨烷基等。杂芳基取代基的示例包括吡咯基、吡咯烷基、吡啶基、喹啉基、吲哚基、嘧啶基、咪唑基、1,2,4
‑
三唑基、四唑基等,含杂原子的脂环族基团的示例为吡咯烷基、吗啉基、哌嗪基、哌啶基等。
[0077]“烃基”是指含有1至约30个碳原子、优选1至约24个碳原子、更优选1至约18个碳原子、最优选约1至12个碳原子的一价烃基,包括直链、带支链、环状、饱和及不饱和基团,例如,烷基、烯基、芳基等。“经取代的烃基”是指被一个或多个取代基取代的烃基,术语“含杂原子的烃基”是指至少一个碳原子被一杂原子取代的烃基。除非另外说明,否则术语“烃基”应被解释为包括经取代的和/或含杂原子的烃基部分。
[0078]
术语“亚烃基”是指含有1至约24个碳原子、最优选1至约12个碳原子的二价烃基部分,包括直链、带支链、环状、饱和及不饱和基团,术语“低级亚烃基”是指含有1
‑
6个碳原子、优选1
‑
4个碳原子的亚烃基。术语“经取代的烃基”是指被一个或多个取代基取代的烃基,术
语“含杂原子的烃基”和“杂烃基”是指至少一个碳原子被一杂原子取代的烃基。类似地,“经取代的亚烃基”是指被一个或多个取代基取代的亚烃基,术语“含杂原子的亚烃基”和“杂亚烃基”是指至少一个碳原子被一杂原子取代的亚烃基。除非另外说明,否则术语“烃基”和“亚烃基”应被分别解释为包括经取代的和/或含杂原子的烃基和亚烃基部分。还设想低聚和聚合亚烃基部分,包括含杂原子的亚烃基,例如,聚(氧化乙烯),及其经取代的类似物。
[0079]
当一官能团被称为“受保护的”或“封端的”时,如在一“封端的”基团中,这意味着该基团为修饰形式以排除不希望的反应和/或促进希望的反应。考虑到本领域的技术水平并参考标准教科书(例如,greene等,protective groups in organic synthesis(纽约:wiley,1991))将从本技术确定用于本发明化合物的合适的保护基团。
[0080]
前述一些定义中简单提及的“经取代的烷基”、“经取代的芳基”等中的“经取代的”是指在烷基、芳基或其它部分中,与一碳(或其它)原子结合的至少一个氢原子被一个或多个非氢取代基取代。这类取代基的示例包括,但不限于:官能团如卤素、羟基、巯基、c1‑
c
24
烷氧基、c2‑
c
24
烯氧基、c2‑
c
24
炔氧基、c5‑
c
24
芳氧基、酰基(包括c2‑
c
24
烷基羰基(
‑
co
‑
烷基)和c6‑
c
24
芳基羰基(
‑
co
‑
芳基))、酰氧基(
‑
o
‑
酰基)、c2‑
c
24
烷氧基羰基(
‑
(co)
‑
o
‑
烷基)、c6‑
c
24
芳氧基羰基(
‑
(co)
‑
o
‑
芳基)、卤代羰基(
‑
(co)
‑
x,其中,x为卤素)、c2‑
c
24
烷基碳酸基(
‑
o
‑
(co)
‑
o
‑
烷基)、c6‑
c
24
芳基碳酸基(
‑
o
‑
(co)
‑
o
‑
芳基)、羧基(
‑
cooh)、羧酸基(carboxylato)(
‑
coo
‑
)、氨基甲酰基(
‑
(co)
‑
nh2)、单
‑
(c1‑
c
24
烷基)取代的氨基甲酰基(
‑
(co)
‑
nh(c1‑
c
24
烷基))、双
‑
(c1‑
c
24
烷基)取代的氨基甲酰基(
‑
(co)
‑
n(c1‑
c
24
烷基)2)、单
‑
(c6‑
c
24
芳基)取代的氨基甲酰基(
‑
(co)
‑
nh
‑
芳基)、双
‑
(c6‑
c
24
芳基)取代的氨基甲酰基(
‑
(co)
‑
n(芳基)2)、双
‑
n
‑
(c1‑
c
24
烷基)、n
‑
(c6‑
c
24
芳基)取代的氨基甲酰基、硫代氨甲酰基(
‑
(cs)
‑
nh2)、脲基(
‑
nh
‑
(co)
‑
nh2)、氰基(
‑
c≡n)、异氰基(
‑
n
+
≡c
‑
‑
)、氰氧基(
‑
o
‑
c≡n)、异氰氧基(
‑
o
‑
n
+
≡c
‑
‑
)、异硫氰基(
‑
s
‑
c≡n)、叠氮基(
‑
n=n
+
≡n
‑
)、甲酰基(
‑
(co)
‑
h)、硫醛基(
‑
(cs)
‑
h)、氨基(
‑
nh2)、单
‑
(c1‑
c
24
烷基)取代的氨基、双
‑
(c1‑
c
24
烷基)取代的氨基、单
‑
(c5‑
c
24
芳基)取代的氨基、双
‑
(c5‑
c
24
芳基)取代的氨基、c2‑
c
24
烷基酰胺基(
‑
nh
‑
(co)
‑
烷基)、c6‑
c
24
芳基酰胺基(
‑
nh
‑
(co)
‑
芳基)、亚氨基(
‑
cr=nh,其中,r为氢、c1‑
c
24
烷基、c5‑
c
24
芳基、c6‑
c
24
烷芳基、c6‑
c
24
芳烷基等)、烷基亚氨基(
‑
cr=n(烷基),其中,r为氢、c1‑
c
24
烷基、c5‑
c
24
芳基、c6‑
c
24
烷芳基、c6‑
c
24
芳烷基等)、芳基亚氨基(
‑
cr=n(芳基),其中,r为氢、c1‑
c
24
烷基、c5‑
c
24
芳基、c6‑
c
24
烷芳基、c6‑
c
24
芳烷基等)、硝基(
‑
no2)、亚硝基(
‑
no)、磺基(
‑
so2‑
oh)、磺酸基(
‑
so2‑
o
‑
)、c1‑
c
24
烷基硫烷基(
‑
s
‑
烷基;也称为“烷硫基”)、芳基硫烷基(
‑
s
‑
芳基;也称为“芳硫基(arylthio)”)、c1‑
c
24
烷基亚磺酰基(
‑
(so)
‑
烷基)、c5‑
c
24
芳基亚磺酰基(
‑
(so)
‑
芳基)、c1‑
c
24
烷基磺酰基(
‑
so2‑
烷基)、c5‑
c
24
芳基磺酰基(
‑
so2‑
芳基)、膦酰基(
‑
p(o)(oh)2)、膦酸基(
‑
p(o)(o
‑
)2)、亚膦酸基(
‑
p(o)(o
‑
))、二氧磷基(
‑
po2)及膦基(
‑
ph2);及烃基部分c1‑
c
24
烷基(优选c1‑
c
18
烷基、更优选c1‑
c
12
烷基、最优选c1‑
c6烷基)、c2‑
c
24
烯基(优选c2‑
c
18
烯基、更优选c2‑
c
12
烯基,最优选c2‑
c6烯基)、c2‑
c
24
炔基(优选c2‑
c
18
炔基、更优选c2‑
c
12
炔基,最优选c2‑
c6炔基)、c5‑
c
24
芳基(优选c5‑
c
14
芳基)、c6‑
c
24
烷芳基(优选c6‑
c
18
烷芳基)、及c6‑
c
24
芳烷基(优选c6‑
c
18
芳烷基)。
[0081]
此外,前述官能团可(如果一特定基团允许的话)进一步被一个或多个外加官能团或一个或多个烃基部分取代,例如前面具体列举的那些官能团和烃基部分。类似地,前述烃基部分可进一步被一个或多个官能团或外加烃基部分取代,例如前面具体列举的那些官能
团和烃基部分。
[0082]
术语“聚合物”用于指包括连接的单体的化合物,可以是直链、带支链或交联的。该术语还包含均聚物、共聚物、三元共聚物、四元共聚物等。任何被确定为含有多于一种类型的重复单元的聚合物(即,共聚物、三元共聚物、四元共聚物等)都不旨在限制构型。也就是说,例如,本文中的共聚物可以是嵌段共聚物、交替共聚物、无规共聚物,三元共聚物可以是嵌段共聚物、无规共聚物等。术语“低聚物”指低分子量线型聚合物,其可与自身或其他化合物(例如,单体和/或其他低聚物)参与一个或多个反应形成较高分子量的聚合物结构。
[0083]
当术语“经取代的”出现在一系列可能被取代的基团的前面时,意指该术语适用于该系列基团中的每个基团。例如,“经取代的烷基、烯基和芳基”应被解释为“经取代的烷基、经取代的烯基和经取代的芳基”。类似地,当术语“含杂原子的”出现在一系列可能的含杂原子基团的前面时,意指该术语适用于该系列基团中的每个基团。例如,“含杂原子的烷基、烯基和芳基”应被解释为“含杂原子的烷基、含杂原子的烯基和含杂原子的芳基”。
[0084]“任选的”或“任选地”意指随后描述的情形可能发生,也可能不发生,因此,该描述包括该情形发生的情况和该情形不发生的情况。例如,“任选地经取代的”意指一非氢取代基可存在于一给定原子上,也可不存在于一给定原子上,因此,该描述包括存在一非氢取代基的结构和一非氢取代基不存在的结构。类似地,如在本文中的化学式中由虚线表示的“任选地存在的”键意指一键可能存在,也可能不存在。
[0085]
在一个实施例中,本发明通过将(i)一封端的且酰亚胺末端预聚物与(ii)可光聚合烯键式单体、(iii)至少一种光引发剂及(iv)一二胺组合提供一可固化树脂组合物;在另一实施例中,本发明提供一种用于合成所述封端的且酰亚胺末端预聚物的方法。所述可固化树脂组合物可用于用于形成固体聚合结构的双固化方法中,例如,在增材制造工艺或其他3d打印方法的背景下。所述双固化方法的第一步骤包括在能有效地使所述烯键式单体聚合并在一支架内提供一聚烯烃的条件下辐照所述可固化树脂组合物,所述支架包括所述预聚物和所述聚烯烃,所述二胺被物理地俘获在所述支架内。在第二步骤中,所述光固化组合物,即,光聚合形成的所述支架,在能有效促进所述预聚物与所述二胺间的转酰亚胺化(transimidization)反应的条件下进行热处理,由此释放所述预聚物的端基,提供最终的具有优异机械性能和最佳表面特性的聚合结构。
[0086]
2.可光固化的树脂组合物
[0087]
a.预聚物:
[0088]
所述封端的端酰亚胺预聚物具有如下式(i)的结构:
[0089]
(i)
[0090]
其中,
[0091]
l包括低聚亚烃基部分,并且可以是未经取代的、被一个或多个如此具体实施方式部分第1节所述的非碳、非氢取代基和/或官能团取代、含杂原子、或即被取代又含杂原子。因此,l可以是亚烷基、经取代的亚烷基、杂亚烷基、经取代的亚烷基,其中,存在的任何杂原
子通常选自于氮、氧和硫,但最通常是氧原子。亚烷基l的一示例为低聚形式的聚乙烯,杂亚烷基l的一示例为低聚形式的聚(氧化乙烯),使得l分别为:
[0092][0093]
其中,n表示包含在l内的单体单元的数量。单体单元的数量通常选择成使所述预聚物具有约500至约5000、通常为约1000至约3000的重均分子量。
[0094]
ar为芳基,如前节所述,包括未经取代的芳基、经取代的芳基、杂芳基和经取代的杂芳基,其中,ar可以单环的、二环的或多环的,在这当中,如果是二环或多环的,则这些环稠合或连接在一起。如式(i)所示的两个ar部分可以相同或不同,但通常相同。当ar为苯基时,所述预聚物具有如下式(ii)的结构:
[0095]
(ii)
[0096]
在式(ii)中,可以看出,两个端基为被r2或r4n
‑
取代的邻苯二甲酰亚胺部分,其中,r2和r4为在转酰亚胺化反应中移除的酰亚胺封端基。也就是说,r2和r4选择成使得下述转酰亚胺化反应可在加热下进行(该反应以简化形式示出用于说明性目的,其中仅示出了预聚物的一个末端和单官能r
‑
nh2反应物而不是二胺):
[0097][0098]
酰亚胺封端基团r2和r4通常相同,以促进预聚物合成,如下文所述。然而,应当理解,本发明并不要求r2与r4相同。
[0099]
r1和r3为任选的非低聚连接基团,只要r和q独立地选自于0和1。与封端基团r2和r4一样,优选(但不一定):r和q相同,并且,当r和q为1时,r1和r3也相同。
[0100]
在一些实施例中,r1和r3包括邻苯二甲酰亚胺基团,使得所述预聚物具有如下式(iii)的结构:
[0101]
(iii)
[0102]
其中,s和t独立地选自于0和1,但在一优选实施例中,s和t相同。x和y独立地选自于o、s和低级亚烷基(例如,经取代或未经取代的亚甲基、亚乙基、正亚丙基或正亚丁基),
但,优选x与y相同。当s和t为0时,式(iii)预聚物具有式(iv)的结构:
[0103]
(iv)
[0104]
当l为聚(氧化乙烯)时,式(iv)预聚物具有如式(v)所示的结构:
[0105]
(v)
[0106]
而当l为聚乙烯时,应当理解,式(iv)预聚物具有式(vi)的结构:
[0107][0108]
当s和t为1且x和y都为o时,式(iii)预聚物具有式(vii)的结构:
[0109][0110]
其中,当l为聚(氧化乙烯)或聚乙烯时,式(vii)预聚物分别具有式(viii)或式(ix)的结构:
[0111][0112][0113]
如上所述的酰亚胺封端基团r2和r4选择成使得能与所述二胺发生转酰亚胺化反应。本文可有利地使用任何此类封端基团,只要他们可促进转酰亚胺化,并且不会与所述可
固化树脂组合物的任何组分不利地相互作用并对最终产物具有不利影响即可。
[0114]
在一些实施例中,r2和r4封端部分相同,为含有1
‑
4个、优选1
‑
3个、最优选1或2个杂原子的五至六元环式基团,其中,至少一个杂原子为氮原子,并且,邻苯二甲酰亚胺基团的环氮与封端部分的碳原子直接结合。这类封端部分的示例包括,但不限于:含氮杂环取代基,例如,吡啶基、二吡啶基、哒嗪基、嘧啶基、二嘧啶基(bipyridaminyl)、吡嗪基、1,3,5
‑
三嗪基、1,2,4
‑
三嗪基、1,2,3
‑
三嗪基、吡咯基、2h
‑
吡咯基、3h
‑
吡咯基、吡唑基、2h
‑
咪唑基、1,2,3
‑
三唑基、1,2,4
‑
三唑基、吲哚基、3h
‑
吲哚基、1h
‑
异吲哚基、环戊二烯并(b)吡啶基(cyclopenta(b)pyridinyl)、吲唑基、喹啉基、二喹啉基(bisquinolinyl)、异喹啉基、二异喹啉基、噌啉基、喹唑啉基、萘啶基、哌啶基、哌嗪基、吡咯烷基、吡唑烷基、奎宁环基(quinuclidinyl)、咪唑烷基、甲基吡啶烷基(picolyliminyl)、嘌呤基、苯并咪唑基、二咪唑基(bisimidazolyl)、吩嗪基(phenazinyl)、吖啶基、及咔唑基。适合作为酰亚胺封端基团的优选含氮杂环为芳基,因此包括吡咯基、咪唑基、吡唑基、三唑基、吡啶基、嘧啶基、哒嗪基和吡嗪基,以及其经取代的类似物。
[0115]
r2和r4为嘧啶基的本发明的代表性预聚物示于下面的式(x)
‑
式(xiii)中:
[0116][0117][0118]
理想地,所述预聚物的重均分子量为约500至约5000,通常为约1000至约3000。
[0119]
b.可光聚合烯键式单体
[0120]
所述可光聚合烯键式单体用作活性稀释剂,在辐照下聚合,由此促进稳定且均匀
的网络或支架(scaffold)的形成,然后可对该网络或支架进行热处理形成最终的聚合产品。在一些实施例中,用作活性稀释剂的可聚合烯键式单体为丙烯酸酯或甲基丙烯酸酯单体。在其他实施例中,所述烯键式单体包括乙烯基酯,如醋酸乙烯酯;氯乙烯;乙烯醇;乙烯基甲苯;苯乙烯;丙烯腈;丙烯;丁二烯;环己烯;或二乙烯基苯。应当理解,前述单体仅是说明性而非限制性的;实际上,任何可光聚合烯键式单体都可有利地与本发明结合。
[0121]
在一些实施例中,如上所述的可聚合烯键式单体为丙烯酸酯或甲基丙烯酸酯单体,其可以是单官能的、双官能的或多官能的。
[0122]“单官能的”是指丙烯酸酯或甲基丙烯酸酯单体具有一个烯基官能团,该官能团为包含在丙烯酸酯部分中的双键(即,羰基碳α碳原子上的=ch2),但该单体可包括一个或多个芳基部分。本文使用的术语“丙烯酸酯单体”包含丙烯酸酯和甲基丙烯酸酯,即,分别为丙烯酸和甲基丙烯酸的酯,还包含高级丙烯酸酯,例如,丙烯酸乙酯、丙烯酸丁酯等。然而,在一些实施例中,甲基丙烯酸酯相较于丙烯酸酯是优选的,只要光聚合反应(1)相较于丙烯酸酯单体用甲基丙烯酸酯单体会以更受控的方式进行,并且(2)可最终产生具有更理想的机械特性和表面光洁度的产品。
[0123]
在一个实施例中,可光聚合的单官能丙烯酸酯单体具有如下式(xiv)的结构:
[0124]
(xiv)
[0125]
其中,r5为h或甲基,使得该单体分别为丙烯酸酯或甲基丙烯酸酯;r6为c1‑
c
36
烃基、经取代的c1‑
c
36
烃基、含杂原子的c1‑
c
36
烃基或经取代且含杂原子的c1‑
c
36
烃基,并且通常为c1‑
c
24
烃基、经取代的c1‑
c
24
烃基、含杂原子的c1‑
c
24
烃基或经取代且含杂原子的c1‑
c
24
烃基,例如,c2‑
c
16
烃基、经取代的c2‑
c
16
烃基、含杂原子的c2‑
c
16
烃基或经取代且含杂原子的c2‑
c
16
烃基;或者,c4‑
c
12
烃基、经取代的c4‑
c
12
烃基、含杂原子的c4‑
c
12
烃基或经取代且含杂原子的c4‑
c
12
烃基。作为示例,在上述种类中,r5可以是c1‑
c
36
烷基、经取代的c1‑
c
36
烷基、含杂原子的c1‑
c
36
烷基或者经取代且含杂原子的c1‑
c
36
烷基,并且通常为c1‑
c
24
烷基、经取代的c1‑
c
24
烷基、含杂原子的c1‑
c
24
烷基或经取代且含杂原子的c1‑
c
24
烷基,例如,c2‑
c
16
烷基、经取代的c2‑
c
16
烷基、含杂原子的c2‑
c
16
烷基或经取代且含杂原子的c2‑
c
16
烷基,或者,c4‑
c
12
烷基、经取代的c4‑
c
12
烷基、含杂原子的c4‑
c
12
烷基或经取代且含杂原子c4‑
c
12
烷基。如上所述,r5部分还可以是芳基,包括未经取代的芳基、经取代的芳基、杂芳基、经取代的杂芳基、未经取代的芳烷基、经取代的芳烷基、杂芳烷基、经取代的杂芳烷基,例如,c5‑
c
36
未经取代的芳基、经取代的c5‑
c
36
芳基、c2‑
c
36
杂芳基、经取代的c2‑
c
36
杂芳基、未经取代的c6‑
c
36
芳烷基、经取代的c6‑
c
36
芳烷基、c3‑
c
36
杂芳烷基、经取代的c3‑
c
36
杂芳烷基,通常为c5‑
c
24
未经取代的芳基、经取代的c5‑
c
24
芳基、c2‑
c
24
杂芳基、经取代的c2‑
c
24
杂芳基、未经取代的c6‑
c
24
芳烷基、经取代的c6‑
c
24
芳烷基、c3‑
c
24
杂芳烷基、经取代的c3‑
c
24
杂芳烷基,例如,c5‑
c
16
未经取代的芳基、经取代的c5‑
c
16
芳基、c2‑
c
16
杂芳基、经取代的c2‑
c
16
杂芳基、未经取代的c6‑
c
16
芳烷基、经取代的c6‑
c
16
芳烷基、c3‑
c
16
杂芳烷基、经取代的c3‑
c
16
杂芳烷基,或者,c5‑
c
12
未经取代的芳基、经取代的c5‑
c
12
芳基、c2‑
c
12
杂芳基、经取代的c2‑
c
12
杂芳基、未经取代的c6‑
c
12
芳烷基、经取代的c6‑
c
12
芳烷基、c3‑
c
12
杂芳烷基、经取代的c3‑
c
12
杂芳烷基。任意杂原子通常为n或o,并
且,芳基通常(但不一定)为单环的,也可设想稠合及连接在一起的二环或三环基团。
[0126]
在一些实施例中,r6包括c6
‑
c36脂环部分,通常为桥接(二环或多环的)c6‑
c
36
脂环部分,并且可以是经替代的和/或含杂原子。因此,r6包括任选地经取代的和/或含杂原子的c6
‑
c24脂环基团和c6
‑
c16脂环基团。适合作为r5的这类基团的非限制性示例包括金刚烷基、2
‑
甲基
‑2‑
金刚烷基、2
‑
乙基
‑2‑
金刚烷基、5
‑
羟基
‑2‑
甲基
‑2‑
金刚烷基、5
‑
羟基
‑2‑
乙基
‑2‑
金刚烷基、1
‑
甲基
‑1‑
金刚烷基乙基、2
‑
甲基
‑2‑
降冰片基、2
‑
乙基
‑2‑
降冰片基、l,2,7,7
‑
四甲基
‑2‑
降冰片基、异冰片基(isobonyl)等。
[0127]
因此,可光固化单官能丙烯酸酯和甲基丙烯酸酯单体的具体示例包括,但不限于:丙烯酸异冰片酯、甲基丙烯酸异冰片酯、丙烯酸金刚烷酯、甲基丙烯酸金刚烷酯、丙烯酸异癸酯、甲基丙烯酸异癸酯、丙烯酸月桂酯、甲基丙烯酸月桂酯、3,3,5
‑
三甲基环己烷丙烯酸酯(3,3,5
‑
trimethylcyclohexane acrylate)、3,3,5
‑
三甲基环己烷甲基丙烯酸酯、2
‑
(2
‑
乙氧基乙氧基)乙基丙烯酸酯、2
‑
(2
‑
乙氧基乙氧基)乙基甲基丙烯酸酯、环状三羟甲基丙烷缩甲醛丙烯酸酯(cyclic trimethylolpropane formal acrylate)、环状三羟甲基丙烷缩甲醛甲基丙烯酸酯、丙烯酸四氢呋喃酯、甲基丙烯酸四氢呋喃酯、丙烯酸十三烷酯、甲基丙烯酸十三烷酯、2
‑
苯氧基乙基丙烯酸酯及2
‑
苯氧基乙基甲基丙烯酸酯。其他示例对本领域技术人员来说是明显的,或者可在相关文本和文献中找到。例如,参见专利号为7,041,846的watanabe等人的美国专利,其关于单官能丙烯酸酯和甲基丙烯酸酯单体的公开内容并入本文中。
[0128]
可用于与本发明的方法和组合物结合的双官能丙烯酸酯和甲基丙烯酸酯部分包括二缩三丙二醇二丙烯酸酯(tripropyleneglycol diacrylate)、1,6
‑
己二醇二丙烯酸酯、三环癸烷二甲醇二丙烯酸酯、二乙二醇二甲基丙烯酸酯、二丙二醇二丙烯酸酯、双官能乙二醇丙烯酸酯(difunctional glycol acrylate)、乙氧化双酚a二丙烯酸酯(ethoxylated bisphenol a diacrylates)、丙氧基化新戊二醇二丙烯酸酯(propyoxylated neopentylglycol diacrylates)、新戊二醇二丙烯酸酯(neopentylglycol diacrylate)、及乙二醇二甲基丙烯酸酯,而适于用于本文中的多官能丙烯酸酯和甲基丙烯酸酯的示例包括三甲基丙烷三丙烯酸酯(trimethylpropane triacrylate)、三甲基丙烷三甲基丙烯酸酯(trimethylpropane trimethacrylate)、乙氧基化三甲基丙烷三丙烯酸酯、丙氧化甘油三丙烯酸酯、三(2
‑
羟乙基)异氰脲酸三丙烯酸酯(tris
‑
(2
‑
hydroxyethyl)isocyanurate triacrylate)、季戊四醇三丙烯酸酯、乙氧基化季戊四醇四丙烯酸酯、三羟甲基丙烷三丙烯酸酯(tmpta)、双三羟甲基丙烷四丙烯酸酯(di(trimethylolpropane)tetraacrylate)、二季戊四醇六丙烯酸酯、及二季戊四醇六丙烯酸酯。
[0129]
c.聚合引发剂
[0130]
所述可固化树脂组合物的另一组分为光聚合引发剂或“光引发剂”。由于双固化过程中的引发步骤需要所述烯键式单体的光聚合,因此,所述可固化树脂组合物包括至少一种光引发剂,即,自由基型光引发剂。作为示例,该自由基型光引发剂可以是,但不限于:酰基膦氧化物,如,2,4,6
‑
三甲基苯甲酰基乙氧基苯基氧化膦(2,4,6
‑
trimethylbenzoylethoxyphenylphosphine oxide,tepo)、(2,4,6
‑
三甲基苯甲酰基)二苯基氧化膦(tpo)、二(2,4,6
‑
三甲基苯甲酰基)苯基膦氧化物(bis(2,4,6
‑
trimethylbenzoyl)phenylphosphine oxide),等;α
‑
羟基酮,如,2
‑
羟基
‑2‑
甲基
‑1‑
苯基丙酮、1
‑
羟基
‑
环己基二苯甲酮、2
‑
羟基
‑
2
‑
甲基
‑
l
‑
p
‑
羟乙基醚苯基丙酮、等;二芳酮,如,二苯甲酮、2,2
‑
二甲氧基
‑2‑
二苯乙酮(dmpa)、或过氧化苯甲酰;偶氮二异丁腈(aibn);或,肟酯,如,来自于basf的商标名为irgacure的那些。
[0131]
d.二胺
[0132]
选择该二胺反应物或“扩链剂”以在对光固化树脂组合物进行热处理时与酰亚胺封端的预聚物发生转酰亚胺化。该二胺具有如下式(xv)的结构:
[0133]
(xv)h2n
‑
l1‑
nh2[0134]
其中,l1为c2‑
c
14
亚烃基,包括未经取代的、经取代的、含杂原子的、及经取代且含杂原子的c2‑
c
14
亚烃基。通常,l1为未经取代的c2‑
c
14
亚烷基,使得该二胺为1,3
‑
丙二胺、1,2
‑
丙二胺、1,4
‑
丁二胺、1,5
‑
戊二胺、1,6
‑
己二胺、1,7
‑
庚二胺、1,8
‑
辛二胺、1,9
‑
壬二胺、1,10
‑
癸二胺、1,11
‑
十一烷二胺、1,12
‑
十二烷二胺、1,2
‑
环己二胺、1,4
‑
环己二胺、或4,4
’‑
二氨基二环己基甲烷。
[0135]
e.添加剂:
[0136]
所述可固化树脂组合物可包括任何添加剂来促进固化过程,并使最终产品具有一种或多种有利特性。作为示例,此类添加剂包括:增韧剂;填充剂;稳定剂;非反应性光吸收剂;阻聚剂;着色剂,包括染料和颜料;增稠剂;可检测化合物(如,放射性或发光化合物);金属粉末或纤维或其他导电材料;半导体微粒或纤维;磁性材料;阻燃剂;等等。优选的添加剂是专利号为9,598,608的rolland等人的美国专利中所描述的那些,通过引用将这部分内容并入本文中。
[0137]
3.新的物质组合物
[0138]
在一个实施例中,本发明提供一种作为新的物质组合物的可固化树脂组合物,其中,该组合物包括:(i)一封端的且酰亚胺末端预聚物;(ii)至少一种可光聚合烯键式单体;(iii)至少一种光引发剂;及(iv)一二胺,其中,每个组分如上文中a至e所定义。
[0139]
在另一实施例中,本发明提供一种光固化的组合物,其通过用波长能有效固化所述可光聚合烯键式单体的光化辐射来辐照所述可固化树脂组合物来制备。
[0140]
在还一实施例中,本发明提供一种固体物质组合物,其通过以下步骤来制备:(a)用波长能有效固化所述可光聚合烯键式单体的光化辐射来照射所述可固化树脂组合物;及(b)通过加热在促进所述封端的端酰亚胺预聚物与所述二胺间的转酰亚胺化反应的条件下对步骤(a)提供的光固化的组合物进行热处理。
[0141]
4.其他实施例
[0142]
本发明还包含其他实施例,在该些实施例中,采用除封端的端酰亚胺预聚物之外的预聚物。由此类其他预聚物形成一3d样品的方法与前述有关所述封端的端酰亚胺预聚物的方法类似。即,将所选的预聚物与至少一种可光聚合烯键式单体、至少一种光引发剂、及一二胺组合,形成一可固化的树脂组合物。在能有效使所述至少一种可光聚合烯键式单体聚合的条件下辐照该组合物,形成一包括所述预聚物和聚烯烃的支架,其中,所述二胺被物理地俘获在该支架内。然后,在能有效使所述预聚物与所述二胺发生交联反应的温度下对所述受辐照的支架进行热处理,提供固体聚合结构。
[0143]
一个这样的实施例在本文实验部分的实例3中进行了例示。该实例描述了可光固化的酯酰胺预聚物的制备,之后使其与作为可光聚合单体的环状三羟甲基丙烷缩甲醛丙烯
酸酯和光引发剂tpo混合。在完全混均后,加入所选的二胺和4,4
’‑
二氨基环己基甲烷。然后,对由此获得的搅拌后的树脂混合物进行辐照,使丙烯酸酯单体聚合,之后进行加热,使预聚物与二胺交联。
[0144]
所述可光固化的酯酰胺预聚物可由如下式(xvi)的结构表示:
[0145]
(xvi)
[0146]
其中,各取代基如下:
[0147]
r8和r
11
为体积较大的取代基,通常包括任选地经取代的、任选地含杂原子的烃基,可以是烷基、芳基等。在一些实施例中,r8和r
11
包括3
‑
12个碳原子的烃基,例如,异丙基、叔丁基、环己基等。r8和r
11
可以相同,也可以不同,但通常相同,因为这样可采用更简单的合成方法。
[0148]
r9和r
10
为包括1至24个、通常为2至12个碳原子的双官能烃基,并且可以是经取代的和/或含杂原子的。例如,r9和r
10
可以是经取代的或未经取代的低级亚烷基或亚苯基;如果是亚苯基的话,则该连接基通常为对亚苯基连接的形式。
[0149]
l2为低聚烃基连接基团,可以是经取代的或未经取代的。l2为如为l所定义的,参见该具体实施方式部分的第2(a)节。因此,适于用作l2的部分与适于用作l的那些相同。
[0150]
作为另一示例,所述预聚物可以是酸酐封端的,具有如下式(xvii)的结构:
[0151]
(xvii)
[0152]
其中,l3为如为l所定义的。
[0153]
本发明还包含一种用于形成固体聚合结构的双固化方法,其中,该方法包括:
[0154]
(a)将(i)一具有在加热时与一胺发生共价反应的端基的预聚物与(ii)至少一种可光聚合烯键式单体、(iii)至少一种光引发剂及(iv)一二胺组合,形成一可固化的树脂组合物;
[0155]
(b)在能有效使所述至少一种烯键式单体聚合并在一支架内提供聚烯烃的条件下辐照所述树脂组合物,所述支架包括所述预聚物和所述聚烯烃,其中,所述二胺被物理地俘获地该支架内;及
[0156]
(c)在能有效使所述预聚物端基与所述二胺之间发生反应的温度下对所述受辐照的组合物进行热处理。
[0157]
应当理解,所述预聚物端基与所述二胺之间的反应会产生交联结构。热处理和辐照按上文中关于封端的端酰亚胺预聚物所述的进行。
[0158]
因此,所述预聚物可通常由如下式(xviii)的结构表示:
[0159]
(xviii)
[0160]
其中,l4为如为l所定义的,r
12
和r
13
为与一胺发生共价反应的官能团,该胺通常为伯胺或仲胺,优选伯胺,例如,含有两个伯胺基团的二胺。
[0161]
在一些实施例中,在进行辐照之前将所述可固化的树脂组合物加入到一构建区域,该构建区域在尺寸上对应于待制造3d结构的预定形状和尺寸。
[0162]
在其他实施例中,所述方法在改进的增材制造工艺的背景下实施,该增材制造工艺包括尺寸与一3d数据图像对应的各层的计算机控制的连续形成,该改进包括通过如下步骤形成所述各层:
[0163]
(a)在一基底上提供一初始可固化层,其中,该层包括通过所述预聚物与至少一种可光聚合烯键式单体、至少一种光引发剂及一二胺制备的一可固化树脂组合物;
[0164]
(b)在能有效使所述烯键式单体聚合并在第一支架层内提供聚烯烃的条件下辐照所述初始层,其中,该第一支架层包括所述预聚物和所述聚烯烃,所述二胺被物理地俘获在该支架层内;
[0165]
(c)重复步骤(a),在所述第一支架层上提供一额外的层;
[0166]
(d)在能有效使所述烯键式单体聚合并提供一额外的支架层的条件下辐照所述额外的层;
[0167]
(e)重复步骤(c)和(d)直至完全形成所述3d物体;及
[0168]
(f)在能有效使所述预聚物与所述二胺之间发生一共价反应的温度下对所述3d物体进行热处理。
[0169]
应当理解,虽然上文中结合诸多具体实施例对本发明进行了说明,但前述说明及下文中的实例仅是说明性的,并不旨在限制本发明的范围。
[0170]
实例1
[0171]
合成端n
‑
(2
‑
嘧啶基)邻苯二甲酰亚胺酰亚胺预聚物:
[0172]
将100.00g(0.05mol)熔融的无水端酰亚胺聚乙二醇(分子量为2000g/mol)加入到500ml的四颈烧瓶中,该四颈烧瓶配备有顶置式搅拌器、氮入口、温度计和具有冷凝器的逆式迪安
‑
斯脱克分水器(reverse dean
‑
stark trap)。然后,将31.02g(0.10mol)4,4
’‑
氧双邻苯二甲酸酐加入到该烧瓶中,随后又加入39ml环己基吡咯烷酮。搅拌溶液4小时,观察到溶液粘度增加。然后,将温度升高至175℃,再搅拌12小时。之后,向溶液中加入9.51g(0.10mol)2
‑
氨基嘧啶,在175℃下再搅拌12小时。冷却粘性液体,倒出,得最终产品。
[0173]
此两步反应如下面的反应路线1(scheme 1)所示:
[0174]
scheme 1:
[0175][0176]
实例2
[0177]
使用端邻苯二甲酰亚胺酰亚胺预聚物形成试样并测试
[0178]
(a)一般步骤
[0179]
将实例1合成的预聚物聚合、固化,形成如下所述的3d结构。根据如下iso标准评估拉伸性能:iso 37(2017)硫化或热塑性橡胶
‑
非刚性材料的拉伸应力
‑
应变特性的测定(iso 37(2017)rubber,vulcanized or thermoplastic
‑
determination of tensile stress
‑
strain properties for non
‑
rigid materials);及iso 527(2017)塑料
‑
刚性材料拉伸性能的测定(国际标准化组织,bibc ii chemin de blandonnet 8,cp 401,1214游标,瑞士日内瓦)(iso 527(2017)plastics
‑
determination oftensile properties for rigid materials(international organization for standardization,bibc ii chemin de blandonnet 8,cp 401,1214 vernier,geneva,switzerland))。
[0180]
将拉伸试样装载到ncs(中国北京市海淀区高粱桥斜街13号钢研纳克(ncs)检测技术股份有限公司,邮编100081)的gnt5万能试验机上,将试样垂直定向并平行于测试方向。使用led uv室(uv波长=405
±
5nm,强度=130
‑
150
×
l02μm/cm2)完全固化浇注样品60s。然后,在对流式烘箱中对这些样品进行热处理,具体处理条件如下所述。表1给出了进行测试的拉伸试样的类型、一般材料特性和相关的应变率。
[0181]
将被测的在中央矩形段不会断裂的狗骨头样品排除。在夹具中或测试前破裂的样品不代表预期故障模式,也将其从数据中排除。
[0182]
为了确保样品的应变速率足以捕捉变形,对样品进行时长30s至5min的拉伸断裂测试。
[0183]
根据材料的类型并依据iso 37和iso 527,测得杨氏模量(应力
‑
应变图在0.05%
‑
0.25%应变下的斜率)、断裂拉伸强度、屈服拉伸强度、断裂伸长百分比、屈服伸长百分比及极限抗拉强度。
[0184]
对于具有高断裂伸长率的弹性材料,需要高速应变速率来使其在指定测试的通常范围内断裂。对于刚性材料,iso标准推荐使用1mm/min的弹性模量测试速率,以确保在5min内发生最低断裂应变。
[0185]
表1
[0186][0187]
(b)试样形成与评估
[0188]
使用顶置式搅拌器将实例1制备的uv可固化的端n
‑
(2
‑
嘧啶基)邻苯二甲酰亚胺酰亚胺预聚物与甲基丙烯酸异冰片酯、三羟甲基丙烷三甲基丙烯酸酯(tmptma)及tpo充分混合,得到一均质树脂。将该树脂浇注到一150mm
×
100mm
×
4mm模具中,uv固化1min。然后,在100℃下加热1h对该试样进行热固化,随后在220℃下加热4h。将如此形成的固化后的弹性片材切割成尺寸为150mm
×
10mm
×
4mm的矩形条。根据iso527,在ncs的万能测试机上测试各试样的机械性能(具体如上所述)。
[0189]
平均拉伸强度(mpa)和断裂伸长率(%)示于表2中,该表中还示出了转酰亚胺化反应混合物中的每个组分的重量百分比。
[0190]
表2
[0191]
组分重量%端n
‑
(2
‑
嘧啶基)邻苯二甲酰亚胺酰亚胺预聚物50.0甲基丙烯酸异冰片酯35.0tmptma10.2tpo1.04,4
’‑
二氨基二环己基甲烷3.8拉伸强度(mpa)16.4断裂伸长率(%)218
[0192]
实例3
[0193]
合成uv可固化聚酰胺预聚物:
[0194]
将100.0g(0.05mol)熔融的无水聚四亚甲基氧化物(polytetramethylene oxide,分子量为2000g/mol)加入到500ml的三颈烧瓶中,该三颈烧瓶配备有顶置式搅拌器、氮入口和温度计。然后,将20.3g(0.1mol)对苯二甲酰氯加入到该烧瓶中,搅拌,形成含有聚四亚甲基氧化物的均相溶液。将温度升高至80℃,搅拌溶液4小时。4小时后,施加真空。在气泡从溶液中消失30min后,去除真空。将反应温度逐渐降低至40℃。然后,加入37.0g(0.2mol)甲基丙烯酸2
‑
(叔丁基氨基)乙酯(2
‑
(t
‑
butylamino)ethyl methacrylate),将温度升高至50℃,在该温度下保持2h。然后,倒出所得粘性液体,作为反应产物。
[0195]
将此两步式反应示出于下面的反应路线3(scheme 3,其中,“ph”表示苯基,“t
‑
bu”表示叔丁基)中。
[0196][0197]
实例4
[0198]
使用聚酰胺预聚物形成试样并测试
[0199]
由实例3制备的uv可固化聚酰胺预聚物形成试样并对其评估在此遵循实例2的一般步骤。
[0200]
使用顶置式搅拌器将实例3制备的预聚物与环状三羟甲基丙烷缩甲醛丙烯酸酯及(2,4,6
‑
三甲基苯甲酰基)二苯基氧化膦(tpo)光引发剂充分混合,得到一均质树脂。然后,加入4,4
′‑
二氨基环己基甲烷,继续混合10min。将该树脂浇注到一150mm
×
100mm
×
4mm模具中,uv固化1min。然后,在100℃下加热1h对该试样进行热固化,随后在220℃下加热4h。将如此形成的固化后的弹性片材切割成尺寸为150mm
×
10mm
×
4mm的矩形条。根据iso527,在ncs的万能测试机上测试各试样的机械性能(具体如上所述)。
[0201]
平均拉伸强度(mpa)和断裂伸长率(%)示于表3中,该表中还示出了转酰亚胺化反应混合物中的每个组分的重量百分比。
[0202]
表3
[0203]
组分重量%uv可固化聚酰胺预聚物70.0环状三羟甲基丙烷缩甲醛丙烯酸酯24.2tpo1.04,4'
‑
二氨基二环己基甲烷4.8
拉伸强度(mpa)4.6断裂伸长率(%)236
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1