3-溴-6-氯吡啶甲酰胺的合成方法与流程
本发明属于药物中间体合成技术领域,具体涉及一种3-溴-6-氯吡啶甲酰胺的合成方法。
背景技术:
3-溴-6-氯吡啶甲酰胺是关键医药中间体,在药物设计中起到广泛作用。其中吡啶环作为含氮杂环的一种,在药物设计中常用于对苯环的替代,对配体有效构象或有利的静电相互作用产生积极影响,或者有利于配体与受体之间形成活性所需的氢键作用,进而优化化合物的生化活性、细胞活性和靶标选择性,甚至可能改变药理功能。因此3-溴-6-氯吡啶甲酰胺被广泛使用。例如bradnerje等人合成了一种新型的异恶唑并吡啶类化合物,作为溴结构域抑制剂。研究发现溴结构域在肿瘤治疗中有潜在的应用价值,因此得到广泛关注,溴结构域抑制剂在不同癌症疾病例如非小细胞肺癌、神经母细胞瘤甲状腺癌、卵巢癌和前列腺癌都有非常好的抑制作用。同时在白血病、淋巴瘤、骨髓瘤和恶性胶质瘤等非实体瘤也有良好抑制活性。其中3-溴-6-氯吡啶甲酰胺作为合成溴结构域抑制剂的关揵医药中间体,起到了非常重要的作用。(wo2012075383a2)因此3-溴-6-氯吡啶甲酰胺在有机合成界尤其是药物化学界引起了广泛的关注。从3-溴-6-氯吡啶甲酰胺出发,合成具有特定结构的分子,然后通过此分子的结构修饰进行sar的研究以其得到高效的药物候选分子的研究开发工作具有重要的意义。
现有3-溴-6-氯吡啶甲酰胺的合成困难,市场价格昂贵,缺少文献以及相关专利报道。
技术实现要素:
本发明的目的在于提供一种全新的3-溴-6-氯吡啶甲酰胺的合成方法,为3-溴-6-氯吡啶甲酰胺的合成路线开发填补了空白。
为了实现上述目的,本发明提供以下技术方案:
3-溴-6-氯吡啶甲酰胺的合成方法,包括以下步骤:
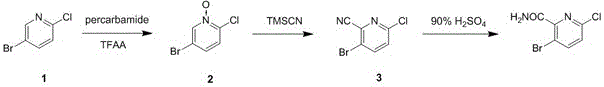
(1)5-溴-2-氯吡啶一氧化物的合成
以5-溴-2-氯吡啶为原料进行氧化反应得到5-溴-2-氯吡啶一氧化物;
(2)3-溴-6-氯吡啶-2-腈的合成
5-溴-2-氯吡啶一氧化物进行氰基化反应得到3-溴-6-氯吡啶-2-腈;
(3)3-溴-6-氯吡啶甲酰胺的合成
3-溴-6-氯吡啶-2-腈在酸性条件下水解得到3-溴-6-氯吡啶甲酰胺。
进一步的,所述步骤(1)的具体合成步骤如下:
在三口瓶中分别加入溶剂三氯甲烷、化合物5-溴-2-氯吡啶、过碳酸钠和tfaa,反应体系常温搅拌过夜;tlc监测反应,待反应结束后,反应液加入饱和连二亚硫酸钠溶液搅拌,三氯甲烷萃取混合液,混合液分离,有机相再加入饱和碳酸氢钠水溶液进行洗涤,浓缩,干燥;乙酸乙酯溶解固体,过滤出不溶固体,乙醚洗涤固体,干燥,得到黄色固体。
进一步的,所述步骤(2)的具体合成步骤如下:
三口瓶中加入乙腈、化合物5-溴-2-氯吡啶一氧化物、tmscn和tea,进行回流搅拌反应;反应用tlc监测,待反应完全后,浓缩反应液,得到粗品,柱层析纯化得到3-溴-6-氯吡啶-2-腈。
进一步的,所述步骤(2)中回流搅拌反应的时间为10-15h。进一步的,所述步骤(3)的具体合成步骤如下:
在三口瓶中加入90%硫酸、3-溴-6-氯吡啶-2-腈,将反应体系加热并搅拌反应,tlc监测反应,待反应结束后,带反应体系冷却至室温导入冰水中,过滤不溶固体,水洗并干燥得到3-溴-6-氯吡啶甲酰胺。
进一步的,所述步骤(3)中反应体系加热的温度是120-130℃,反应时间是4-6h。
有益效果:本发明提供了一种3-溴-6-氯吡啶甲酰胺的合成方法,该方法解决了现有合成方法中原料昂贵、收率低、使用微波反应等条件、不易放大等缺点,本发明所述合成路线简单、工艺选择合理、原料成本低、原料简单易得、操作和后处理方便、总收率高、不使用剧毒试剂、易于放大,可进行大规模生产。
附图说明
图1为本发明所述3-溴-6-氯吡啶甲酰胺合成的化学式。
具体实施方式
下面结合具体实施例来进一步描述本发明,但实施例仅是范例性的,并不对本发明的范围构成任何限制。本领域技术人员应该理解的是,在不偏离本发明的精神和范围下可以对本发明技术方案的细节和形式进行修改或替换,但这些修改和替换均落入本发明的保护范围内。
本发明所述3-溴-6-氯吡啶甲酰胺的合成工艺以5-溴-2-氯吡啶为原料进行氧化反应得到5-溴-2-氯吡啶一氧化物,再进行氰基化反应得到3-溴-6-氯吡啶-2-腈,最后在酸性条件下水解得到3-溴-6-氯吡啶甲酰胺。反应式如下:
具体合成步骤如下所示:
第一步:5-溴-2-氯吡啶一氧化物的合成
在500ml的三口瓶中,分别加入溶剂三氯甲烷200ml,化合物5-溴-2-氯吡啶(32.4g,169mmol),过碳酸钠(33.2g,350mmol)和tfaa(47.6ml,336mmol)。反应体系常温搅拌过夜。tlc监测反应,待反应结束后,反应液加入饱和连二亚硫酸钠溶液搅拌40min,三氯甲烷萃取混合液,混合液分离,有机相再加入饱和碳酸氢钠水溶液进行洗涤,浓缩,干燥。乙酸乙酯(200ml)溶解固体,过滤出不溶固体,乙醚洗涤固体,干燥,得到黄色固体(24.1g得率为68%),无需进一步纯化即可用于下一步反应。
第二步:3-溴-6-氯吡啶-2-腈的合成
1l的三口瓶中,加入575ml乙腈,化合物5-溴-2-氯吡啶一氧化物(24g,116mmol),tmscn(45.7g,461mmol)和tea(34.8g,345mmol)。反应回流搅拌反应12h。反应用tlc监测,待反应完全后,浓缩反应液,得到粗品,柱层析纯化得到3-溴-6-氯吡啶-2-腈(10g,40%)。
第三步:3-溴-6-氯吡啶甲酰胺的合成
在250ml三口瓶中,加入90%硫酸100ml,3-溴-6-氯吡啶-2-腈(10.1g,46.8mmol)反应体系加热至120℃并搅拌反应4h。tlc监测反应,待反应结束后,带反应体系冷却至室温导入冰水(500ml)中,过滤不溶固体,水洗并干燥得到3-溴-6-氯吡啶甲酰胺(8.1g,73.1%)
1hnmr(400mhz,dmso-d6)δ8.21(d,j=7.6hz,1h),8.09(s,1h),7.85(s,1h),7.56(d,j=8.0,1h);esi+-ms,m/z:236.9[m+h]+。
技术特征:
1.3-溴-6-氯吡啶甲酰胺的合成方法,其特征在于,包括以下步骤:
(1)5-溴-2-氯吡啶一氧化物的合成
以5-溴-2-氯吡啶为原料进行氧化反应得到5-溴-2-氯吡啶一氧化物;
(2)3-溴-6-氯吡啶-2-腈的合成
5-溴-2-氯吡啶一氧化物进行氰基化反应得到3-溴-6-氯吡啶-2-腈;
(3)3-溴-6-氯吡啶甲酰胺的合成
3-溴-6-氯吡啶-2-腈在酸性条件下水解得到3-溴-6-氯吡啶甲酰胺。
2.根据权利要求1所述的3-溴-6-氯吡啶甲酰胺的合成方法,其特征在于,所述步骤(1)的具体合成步骤如下:
在三口瓶中分别加入溶剂三氯甲烷、化合物5-溴-2-氯吡啶、过碳酸钠和tfaa,反应体系常温搅拌过夜;tlc监测反应,待反应结束后,反应液加入饱和连二亚硫酸钠溶液搅拌,三氯甲烷萃取混合液,混合液分离,有机相再加入饱和碳酸氢钠水溶液进行洗涤,浓缩,干燥;乙酸乙酯溶解固体,过滤出不溶固体,乙醚洗涤固体,干燥,得到黄色固体。
3.根据权利要求1所述的3-溴-6-氯吡啶甲酰胺的合成方法,其特征在于,所述步骤(2)的具体合成步骤如下:
三口瓶中加入乙腈、化合物5-溴-2-氯吡啶一氧化物、tmscn和tea,进行回流搅拌反应;反应用tlc监测,待反应完全后,浓缩反应液,得到粗品,柱层析纯化得到3-溴-6-氯吡啶-2-腈。
4.根据权利要求3所述的3-溴-6-氯吡啶甲酰胺的合成方法,其特征在于,所述步骤(2)中回流搅拌反应的时间为10-15h。
5.根据权利要求1所述的3-溴-6-氯吡啶甲酰胺的合成方法,其特征在于,所述步骤(3)的具体合成步骤如下:
在三口瓶中加入90%硫酸、3-溴-6-氯吡啶-2-腈,将反应体系加热并搅拌反应,tlc监测反应,待反应结束后,带反应体系冷却至室温导入冰水中,过滤不溶固体,水洗并干燥得到3-溴-6-氯吡啶甲酰胺。
6.根据权利要求5所述的3-溴-6-氯吡啶甲酰胺的合成方法,其特征在于,所述步骤(3)中反应体系加热的温度是120-130℃,反应时间是4-6h。
技术总结
本发明公开了一种3‑溴‑6‑氯吡啶甲酰胺的合成方法,该方法以5‑溴‑2‑氯吡啶为原料进行氧化反应得到5‑溴‑2‑氯吡啶一氧化物,再进行氰基化反应得到3‑溴‑6‑氯吡啶‑2‑腈,最后在酸性条件下水解得到3‑溴‑6‑氯吡啶甲酰胺。该方法解决了现有合成方法中原料昂贵、收率低、使用微波反应等条件、不易放大等缺点,本发明所述合成路线简单、工艺选择合理、原料成本低、原料简单易得、操作和后处理方便、总收率高、不使用剧毒试剂、易于放大,可进行大规模生产。
技术研发人员:晋浩文;徐卫良;徐炜政
受保护的技术使用者:苏州康润医药有限公司
技术研发日:2020.09.03
技术公布日:2021.03.16
- 还没有人留言评论。精彩留言会获得点赞!