一种化合物、其制备方法及作为荧光探针的应用
1.本技术涉及一种化合物、其制备方法及作为荧光探针的应用,属于荧光探针领域。
背景技术:
2.致病蛋白质分子的错误折叠、变性与聚集会导致多种人类疾病,包括阿尔兹海默症(aβ肽段)、疯牛病(朊病毒蛋白)、亨丁顿舞蹈症(polyq蛋白)、渐冻人症(tdp43和sod1蛋白等)、糖尿病(iapp肽段)、帕金森症(α-突触核蛋白)以及淀粉样化病变(轻链蛋白等)。尽管研究人员在蛋白质构型疾病领域耕耘数十年,大部分此类疾病的发病机理仍不十分清晰,致使早期诊断方法和特效治疗手段相对匮乏。其中一个重要原因是,领域内缺乏精准的实验工具在活体细胞中实时实地跟踪观测致病蛋白质的整个错误折叠进程,进而无法确定致病机理。此外,识别蛋白质错误折叠与聚集的荧光分子和相关检测方法也常用于建立药物筛选方法,如热位移测定法(thermal shift assay)。因此,发展探测蛋白质聚集过程的方法具有广阔的应用前景。
3.荧光染料分子常用于探测解析蛋白质分子的构型变化,尤其常用于蛋白质淀粉样化病变(amyloid)的临床诊断。如刚果红和硫磺素t常用于对阿尔兹海默症等蛋白质构型疾病的病理确诊。此类分子的设计与发光机理依托于蛋白错误折叠过程通常伴随分子内部疏水氨基酸残基的曝露和蛋白质分子间的堆积聚集。这一系列的物理变化过程导致局部环境的极性和流动性下降。荧光分子对这种微环境的细微变化十分敏感,常发生荧光量子产率、荧光寿命、荧光偏振和荧光光谱等性质的剧烈改变,常被科学家利用实现探测和识别目的。但上述分子因不具备选择性,无法用于复杂生物体系中,均不适合用于细胞内蛋白质聚集态的识别与探测。
4.现有商用试剂盒(us 2018/0273759 a1)因其对错误折叠的变性蛋白质具有高度的特异性结合,因此可用于细胞内探测蛋白质聚集态。然而这种方法因其所用荧光分子带有电荷,不具备细胞膜穿透力,因此无法渗透到活细胞的细胞质内与聚集态蛋白质结合并发出荧光。使用技术探测胞内蛋白质聚集态必须使用福尔马林等试剂固定并破坏细胞膜的完整性,使其荧光分子进入细胞结合聚集态的蛋白质并发出荧光。
5.针对活细胞原位探测蛋白质聚集态的荧光分子,领域内报道甚少。通过在聚集诱导荧光(aie)类分子上加装马来酸酐可以有效穿透活细胞的细胞膜(angew.chem.int.ed.2020,59,2
–
9),在蛋白质发生聚集时,激活荧光。然后此类分子不具备对聚集蛋白的特异性和选择性。其发光机理在于利用马来酸酐对半胱氨酸的反应活性普遍性的标记蛋白质组,当蛋白质组某一或某些蛋白发生聚集时激活各自标记的荧光分子,其他未聚集蛋白上的荧光分子部发出荧光。因此上述办法虽然解决了细胞穿透性的问题,但同时也失去了其胞内复杂环境下的选择性。
6.因此,发展可以穿透活细胞细胞膜且具有胞内聚集蛋白质选择性的荧光分子及其相关检测方法,对研究蛋白质聚集所导致的疾病有着重大的科学意义和临床价值。
技术实现要素:
7.根据本技术的一个方面,提供一种化合物i,该化合物i是一类结构新颖的新型仿生环境敏感类荧光分子。该类分子在蛋白质具备完整三维结构时不发出荧光;当蛋白质错误折叠、变性并聚集时,分子可通过非共价键选择性高效结合,结合之后发出强烈荧光。该荧光分子的上述性质可用于荧光方法探测活细胞内的聚集态蛋白质。该发明是首个通过非共价键识别聚集态蛋白质的荧光分子,且可用于活细胞原位荧光探测胞内蛋白质的聚集过程。
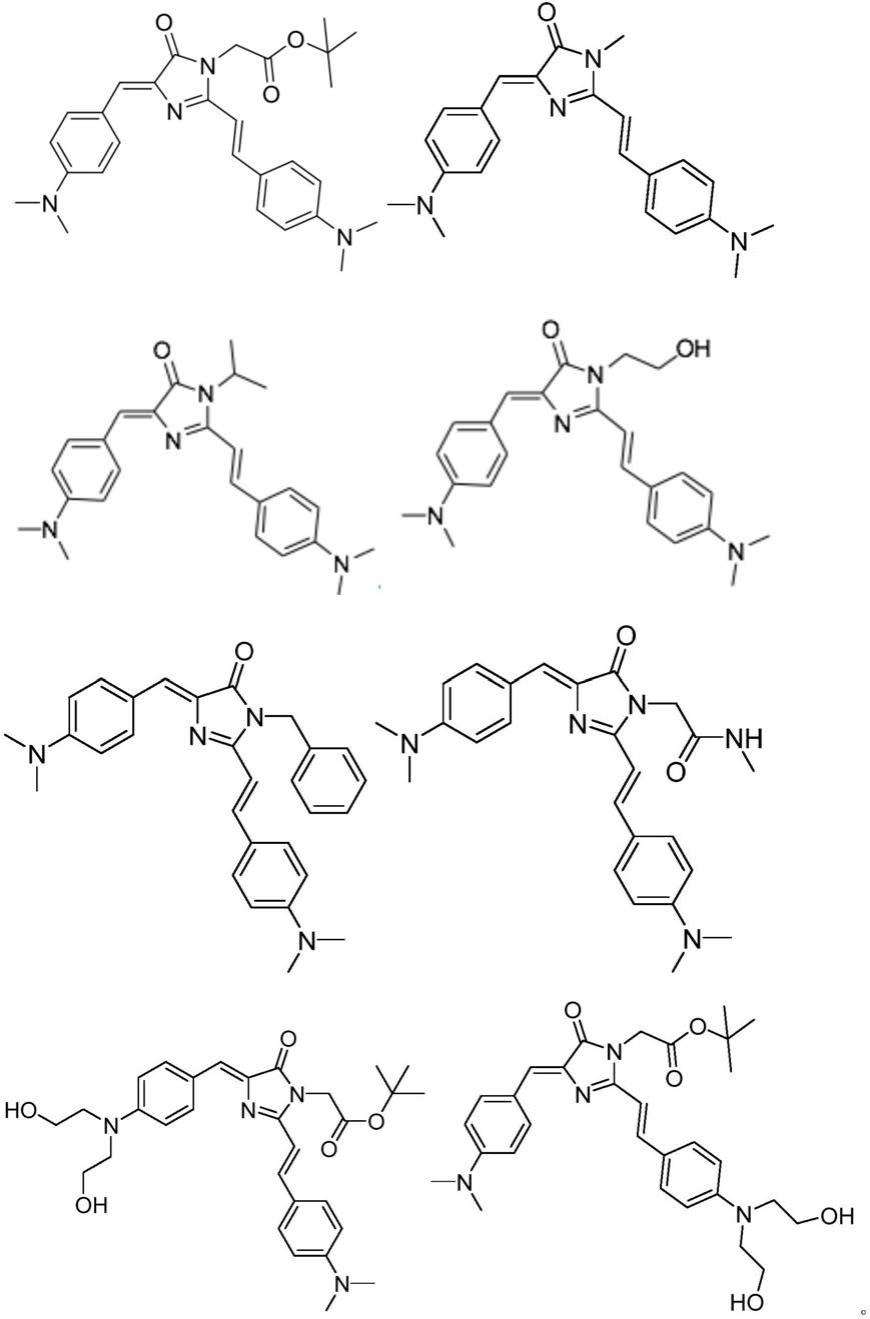
8.根据本技术的一方面,提供了一种化合物i,所述化合物i选自具有式i所示化学式的化合物中的至少一种;
[0009][0010]
式i中,r1、r2独立地选自c
1-c5的烷基、c
1-c5的烷氧基、取代的c
1-c5的烷基i中的至少一种;
[0011]
r3选自c
1-c5的烷基、取代的c
1-c5的烷基ii中的至少一种。
[0012]
可选的,所述取代的c
1-c5的烷基ii中的取代基选自羟基、芳基、取代的羰基中的至少一种。
[0013]
可选的,所述取代的c
1-c5的烷基i中的取代基选自羟基、芳基中的任一种。
[0014]
可选的,所述取代的羰基中的取代基选自取代的氨基、c
1-c5的烷氧基中的至少一种。
[0015]
可选的,所述取代的氨基中的取代基选自c
1-c5的烷基中的至少一种。
[0016]
可选的,所述化合物i选自以下化合物中的至少一种;
[0017]
[0018][0019]
可选地,所述化合物i的激发波长为450nm~550nm;发射波长为550nm~700nm。
[0020]
根据本技术的另一方面,还提供了一种上述化合物i的制备方法,至少包括如下步骤:
[0021]
(1)将含有化合物ii和化合物iii的溶液,反应i,得到中间产物i;
[0022]
所述化合物ii选自具有式ii所示化学式的化合物中的至少一种:
[0023][0024]
其中,r3选自c
1-c5的烷基、取代的c
1-c5的烷基ii中的至少一种。
[0025]
可选的,所述取代的c
1-c5的烷基ii中的取代基选自羟基、芳基、取代的羰基中的至少一种。
[0026]
可选的,所述取代的羰基中的取代基选自取代的氨基、c
1-c5的烷氧基中的至少一种。
[0027]
可选的,所述取代的氨基中的取代基选自c
1-c5的烷基中的至少一种。
[0028]
所述化合物iii选自具有式iii所示化学式的化合物中的至少一种:
[0029][0030]
其中,r1选自c
1-c5的烷基、c
1-c5的烷氧基、取代的c
1-c5的烷基i中的至少一种。
[0031]
可选的,所述取代的c
1-c5的烷基i中的取代基选自羟基、芳基中的任一种。
[0032]
优选地,r1表示-ch3、-ch(ch3)2、-ch2ch2oh、-ch2ph中的任一种。
[0033]
优选地,r2表示-ch3,-ch(ch3)2,-ch2ch2oh、-ch2ph中的任一种。
[0034]
优选地,r3表示-(co)nhch3或-(co)oc(ch3)3。
[0035]
(2)向中间产物i中加入n-(1-乙氧基乙二烯)-甘氨酸甲酯,反应ii,淬灭,萃取,柱色谱分离,得到中间产物ii;
[0036]
(3)向中间产物ii中加入化合物iv和溶剂i,在路易斯酸催化剂的存在下,反应iii,除去溶剂i,柱色谱分离,得到化合物i;
[0037]
所述化合物iv选自具有式iv所示化学式的化合物中的至少一种:
[0038][0039]
优选地,r2独立地选自c
1-c5的烷基、c
1-c5的烷氧基、取代的c
1-c5的烷基i中的至少一种。
[0040]
可选的,所述取代的c
1-c5的烷基i中的取代基选自羟基、芳基中的任一种。
[0041]
可选地,在所述步骤(1)中,反应i的条件为:温度为20~30℃;时间为12~24h。
[0042]
可选地,在所述步骤(1)中,所述化合物ii和化合物iii的摩尔比为1:1~2。
[0043]
优选地,在所述步骤(1)中,还包括碱性物质;所述碱性物质选自氢氧化钠、氢氧化钾、氢氧化锂中的至少一种。
[0044]
可选地,在所述步骤(2)中,反应ii的条件为:温度为20~30℃;时间为12~24h。
[0045]
可选地,在所述步骤(2)中,在所述步骤(2)中,所述中间产物i和n-(1-乙氧基乙二烯)-甘氨酸甲酯的摩尔比为1:1.0~2.0。
[0046]
可选地,在所述步骤(3)中,反应iii的条件为:温度为20~30℃;时间为12~24h。
[0047]
可选地,在所述步骤(3)中,所述中间产物ii和化合物iv的摩尔比为1:1~10。
[0048]
可选地,在所述步骤(3)中,所述中间产物ii和催化剂的摩尔比为1:0.01~1;
[0049]
优选地,在所述步骤(3)中,所述催化剂选自氯化锌、氯化铝、氯化镍、四氯化钛中的至少一种。
[0050]
可选地,在所述步骤(3)中,所述溶剂i选自1,4-二氧六环、四氢呋喃、甲苯中的至少一种。
[0051]
可选地,所述步骤(1)至少包括:将含有化合物ii的溶液,搅拌1~5h,加入化合物iii,反应i,得到中间产物i。
[0052]
根据本技术的另一方面,还提供了一种荧光探针,包括上述化合物i、根据上述方法制备得到的化合物i中的至少一种。
[0053]
本技术还提供了一种试剂盒,包括上述化合物i、根据上述方法制备得到的化合物i中的至少一种。
[0054]
本发明的化合物i是一类具有新颖化学结构和特异性荧光性质的荧光分子。其次,此类分子的特殊性质使其可实现现有方法无法完成的多种生物医学的重要应用场景。
[0055]
本技术还提供了上述化合物i、根据上述方法制备得到的化合物i中的至少一种在缓冲液中探测重组蛋白质聚集态的应用。
[0056]
可选地,本技术中的缓冲液是由多种无机盐组成的水溶液,用于各种蛋白质的溶解、保存、加热聚集等实验。缓冲液所用无机盐包括磷酸钠盐或钾盐(1mm-100mm)或氨基丁三醇(1mm-100mm),氯化钠或氯化钾盐(1mm-500mm),或乙酸钠(1mm-500mm),ph=4.0-8.5。
[0057]
本技术还提供了上述化合物i、根据上述方法制备得到的化合物i中的至少一种在活细胞中探测蛋白质聚集态的应用;
[0058]
所述蛋白质选自致病蛋白质、药物导致蛋白质中的任一种。
[0059]
本技术还提供了上述化合物i、根据上述方法制备得到的化合物i中的至少一种在筛选具有药物活性的小分子中的应用;
[0060]
所述具有药物活性的小分子选自蛋白质的小分子抑制剂或者激活剂。
[0061]
本技术要解决的技术问题为利用荧光分子的环境敏感性和结构特异性,实现活细胞内对聚集态蛋白质有选择性的荧光识别和探测。在胞内蛋白质的三维结构正常时不发出荧光,当蛋白质发生错误折叠、变性并聚集时发出强烈荧光。且该荧光激活类型探针无须共价键广谱修饰胞内蛋白质组,因此大幅降低对细胞的干扰和毒性。因此,该发明所述荧光探针可用于活细胞内非共价键探测聚集态的蛋白质分子。
[0062]
本发明的荧光探针由一族基于荧光蛋白发光基团的仿生衍生物组成。该类衍生物的荧光量子产率和荧光强度对外界微环境敏感。具有良好的生物兼容性和优异的荧光性质。制备成本低廉,制备方法简单,可进行大规模量产。该荧光分子由荧光发光基团、非共价键结合聚集态蛋白基团、亲水嵌段组成。化合物ⅰ中,非共价键结合基团为r3,亲水嵌合段为r1和r2,分子结构中除了r1、r2、r3之外的结构为发光基团。
[0063]
本发明所述探测活细胞内蛋白质聚集态的荧光探针,其发光机理是,在蛋白质三维结构不被破坏,功能完整,正确折叠态时,荧光分子内部的化学键在荧光激发态时可自由旋转,因此能量以热能形式释放,发生荧光淬灭。当蛋白质错误折叠、变性、聚集时,荧光分子可快速的、有选择性的结合聚集态蛋白质。由于分子与蛋白质聚集态的分子间相互作用,使得荧光分子激发态自由旋转的化学键受到禁阻能量以荧光形式释放,发出强烈荧光。利用荧光强度的大幅跃升,探测活细胞内聚集态蛋白质的形成。
[0064]
本技术中的荧光分子可发出红色荧光,此外,其发射光谱也覆盖了绿色发光区间,可同时使用两个发光光路,即绿色和红色。
[0065]
本技术中的重组蛋白质的含义是:任一通过大肠杆菌、酵母菌、昆虫细胞或者哺乳动物细胞为载体,表达并纯化出的模型蛋白质均可称为重组蛋白质。
[0066]
以下介绍一种具有式i-1所示的化学式的化合物i的制备方法:
[0067]
s1.将甘氨酸叔丁酯盐酸盐和氢氧化钠在有机溶剂中混合搅拌,然后加入4-二甲氨基苯甲醛,室温搅拌后加入制备好的n-(1-乙氧基乙二烯)-甘氨酸甲酯,室温下搅拌,反应结束后将反应用水淬灭,加入有机溶剂萃取,收集有机相成分浓缩,最后利用柱色谱层析法将其纯化。
[0068]
s2.将上述步骤所得产物与4-二甲氨基苯甲醛在有机溶剂中混合,加入极少量氯化锌后回流,除去溶剂后用柱色谱层析法纯化,即得化合物i。
[0069]
在其中一优选实例中,s1第一次搅拌时间为1~5小时,第二次搅拌时间为12~24小时,第三次搅拌时间为12~24小时。
[0070]
在其中一优选实例中,s2回流时间为12~24小时。
[0071]
在其中一优选实例中,4-二甲氨基苯甲醛与甘氨酸叔丁酯盐酸盐的摩尔比为1:1~5。
[0072]
在其中一优选实例中,s1所得产物与4-二甲氨基苯甲醛的摩尔比为1:1~5。
[0073]
在其中一优选实例中,s1所得产物与加入的氯化锌摩尔比为1:0.01~1。
[0074]
在其中一优选实例中,s1反应有机溶剂为乙醇,萃取有机溶剂为二氯甲烷,柱色谱层析所用溶剂为石油醚与乙酸乙酯混合溶剂,乙酸乙酯和石油醚的体积比为1:0.5~2。
[0075]
在其中一优选实例中,s2反应有机溶剂为1,4-二氧六环,柱色谱层析所用溶剂为石油醚与乙酸乙酯混合溶剂,乙酸乙酯和石油醚的体积比为1:1~5。
[0076]
本发明所述荧光分子在缓冲液中对蛋白质聚集态的检测方法,包括:向缓冲液中加入研究目标蛋白待测样本和本专利所述荧光分子,在25℃下进行孵育处理。孵育后,加热或加入蛋白质聚集引发剂后,引发蛋白质聚集后进行荧光定量检测。荧光定量检测可用仪器包括:荧光光谱仪、荧光高通量微孔板酶标仪、荧光凝胶成像仪等。所荧光分子与聚集态二氢叶酸还原酶结合后发出强烈荧光,荧光强度在蛋白质聚集前后发生33倍增益。
[0077]
本发明所述荧光分子对药物分子与目标蛋白结合后稳定性的检测办法,用于建立热位移药物筛选平台,包括:向缓冲液中加入研究目标蛋白(0.1μm-100μm)待测样本和本专利所述荧光分子,在25℃-37℃下进行孵育处理。孵育后,样本进行37℃-95℃温度梯度加热引发蛋白质聚集后进行实时荧光定量跟踪测量。所产生的荧光曲线与加入小分子药物(0.1μm-1000μm)的曲线进行对比,可检测药物分子是否与目标蛋白发生结合,稳定目标蛋白。
[0078]
本发明所述荧光分子在活细胞中选择性结合蛋白质的聚集态,发出强烈荧光,用于荧光成像检测,具体包括:向活细胞中加入蛋白质聚集引发剂(1nm-1mm),或表达可发生聚集的致病蛋白质,同时引入本发明所述荧光分子(0.1μm-20μm)进行原位孵育。待蛋白质发生聚集后,直接进行荧光成像,观测活细胞内的蛋白质聚集态的形貌和胞内位置。
[0079]
本技术的合成路线如下式所示:
[0080][0081]
本技术中n-(1-乙氧基乙二烯)-甘氨酸甲酯按以下方法制备得到:将甘胺酸甲酯盐酸盐,乙基乙酰亚胺盐酸盐和碳酸钾加入到放有乙醚/水的混合溶液的瓶子中,剧烈摇晃使反应物溶解,并不断加入水,最终溶液澄清反应结束,将有机层分离浓缩即得n-(1-乙氧基乙二烯)-甘氨酸甲酯。
[0082]
本技术中,c1~c
10
和c
1-c5指所包含的碳原子数。对所述“取代的c
1-c5的烷基i”、“取代的c
1-c5的烷基ii”的碳原子数限定,是指烷基本身所含的碳原子数,而非取代后的碳原子数。如取代的c
1-c5的烷基ii,指碳原子数为1~5的烷基上,至少一个氢原子被取代基取代。
[0083]
本技术中,“烷基”是由烷烃化合物分子上失去任意一个氢原子所形成的基团。所述烷烃化合物包括直链烷烃、支链烷烃、环烷烃、带有支链的环烷烃。
[0084]
本技术中,“苄基”是由甲苯分子上失去任意一个氢原子所形成的基团。
[0085]
本技术中,所述“烷氧基”指r
501-o-*,其中r501为烷基。
[0086]
本技术的有益效果包括但不限于:
[0087]
(1)本发明提供的荧光探针可以非共价键结合聚集态蛋白质,具体地,整个分子结构与聚集态蛋白质作为一个整体发生非共价键结合,其中分子的r3基团对结合强度起到重要作用;
[0088]
(2)本发明提供的荧光探针可以在胞内复杂生物环境下特异性的结合聚集态蛋白质;
[0089]
(3)本发明提供的荧光探针可以在活细胞内通过荧光成像探测蛋白质聚集态的形貌和位置。
附图说明
[0090]
图1为荧光分子探针的荧光激活机理示意图;
[0091]
图2为荧光分子缓冲液中对聚集态蛋白质的荧光光谱扫描;
[0092]
图3为基于荧光分子的热位移测定法测定大肠杆菌二氢叶酸还原酶结合抗生素甲氧苄胺嘧啶后的稳定性提升;
[0093]
图4为基于荧光分子的荧光显微镜成像效果;在人胚胎肾细胞293细胞系(hek293)中表达导致亨丁顿舞蹈症的致病蛋白htt97后,通过本发明提供的荧光探针识别聚集态htt97在活细胞中聚集的位置;
[0094]
图5为基于荧光分子的荧光显微镜成像效果;在人胚胎肾细胞293细胞系(hek293)中加入蛋白酶体抑制剂mg132,通过本发明提供的荧光探针广谱识别聚集态蛋白质在活细胞中聚集的位置;
[0095]
图6为基于荧光分子的荧光显微镜成像效果。在人宫颈癌细胞系(hela)中加入热休克蛋白90抑制剂17aag,通过本发明提供的荧光探针广谱识别聚集态蛋白质在活细胞中聚集的位置;
[0096]
图7为商用试剂盒与本发明所述荧光探针活细胞探测聚集蛋白质的成像对比图。
具体实施方式
[0097]
以下结合具体实施例来进一步说明本发明,但实施例并不对本发明做任何形式的限定。在不背离本发明精神和实质的情况下,对本发明方法、步骤或条件所作的简单修改或替换,均属于本发明的范围;若未特别指明,实施例中所用的技术手段为本领域技术人员所熟知的常规手段。
[0098]
如无特殊说明,本技术所用原料和试剂均来自商业购买,未经处理直接使用,所用仪器设备采用厂家推荐的方案和参数。
[0099]
本发明实施例中的n-(1-乙氧基乙二烯)-甘氨酸甲酯按以下方法制备得到:将2.50g甘胺酸甲酯盐酸盐,2.48g乙基乙酰亚胺盐酸盐和5.52g碳酸钾加入到放有200ml乙醚/水的混合溶液的瓶子中,剧烈摇晃使反应物溶解,并不断加入水,最终溶液澄清反应结束,将有机层分离浓缩即得n-(1-乙氧基乙二烯)-甘氨酸甲酯。
[0100]
本技术实施例中的核磁数据采用核磁共振波谱仪bruker avance iii400mhz进行;
[0101]
本技术实施例使用的共聚焦荧光显微镜型号为olympus fv1000fluoview
tm confocal microscope。
[0102]
图1为本技术实施例中荧光分子探针的荧光激活机理示意图。
[0103]
实施例1
[0104]
(1)将1.67g甘氨酸叔丁酯盐酸盐和0.40g氢氧化钠在50ml乙醇溶液中混合搅拌3小时,然后加入1.49g 4-二甲氨基苯甲醛,室温搅拌过夜12h。
[0105]
(2)向上述反应溶液中加入制备好的1.58g n-(1-乙氧基乙二烯)-甘氨酸甲酯,室温下搅拌过夜12h,反应结束后将反应用水淬灭,加入二氯甲烷萃取,收集有机相成分浓缩,最后利用柱色谱层析法将其纯化。
[0106]
(3)将上述步骤所得产物1.72g与7.5g 4-二甲氨基苯甲醛在5ml1,4-二氧六环溶液中混合,加入少量氯化锌后回流过夜,除去溶剂后用柱色谱层析法纯化,即得最终产物。
[0107]
将最终产物使用核磁共振波谱仪进行测试,液相色谱高分辨飞行时间质谱q-tof 6540进行结构表征和纯度测定,结果如下:1h nmr(400mhz,cdcl3,25℃):δ8.09(d,j=8hz,2h),7.89(d,j=16hz,1h),7.40(d,j=8hz,2h),7.01(s,1h),6.63(m,4h),6.33(d,j=
16hz,1h),4,35(s,2h),2.94(br,d,12h),1.37(s,9h)ppm.
13
c nmr(100mhz,cdcl3,25℃):δ14.37,26.94,39.13,41.34,81.72,106.19,110.83,121.99,122.42,126.58,128.50,133.32,139.21,150.45,155.50,157.01,166.00,169.04.hrms(m/z)anal.calc’d for c
28
h
34
n2o3(m+h)
+
:475.2704,found(m+h)
+
:475.2689.
[0108]
本实施例的合成路线如下:
[0109][0110]
实施例2
[0111]
(1)将40wt%甲胺水溶液4.3ml和1.49g 4-二甲氨基苯甲醛50ml乙醇溶液中混合搅拌3小时,然后继续室温搅拌过夜15h。
[0112]
(2)向上述反应溶液中加入制备好的1.58g n-(1-乙氧基乙二烯)-甘氨酸甲酯,室温下搅拌过夜12h,反应结束后将反应用水淬灭,加入二氯甲烷萃取,收集有机相成分浓缩,最后利用柱色谱层析法将其纯化。
[0113]
(3)将上述步骤所得产物与1.5g 4-二甲氨基苯甲醛在5ml 1,4-二氧六环溶液中混合,加入1m氯化锌10ml后回流过夜,除去溶剂后用柱色谱层析法纯化,即得最终产物。
[0114]
将最终产物使用核磁共振波谱仪进行测试,液相色谱高分辨飞行时间质谱q-tof 6540进行结构表征和纯度测定,结果如下:1h nmr(400mhz,cdcl3,25℃):δ8.18(d,j=8hz,2h),8.02(d,j=16hz,1h),7.54(d,j=8hz 2h),7.09(s,1h),6.73(m,4h),6.61(d,j=16hz 1h),3.32(s,3h),3.06(br,d,12h)ppm.
13
c nmr(100mhz,cdcl3,25℃):δ26.64,40.11,40.17,107.11,111.88,111.93,123.08,123.49,127.03,129.65,134.25,140.45,151.32,151.54,157.50,170.68ppm.hrms(m/z)anal.calc’d for c
23
h
26
n4o(m+h)
+
:375.2179,found(m+h)
+
:375.2176.
[0115]
本实施例的合成路线如下:
[0116][0117]
实施例3
[0118]
(1)将1.20g异丙胺和3.00g 4-二甲氨基苯甲醛在50ml乙醇溶液中混合,室温搅拌过夜24h。
[0119]
(2)向上述反应溶液中加入制备好的3.20g n-(1-乙氧基乙二烯)-甘氨酸甲酯,室温下搅拌过夜24h,反应结束后将反应用水淬灭,加入二氯甲烷萃取,收集有机相成分浓缩,最后利用柱色谱层析法将其纯化。
[0120]
(3)将上述步骤(2)中所得产物与4.5g 4-二甲氨基苯甲醛在5ml1,4-二氧六环溶液中混合,加入1m氯化锌5ml后回流过夜,除去溶剂后用柱色谱层析法纯化,即得最终产物。
[0121]
将最终产物使用核磁共振波谱仪进行测试,液相色谱高分辨飞行时间质谱q-tof 6540进行结构表征和纯度测定,结果如下:1h nmr(400mhz,(cd3)2so,25℃):δ8.18(d,j=8hz,2h),8.08(d,j=12hz,1h),7.55(d,j=8hz,2h),7.05(s,1h),6.74(m,5h),4.56(hept,j=8hz,1h),3.10(br,d,12h),1.55(d,j=8hz,6h)ppm.
13
c nmr(100mhz,(cd3)2so,25℃):δ20.95,44.54,108.50,112.28,112.33,123.01,123.42,124.82,130.28,134.22,136.34,140.15,151.43,151.82,158.02,170.50ppm.hrms(m/z)anal.calc’d for c
23
h
26
n4o(m+h)
+
:403.2492,found(m+h)
+
:403.2505.
[0122]
本实施例的合成路线如下:
[0123][0124]
实施例4
[0125]
(1)将0.62g乙醇胺和1.49g 4-二甲氨基苯甲醛0在50ml乙醇溶液中混合搅拌,然后室温搅拌过夜12h。
[0126]
(2)向上述反应溶液中加入制备好的1.58g n-(1-乙氧基乙二烯)-甘氨酸甲酯,室温下搅拌过夜8h,反应结束后将反应用水淬灭,加入二氯甲烷萃取,收集有机相成分浓缩,最后利用柱色谱层析法将其纯化。
[0127]
(3)将上述步骤(2)所得产物与7.5g 4-二甲氨基苯甲醛在5ml1,4-二氧六环溶液中混合,加入1m氯化锌1ml后回流过夜,除去溶剂后用柱色谱层析法纯化,即得最终产物。
[0128]
将最终产物使用核磁共振波谱仪进行测试,液相色谱高分辨飞行时间质谱q-tof 6540进行结构表征和纯度测定,结果如下:1h nmr(400mhz,cdcl3,25℃):δ8.14(d,j=8hz,2h),7.95(d,j=16hz,1h),7.50(d,j=8hz,2h),7.03(s,1h),6.69(m,5h),3.86(m,4h),3.04(br,d,12h)ppm.
13
c nmr(100mhz,cdcl3,25℃):δ40.08,40.15,43.24,53.48,61.33,72.78,107.33,111.83,111.88,122.99,123.59,127.49,129.70,134.37,140.58,151.36,157.44,171.30ppm.hrms(m/z)anal.calc’d for c
24
h
28
n4o2(m+h)
+
:404.2285,found(m+h)
+
:405.2293.
[0129]
本实施例的合成路线如下:
[0130][0131]
实施例5
[0132]
(1)将2.14g苄胺和3.00g 4-二甲氨基苯甲醛在50ml乙醇溶液中混合搅拌3小时,然后继续室温搅拌过夜12h。
[0133]
(2)向上述反应溶液中加入制备好的3.3g n-(1-乙氧基乙二烯)-甘氨酸甲酯,室温下搅拌过夜24h,反应结束后将反应用水淬灭,加入二氯甲烷萃取,收集有机相成分浓缩,最后利用柱色谱层析法将其纯化。
[0134]
(3)将步骤(2)所得产物与7.5g 4-二甲氨基苯甲醛在5ml1,4-二氧六环溶液中混合,加入1m氯化锌1ml后回流过夜,除去溶剂后用柱色谱层析法纯化,即得最终产物。
[0135]
将最终产物使用核磁共振波谱仪进行测试,液相色谱高分辨飞行时间质谱q-tof 6540进行结构表征和纯度测定,结果如下:1h nmr(400mhz,cdcl3,25℃):δ8.11(d,j=8hz,2h),7.94(d,j=16hz,1h),7.33(d,j=8hz,2h),7.21(m,6h),7.07(s,1h),6.68(d,j=12hz,2h),6.57(d,j=12hz,2h),6.42(d,j=16hz,2h),4,91(s,2h),3.00(s,6h),2.94(s,6h)ppm.
13
c nmr(100mhz,cdcl3,25℃):δ39.10,39.14,42.58,110.89,122.46,125.88,126.48,126.59,127.82,128.14,128.69,133.38,135.73,150.42,156.17,161.67ppm.hrms(m/z)anal.calc’d for c
29
h
31
n4o(m+h)
+
:451.2492.found(m+h)
+
:451.2499.
[0136]
本实施例的合成路线如下:
[0137][0138]
实施例6
[0139]
(1)将0.88g 2-氨基-n-甲基乙酰胺和1.49g 4-二甲氨基苯甲醛在50ml乙醇溶液中混合搅拌,然后室温搅拌过夜12h。
[0140]
(2)向上述反应溶液中加入制备好的1.58g n-(1-乙氧基乙二烯)-甘氨酸甲酯,室温下搅拌过夜12h,反应结束后将反应用水淬灭,加入二氯甲烷萃取,收集有机相成分浓缩,最后利用柱色谱层析法将其纯化。
[0141]
(3)将步骤(2)所得产物与15.00g 4-二甲氨基苯甲醛在5ml1,4-二氧六环溶液中混合,加入1m氯化锌0.1ml后回流过夜,除去溶剂后用柱色谱层析法纯化,即得最终产物。
[0142]
将最终产物使用核磁共振波谱仪进行测试,液相色谱高分辨飞行时间质谱q-tof 6540进行结构表征和纯度测定,结果如下:1h nmr(400mhz,cdcl3,25℃):δ8.02(m,3h),7.48(d,j=8hz,2h),6.99(s,1h),6.68(d,j=8hz,2h),6.55(d,j=20hz,2h),6.50(d,j=16hz,1h),4.38(s,2h),3.01(s,6h),2.97(s,6h),2.72(d,j=4hz,3h)ppm.
13
c nmr(100mhz,cdcl3,25℃):26.35,40.09,40.17,40.23,44.14,111.92,111.94,120.73,123.06,128.14,130.42,134.60,151.79,151.99,156.01,167.86ppm.hrms(m/z)anal.calc’d for c
25
h
29
n5o2(m+h)
+
:432.2395,found(m+h)
+
:432.2403.
[0143]
本实施例的合成路线如下:
[0144][0145]
实施例7
[0146]
(1)将3.34g甘氨酸叔丁酯盐酸盐和0.80g氢氧化钠在50ml乙醇溶液中混合搅拌3小时,然后加入2.10g 4-[n,n-双(2-羟乙基)氨基]苯甲醛,室温搅拌过夜12h。
[0147]
(2)向上述反应溶液中加入制备好的3.16g n-(1-乙氧基乙二烯)-甘氨酸甲酯,室温下搅拌过夜24h,反应结束后将反应用水淬灭,加入二氯甲烷萃取,收集有机相成分浓缩,最后利用柱色谱层析法将其纯化。
[0148]
(3)将步骤(2)所得产物与3.0g 4-二甲氨基苯甲醛在5ml1,4-二氧六环溶液中混合,加入1m氯化锌1.0ml后回流过夜,除去溶剂后用柱色谱层析法纯化,即得最终产物。
[0149]
将最终产物使用核磁共振波谱仪进行测试,液相色谱高分辨飞行时间质谱q-tof 6540进行结构表征和纯度测定,结果如下:1h nmr(400mhz,(cd3)2so,25℃):δ8.13(d,j=8hz,2h),7.82(d,j=16hz,2h),7.62(d,j=8hz,2h),6.82(s,1h),6.79(m,5h),4.82(t,j=4hz,2h),4.55(s,1h),3.57(m,8h),3.00(s,6h),1.41(s,9h)ppm.
13
c nmr(100mhz,(cd3)2so,25℃):δ28.11,42.19,53.66,58.65,82.21,108.31,111.98,112.36,122.35,123.42,125.78,128.66,129.36,130.15,134.58,135.27,139.62,150.00,151.83,157.09,167.94,169.99ppm.hrms(m/z)anal.calc’d for c
30
h
38
n4o5(m+h)
+
:535.2915,found(m+h)
+
:535.2917.
[0150]
本实施例的合成路线如下:
[0151][0152]
实施例8
[0153]
(1)将3.34g甘氨酸叔丁酯盐酸盐和0.80g氢氧化钠在50ml乙醇溶液中混合搅拌3小时,然后加入1.49g 4-二甲氨基苯甲醛,室温搅拌过夜12h。
[0154]
(2)向上述反应溶液中加入制备好的3.16g n-(1-乙氧基乙二烯)-甘氨酸甲酯,室温下搅拌过夜24h,反应结束后将反应用水淬灭,加入二氯甲烷萃取,收集有机相成分浓缩,最后利用柱色谱层析法将其纯化。
[0155]
(3)将步骤(2)所得产物与4.20g 4-[n,n-双(2-羟乙基)氨基]苯甲醛在5ml1,4-二氧六环溶液中混合,加入1m氯化锌1.0ml后回流过夜,除去溶剂后用柱色谱层析法纯化,即得最终产物。
[0156]
将最终产物使用核磁共振波谱仪进行测试,液相色谱高分辨飞行时间质谱q-tof 6540进行结构表征和纯度测定,结果如下:1h nmr(400mhz,(cd3)2so,25℃):δ8.17(d,j=8hz,2h),7.81(d,j=16hz,2h),7.58(d,j=8hz,2h),6.86(s,1h),6.77(m,5h),4.81(t,j=4hz,2h),4.56(s,1h),3.57(m,8h),3.04(s,6h),1.42(s,9h)ppm(s,1h).
13
c nmr(100mhz,(cd3)2so,25℃):δ28.11,42.21,53.57,55.37,58.59,82.22,107.79,111.89,112.27,122.81,125.60,130.34,134.37,135.58,139.74,150.08,151.58,157.42,167.91,170.03ppm.hrms(m/z)anal.calc’d for c
30
h
38
n4o5(m+h)
+
:535.2915,found(m+h)
+
:535.2917.
[0157]
本实施例的合成路线如下:
[0158][0159]
实施例9
[0160]
(1)将3.34g甘氨酸叔丁酯盐酸盐和0.80g氢氧化钠在50ml乙醇溶液中混合搅拌3小时,然后加入1.49g 4-二甲氨基苯甲醛,室温搅拌过夜12h。
[0161]
(2)向上述反应溶液中加入制备好的3.16g n-(1-乙氧基乙二烯)-甘氨酸甲酯,室温下搅拌过夜24h,反应结束后将反应用水淬灭,加入二氯甲烷萃取,收集有机相成分浓缩,最后利用柱色谱层析法将其纯化。
[0162]
(3)将步骤(2)所得产物与3.0g 4-二甲氨基苯甲醛在5ml1,4-二氧六环溶液中混合,加入1m氯化锌1.0ml后回流过夜,除去溶剂后用柱色谱层析法纯化,即得最终产物。
[0163]
将最终产物使用核磁共振波谱仪进行测试,液相色谱高分辨飞行时间质谱q-tof 6540进行结构表征和纯度测定,结果如下:1h nmr(400mhz,cdcl3,25℃):δ8.09(d,j=8hz,2h),7.89(d,j=16hz,1h),7.40(d,j=8hz,2h),7.01(s,1h),6.63(m,4h),6.33(d,j=16hz,1h),4,35(s,2h),2.94(br,d,12h),1.37(s,9h)ppm.
13
c nmr(100mhz,cdcl3,25℃):δ14.37,26.94,39.13,41.34,81.72,106.19,110.83,121.99,122.42,126.58,128.50,133.32,139.21,150.45,155.50,157.01,166.00,169.04.hrms(m/z)anal.calc’d for c
28
h
34
n2o3(m+h)
+
:474.2704,found(m+h)
+
:475.2689.
[0164]
实施例10仿生荧光探针在缓冲液中通过荧光激活探测重组蛋白质聚集态的方法
[0165]
对实施例1至实施例9中制备得到的仿生荧光探针进行在缓冲液中通过荧光激活探测重组蛋白质聚集态测试,具体操作步骤为:取实施例中制备的仿生荧光探针(10μm),与重组纯化的二氢叶酸还原酶(dhfr)(50μm)在酸诱导聚集缓冲液(naoac 200mm,kcl 100mm,通过冰醋酸酸化至ph=6.23)在25℃-37℃下进行孵育(5min)处理。引发蛋白质聚集的方法为加热(37℃-95℃)5min。蛋白质发生聚集后,利用荧光酶标仪tecan spark进行定量的荧光强度测量,考察荧光激活强度。
[0166]
以实施例1为典型代表,图2(a)为使用630nm作为发射波长,采集400-615nm的激发信号,图2(b)为使用560nm作为激发波长,采集575-800nm的发射信号。如图可看出,荧光分子在dhfr蛋白质未发生错误折叠与聚集时不发出荧光。当dhfr因为热导致聚集时,荧光分子与聚集态dhfr结合并发出强烈荧光,荧光增益为33倍。最大激发为540nm,最大发射为
610nm。
[0167]
实施例11仿生荧光探针在缓冲液中通过热转移荧光曲线测量药物分子与目标靶点蛋白的结合
[0168]
对实施例1至实施例9中制备得到的仿生荧光探针进行通过热转移荧光曲线测量药物分子与目标靶点蛋白的结合,具体操作步骤为:取实施例中制备得到的仿生荧光探针(1nm-100μm),与重组纯化的二氢叶酸还原酶(dhfr)(1μm-1000μm)在25℃-37℃下进行孵育(5min-30min)处理。通过温度梯度加热引发蛋白质聚集。加热的温度梯度方法为:(1)利用pcr或恒温加热装置在37℃-95℃之间每隔1℃-10℃中任意数值温度间隔,进行样本加热,然后分别读取荧光强度,绘制温度荧光曲线;(2)利用实时荧光pcr仪,从37℃到95℃每分钟1℃-10℃中任意数值温度递增,并实时记录荧光强度,绘制实时荧光曲线。通过比较导致蛋白聚集临界温度的变化,考察加入药物分子是否能够迁移荧光曲线,推到分子是否结合目标靶点蛋白。分子如果结合目标靶点蛋白,即证明小分子作用于蛋白质,可用于筛选目标蛋白的小分子抑制剂或者激活剂等具有药物活性的小分子。
[0169]
以实施例1为典型代表,结果如图3所示,由图3可看出,荧光分子在dhfr加热引发聚集时会发出荧光。加热温度上升,引发的聚集程度加剧,因此荧光强度逐步上升。当dhfr蛋白质结合甲氧苄啶tmp小分子时,稳定性上升,需要更高的温度引发其发生聚集并发出荧光。横轴为摄氏温度,纵轴为相对荧光强度。证明了tmp结合dhfr并稳定dhfr蛋白,tmp分子是一个已知的dhfr抑制剂,是常见的抗生素。因此用tmp与dhfr的强结合力,验证我们的方法可用于筛选其他蛋白质的抑制剂。
[0170]
实施例12仿生荧光探针在活细胞中通过荧光成像探测致病蛋白质聚集态的方法
[0171]
对实施例1至实施例9中制备得到的仿生荧光探针进行在活细胞中通过荧光成像探测致病蛋白质聚集态,具体操作步骤为:取实施例中制备的仿生荧光探针(1nm-50μm),放入人胚胎肾细胞293细胞系(hek293)中培养基中,同时在细胞中表达导致亨丁顿舞蹈症的致病蛋白htt97。12小时至72小时后,通过各自荧光显微镜识别聚集态htt97蛋白质在活细胞中聚集的形貌和位置。
[0172]
以实施例1为典型代表,结果如图4和图5所示,由图4可看出,在人胚胎肾细胞293细胞系(hek293)中表达导致亨丁顿舞蹈症的致病蛋白htt97后,通过本发明提供的荧光探针识别聚集态htt97在活细胞中聚集的位置(白色箭头)。由图5可看出,在人胚胎肾细胞293细胞系(hek293)中加入蛋白酶体抑制剂mg132,通过本发明提供的荧光探针广谱识别聚集态蛋白质在活细胞中聚集的位置,其中最大激发为540nm,最大发射为610nm。
[0173]
实施例13仿生荧光探针在活细胞中通过荧光成像探测药物导致蛋白质聚集态的方法
[0174]
对实施例1至实施例9中制备得到的仿生荧光探针进行在活细胞中通过荧光成像探测药物导致蛋白质聚集态,具体操作步骤为:取实施例中制备的仿生荧光探针(1nm-50μm),放入人宫颈癌细胞系(hela)中加入热休克蛋白90抑制剂17aag(1nm-50μm)或者蛋白酶体抑制剂mg132(1nm-50μm)。12小时至72小时后,通过各自荧光显微镜识别聚集态蛋白质在活细胞中聚集的形貌和位置。
[0175]
以实施例1为典型代表,结果如图6所示,图6为在人宫颈癌细胞系(hela)中加入热休克蛋白90抑制剂17aag,通过本发明提供的荧光探针广谱识别聚集态蛋白质在活细胞中
聚集的位置。最大激发为540nm,最大发射为610nm。
[0176]
对比例1
[0177]
取现有商用试剂盒用于探测人宫颈癌细胞系(hela)中加入热休克蛋白90抑制剂17aag(1nm-50μm)所引起的胞内蛋白质聚集。取本发明所述实施例中制备的仿生荧光探针用于同样条件下实验,二者进行同步对比型荧光成像实验。
[0178]
以实施例1为典型代表,结果如图7所示,图7为人宫颈癌细胞系(hela)中加入热休克蛋白90抑制剂17aag后,商用试剂盒与本发明所述荧光探针活细胞探测聚集蛋白质的成像对比图,实验发现,现有商用试剂盒无法进入活细胞,导致荧光背景高,无法观测胞内蛋白质聚集状态。相应的,本发明所述实施案例1中制备的亚苄基咪唑啉酮类仿生荧光探针,可在活细胞中进行聚集蛋白质的直接观测。
[0179]
以上所述,仅是本技术的几个实施例,并非对本技术做任何形式的限制,虽然本技术以较佳实施例揭示如上,然而并非用以限制本技术,任何熟悉本专业的技术人员,在不脱离本技术技术方案的范围内,利用上述揭示的技术内容做出些许的变动或修饰均等同于等效实施案例,均属于技术方案范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1