碱性磷酸酶的下游处理的制作方法
分案说明本申请为申请日为2015年1月26日,申请号为201580015952.3,题目为“碱性磷酸酶的下游处理”的专利申请的分案申请。本发明涉及碱性磷酸酶(ap)的下游处理(dsp)领域。更具体地,本发明涉及用于降低包含ap的组合物中宿主细胞蛋白含量的方法。本发明进一步涉及包含ap和降低含量的宿主细胞蛋白的组合物。
背景技术:
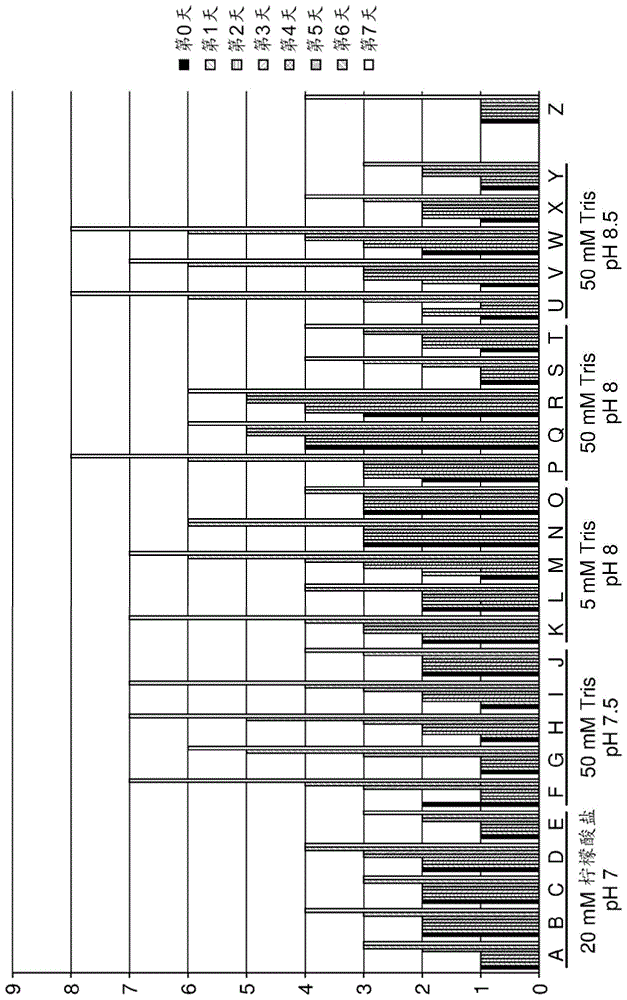
:碱性磷酸酶(ap)ap;根据iubmb酶命名法ec3.1.3.1,常用名为碱性磷酸酶(ap)是一种将磷酸酶单酯和h2o催化成为醇和磷酸盐的反应的酶。ap的其他名称为碱性磷酸单酯酶;磷酸单酯酶;甘油磷酸酶;碱性磷酸水解酶;碱性苯基磷酸酶;正磷酸单酯磷酸水解酶(碱性最佳)。ap的系统命名为磷酸单酯磷酸水解酶(碱性最佳)。ap是一种广谱特异性酶,其也催化转磷酸作用。在人类和其他哺乳动物中,已知存在至少四种不同的,但相关的碱性磷酸酶。其为肠(alpi)、胎盘(alpp;仅在人和灵长目动物中)、胎盘样(gcap)和肝/骨/肾(或组织非特异性)碱性磷酸酶(tnap)。前三种一起位于染色体2上,而组织非特异性形式位于染色体1上。碱性磷酸酶的氨基酸序列以及催化结构域和冠状结构域的相对位置是本领域技术人员公知的。例如,可参照millán的关于哺乳动物碱性磷酸酶的教科书(mammalianalkalinephosphatases,wiley-vch(2006),isbn-13:978-3-527-31079-1),其中示出了四种人碱性磷酸酶的氨基酸序列等。用于药物用途的ap之前已经显示ap作为许多疾病的药物是有益的(急性肾损伤(aki)、败血症、炎性肠病(ibd)等)。这些研究使用天然存在的ap,例如分离的牛ap以及分离和重组人ap。在一些动物模型中,已经使用了重组嵌合的碱性磷酸酶,其包括人肠ap的催化结构域和人胎盘ap的冠状结构域(描述在wo2008133511中)。目前,包含具有图18中所示序列(seqidno:1)的新型和改善的重组嵌合的碱性磷酸酶的药物组合物正在被开发用作药物。在药物组合物的开发过程中,使用蛋白质并且尤其是ap的标准(dsp)方法,例如亲和色谱(mimeticblue)、阴离子交换色谱(poros50)以及混合模式色谱(capto-)。在优化dsp步骤之后并且药物组合物符合之前监管当局设定和要求的规格之后,制备用于临床批次生产的制剂。然而,在最后阶段稳定性测试期间,观察到了颗粒形成。这是一个无法预见的问题,而由于颗粒形成是药物组合物中有害的方面,必须找到解决方案。不清楚颗粒的性质如何,并且通过离心收集颗粒和分析之后,在银染色的非还原性sds-page凝胶中富集了非ap条带。该条带的酶促消化的质谱分析揭示了存在宿主细胞蛋白(hcp)而不是ap。这是尤其不可预见的,因为根据之前对宿主细胞蛋白进行的测试,组合物中如此少量的hcp(<100ppm)无法解释相对大量的颗粒。宿主细胞蛋白(hcp)hcp是由生产过程中使用的细胞或生物体产生或编码,并且与期望的产物不相关的蛋白。一些hcp对于生长、存活和正常细胞加工是必需的,而其他的hcp可能不是必需的。就像期望的产物,hcp也可以通过宿主由一些翻译后修饰而被修饰。无论hcp的用途如何或是否缺少,在最终的药物物质中通常不希望有hcp。虽然通常以少量存在(百万分之一,表示为纳克/毫克期望的蛋白),但是工业上花费了更多的努力和成本以将其去除(wang等人;biotechnologyandbioengineering,vol.103,no.3,june15,2009)。根据美国食品和药品管理局的“考虑要点(pointstoconsider)”文件和欧盟的“指导说明(notesforguidance)”,在批准用于治疗用途的生物产品之前,必须对产品中残留hcp的水平进行定量测量。因此,在dsp期间,通常消除hcp并且必须证实消除。目前用于分析重组生物产品中污染物hcp的分析方法包括sds-page、免疫印记技术和elisa。还有有关除去hcp污染以及利用例如疏水相互作用色谱(shukla等人,biotechnol.prog.2002,18,556-564)、蛋白质a色谱(shukla等人,biotechnol.prog.2008,24,11151121)或耐盐阴离子交换配体(riordan等人,biotechnol.prog.2009,vol.25,no.6)除去的出版物。因此,需要优化的dsp和制剂以提供包含具有降低的hcp含量和改善的物理稳定性的碱性磷酸酶的药物组合物。本发明的发明人已经采用了dsp和最终药物制剂,从而a)降低hcp的含量和b)减少观察到的颗粒形成。采用dsp的主要目的是降低hcp的含量,而采用制剂的主要目的是减少由于任何残留hcp造成的(可见)颗粒的形成。采用dsp和采用制剂在稳定性测试期间协同减少了颗粒形成,因此改善了药物组合物的物理稳定性。至少当组合时,改善的dsp和新剂型得到药物组合物,其中该组合物具有降低的hcp含量并且其中在稳定性测试期间无可见颗粒形成。在第一个实施方式中,本发明提供了用于产生包含分离的碱性磷酸酶和降低含量的宿主细胞蛋白(hcp),优选少于100ppm的hcp的组合物的方法,该方法包括第一纯化步骤,该第一纯化步骤由以下步骤组成:-提供包含具有下式配体的固相:其中r表示将配体与固相连接的间隔分子,-使所述配体与包含与seqidno:1具有至少90%序列同一性的分离的ap和hcp的组合物接触;-实施数个洗涤步骤,其中至少一个洗涤步骤是利用洗涤缓冲液进行的,所述洗涤缓冲液能够将至少部分hcp与配体分离同时保持至少部分ap与配体结合,和-利用能够将至少部分所述ap与配体分离的洗脱缓冲液获得ap。优选地,当进行该最后步骤时,至少部分hcp保持与配体结合或在固相上。氨基酸或核酸序列的同一性百分比,或术语“%序列同一性”在本文中被定义为在必要时为了实现最大百分比同一性,在两个序列进行比对并引入缺口之后,候选氨基酸或核酸序列中的残基与参比序列中残基相同的百分比。在优选的实施方式中,所述至少百分比序列同一性的计算是在不引入缺口的情况下进行的。用于比对的方法和计算机程序是本领域已知的,例如,“align2”或国家生物信息中心(nationalcenterforbiotechnologyinformation,ncbi)提供的blast服务。dsp包括涉及亲和纯化的纯化步骤,其利用具有下式的针对碱性磷酸酶的已知配体:其中r为反应性基团或与固相连接的间隔分子,其中,洗涤步骤已被优化以达到高纯度并且产生降低的hcp含量,从而使所得包含ap的组合物的物理稳定性最优。柱形式的式(i)的配体可以商标名mimeticblueaptm(mmbap;prometic,uk)商购获得,其中r为间隔分子,其用于产生柱子的固相基质和配体之间的距离。该距离确保大的ap分子能够与配体有效结合。式i的配体是本领域中已知的,并且描述在例如lindner等人(jchromatography,473(1989)227-240;图1,化合物iv)中,用于小牛肠碱性磷酸酶(ciap)的纯化。lindner等人已经显示该配体用于肠碱性磷酸酶(alpi)的dsp。然而,lindner并未描述减少hcp等,同时由于lindner并没有纯化ap用于治疗用途,因此他并没有遇到hcp污染的问题。lindner等人利用用于纯化的标准洗涤和洗脱缓冲液实现了ciap的330倍纯化。然而,如本发明人所发现的,lindner所叙述的条件并没有解决在dsp用于药物制造过程中观察到的颗粒形成和hcp污染的问题。对于本申请而言,“宿主细胞”意思是含有活性(可选地修饰的)ap基因的动物或人细胞,并且该ap基因体内(例如,在非人动物中)或体外(细胞培养)在该细胞中转录和翻译。该ap基因可作为外源基因引入该宿主细胞中,优选与作为活性内源性基因或作为内源性非活性基因被激活而已存在于宿主细胞中的调控元件一起被引入。这样的内源性基因的激活能够例如通过将调控元件特异性引入到基因组中来实现,例如通过同源重组。哺乳动物细胞通常被用作宿主细胞。如果引入外源ap基因,中华仓鼠卵巢(cho)或人胚肾(hek)细胞可例如用作宿主细胞。hcp能够存在于获自表达内源性ap的(未修饰的)动物细胞的制剂中,在这种情况下,所获得的ap蛋白通常被称为“纯化的”或“分离的”。相对而言,“重组”蛋白通常认为是如上文所述通过包含外源性(可选地经修饰的)ap基因的细胞表达或者通过同源重组表达ap的细胞表达的蛋白。重组蛋白也可被称为“分离的”,例如当其被纯化时。应理解,根据本发明的方法可用于内源性表达的ap以及重组表达的ap。应注意,术语“分离的”和“纯化的”可以在重组蛋白的dsp的过程中使用,以表示不同的纯化步骤,例如从细胞分离和纯化蛋白,以除去污染物。如本申请中所使用的,术语分离的和纯化的可以具有后者的含义,这取决于这些术语使用的语境。然而,对于包含新型和改善的重组碱性磷酸酶的组合物,标准纯化方法并不能充分除去hcp。另外,用于hcp测量的商业方法低估了hcp含量,尤其是因为ap与hcp共纯化这一事实等,因为二者均具有相似的等电点并且可能是由于在纯化过程中hcp与ap结合。在组合物中hcp的存在进一步导致颗粒形成,这也是药物组合物中不希望发生的,原因是颗粒可能包含不溶性蛋白质聚集体,其比可溶性蛋白具有更大的免疫原性。当面临用于制药用途的组合物含有相对较高浓度的hcp并且当储存时形成颗粒的问题时,发明人找到了一种解决方案。因此,待解决的一个问题是提供包含特定重组碱性磷酸酶的组合物,该组合物包含低含量的hcp并且优选在2-8℃下持续2个月的稳定性测试期间不出现可见的颗粒形成。可见的颗粒形成意思是本领域技术人员可用肉眼,可选地利用放大装置,例如放大镜观察到的颗粒。在监管的情况下,可见的和不可见的颗粒物之间存在差异。可见的颗粒宽松定义为能够用肉眼观察到的任何颗粒。通常,可见物体定义为0.05mm或更大的物体。如本发明中使用的,术语“可见的颗粒”意思是0.05mm或更大,优选0.1mm或更大,更优选0.2mm或更大,更优选0.5mm或更大,最优选1mm或更大的颗粒。也可以用设计为检测这样的可见的颗粒的设备来检测可见的颗粒。在如同根据本发明的(通过本发明方法获得的)组合物的清澈溶液中自动检查颗粒的最常用方法是搅拌该溶液并使该溶液随着时间的推移而成像。成像系统通常由机械视觉相机、光源(在该情况下为背光)和分析图像的视觉处理器组成。一旦已经获得了图像,接着按顺序分析图像与图像之间的差异。该差异能够被解释为在溶液内部运动的物体,例如气泡和颗粒。在较大致密颗粒的情况下,通过从分析中滤除气泡来进行检测,因为随着颗粒的下沉气泡将会上浮。如果目标是找到直径小于约1mm的颗粒,则必须进行更谨慎的搅拌方法以从视野中除去气泡。这可通过旋转搅拌溶液来实现,并且小心注意加速和减速速率。然而,为了本发明的目的,检测方法对于定义“可见的颗粒”的并不重要,只要颗粒具有如上定义的直径即可。如上文所述,重组碱性磷酸酶的标准dsp和制剂不能产生这样的组合物,因为目前hcp含量超过通常使用的标准100ppm并且在稳定性测试期间观察到了颗粒形成。为了满足监管标准,根据本发明的方法优选产生相对于碱性磷酸酶含量包含少于100ppmhcp的组合物。在优选的实施方式中,洗涤缓冲液进一步包含mgcl2、zncl2和tris。在新的和创造性洗涤步骤中,这些组分对于除去hcp不是必要的,但是对于碱性磷酸酶的稳定性特性方面总体上是有利的,因为ap是ph敏感的金属结合酶。优选地,mg以大于0.1mm的浓度存在。优选地,mg浓度不超过100mm。mg浓度优选在0.1-100mm之间,更优选在0.5-20mm之间,更优选在1-5mm之间,最优选为约2mm。zn优选以大于10μm的浓度存在。优选地,zn浓度不超过1mm。zn浓度优选在10-1000μm之间,更优选在20-100μm之间,更优选在40-60μm之间,最优选为约50μm。固相可以为例如树脂并且通常为柱的形式,其是可商购的。优选在根据本发明的方法中使用柱,因为在dsp设定中其可以容易地扩大规模。适合用于本发明方法的柱为,例如,mimeticblue然而,也可以使用例如包含所述配体的微珠,并进行接触步骤和通过离心、倾析和溶解的洗涤步骤。在洗涤步骤中也可以使用磁珠和磁力分离。本领域技术人员熟悉用于亲和纯化的不同方法并且能够容易地将这样的方法应用于根据本发明的方法中。通常,利用由式(i)中的r表示的间隔分子将配体与固相连接。这样的间隔分子是本领域中已知的并且可以是任何适合的间隔分子,例如六甲基二胺(1,6-己二胺)(dye-ligandaffinityabsorbentsforenzymepurificationn.e.labroumolecularbiotechnologyvol20,2002p77-84)或3’,3’-二氨基二丙胺。在优选的实施方式中,本发明提供了根据本发明的方法,其中洗涤缓冲液包含20-100mm之间的arg,优选30-50mm之间的arg,最优选约40mm的arg。发明人出人意料地示出了,通过将上述洗涤步骤与用于ap的标准dsp结合,现可以使最终组合物中hcp含量降低至低于100ppm的值。因此,所述的洗涤步骤在这个方面能够提供符合针对药物组合物设定的规则的组合物,并且具有含量少于100ppm的hcp。在本发明之前不能获得这样的组合物,因为不使用本发明的新的和创造性dsp步骤的标准(最优)dsp步骤并不能得到包含少于100ppmhcp的组合物。此外,当这样的组合物在2–8℃下储存2个月时不显示可见的颗粒形成,而通过标准最优dps步骤制备的组合物则显示可见的颗粒形成。发明人已经进一步显示包含0.5-2m之间的尿素,优选1-2m之间的尿素,更优选约1m的尿素的洗涤缓冲液,当在根据本发明的方法中使用时也产生降低的hcp含量。包含尿素和arg二者的洗涤缓冲液也得到了非常好的结果。因此,进一步提供了根据本发明的方法,其中洗涤缓冲液包含0.5-2m之间的尿素,优选1-2m之间的尿素,更优选约1m的尿素。优选地,该洗涤缓冲液包含如上所述浓度的arg和尿素二者。向洗涤缓冲液中添加5-15%,优选约10%的乙二醇也提供了良好的结果。因此,本发明提供了根据本发明的方法,其中洗涤缓冲液包含5-15%之间的乙二醇,优选约10%的乙二醇。本发明进一步显示,为了增加hcp的下降和/或改善ap产率,洗涤缓冲液优选不含nacl。因此,在优选的实施方式中,提供了根据本发明的方法,其中优选包含如上文浓度的arg、尿素、乙二醇或它们的任意组合的洗涤缓冲液基本上不含nacl。基本上不含nacl意思是洗涤缓冲液优选包含少于1mm,更优选少于100μm,更优选少于10μm,更优选少于1μm,更优选少于100nm,更优选少于10nm,最优选少于1nm的nacl。减少的含量意思是当与其中在洗涤过程中不使用arg、尿素或乙二醇并且其中洗涤缓冲液中存在大量的nacl(例如大于1mm)的类似方法相比较时,在采用根据本发明的方法之后,hcp:ap之比更低。优选地,当将根据本发明的方法与dsp步骤的完整设定结合时,相对于ap含量,得到的hcp含量少于100ppm,更优选少于50ppm,更优选少于20ppm,更优选少于10ppm,更优选少于5ppm,更优选少于2ppm,更优选少于1ppm。类似地,提高的产率意思是相对于不使用根据本发明的洗涤步骤的方法,以apout对apin百分比测量的ap的产率更高,其中apout为处理步骤之后获得的ap量而apin为在洗涤之前与配体接触的ap的量。在根据本发明的方法中的洗涤步骤之后,相对于本发明之前的现有技术已知的方法,还改善了洗脱步骤。根据本发明,在根据本发明的方法中使用的洗脱缓冲液优选包含少于100mm的nacl,从而将至少一部分ap与具有式(i)的配体有效分离。通过利用包含约40mmarg,ph约为8且不含nacl的洗涤缓冲液与优选基本上不含nacl的洗脱缓冲液的结合已经获得了最好的结果。因此,在优选的实施方式中,提供了根据本发明的方法,其中该洗涤缓冲液包含约40mm的arg并且ph是约8,并且其中洗涤缓冲液和洗脱缓冲液基本上都不含nacl。基本上不含nacl意思是洗涤缓冲液和/或洗脱缓冲液优选包含少于1mm,更优选少于100μm,更优选少于10μm,更优选少于1μm,更优选少于100nm,更优选少于10nm,最优选少于1nm的nacl。在上述的亲和dsp步骤之后,用于碱性磷酸酶的完整dsp通常进一步包括用于纯化的步骤。一个这样的步骤是利用以下结构式(ii)描述的配体的所谓混合模式纯化步骤。该配体在本领域中称为“captoadhere”,并且与疏水相互作用和离子交换纯化组合。当进行混合模式纯化步骤时,本发明人已经改变了洗涤条件,从而进一步降低hcp含量,并具有最佳产率。因此,在优选的实施方式中,本发明提供了根据本发明的方法,该方法进一步包括第二纯化步骤,其包括以下步骤:-提供包含第二配体的第二固相,该第二配体具有下式:-使所述第二配体与包含分离的碱性磷酸酶和hcp的组合物接触,以及-进行数个洗涤步骤,其中至少一个洗涤步骤是利用第二洗涤缓冲液进行,该第二洗涤缓冲液的ph在7.5-8.5之间,优选在7.8至8.2之间,更优选为约8.0,并且包含0.05至0.2m之间的nacl,优选约0.1m的nacl,和0.1至0.5m之间的l-arg,优选0.15至0.3m之间的l-arg,更优选约0.2m的l-arg。在优选的实施方式中,所述第二洗涤缓冲液进一步包含1-20%之间,优选2-10%之间,更优选4-6%之间,最优选约5%的甘油。该固相也可以是例如树脂并且通常是柱的形式,其是可商购的。优选在根据本发明的方法中使用柱,因为在dsp设定中其可以容易扩大规模。适合用于本发明的方法的树脂是例如capto-其可以以柱的形式使用。然而,也可能使用例如包含所述配体的微珠,并进行接触以及通过离心、倾析和溶解的洗涤步骤。也可能在洗涤步骤期间使用磁珠和磁力分离。本领域技术人员熟悉用于亲和纯化的不同方法并且可容易改变这样的方法以用于根据本发明的方法。尽管dsp步骤的顺序可以是不同的,以适应有效地处理该组合物,但是在优选的实施方式中,所述第二纯化步骤在先,即,在所述第一纯化步骤之前进行。用于碱性磷酸酶的完整dsp可以并且的确优选包括本领域已知的其他纯化步骤,例如阴离子交换色谱、超滤/渗滤、病毒过滤、尺寸排阻色谱、亲和色谱和疏水相互作用色谱,以及它们的任意组合。在优选的实施方式中,根据本发明的方法包括至少三个,优选至少5个,更优选全部以下的步骤,优选按照如下列出的顺序:-混合模式色谱(例如captoadhere(式ii))-基于去垢剂的病毒灭活(例如tritonx-100)-阴离子交换色谱(例如poros50hq)-亲和色谱(例如mimeticblueap(式i))-疏水相互作用色谱(例如butyl650m)-超滤/渗滤-病毒过滤-散装填充(bulkfill)在一个实施方式中,本发明进一步提供了配制步骤,其可产生具有具有改善的物理稳定性的分离的ap组合物,优选已具有减少的hcp含量,从而在稳定性测试期间具有减少的颗粒形成,优选不出现可见的颗粒形成。优选地,用于生产包含分离的碱性磷酸酶和减少含量的hcp的组合物的根据本发明的方法在根据本发明的所述配制步骤之前。在优选的实施方式中,所述配制步骤包括在缓冲液中溶解或稀释洗脱的ap,从而所得组合物的ph在6.5至7.5之间,优选在6.8至7.2之间,更优选为约7.0,优选包含200-300mm的山梨糖醇,更优选包含225-275mm的山梨糖醇,最优选包含约250mm的山梨糖醇,和/或包含10-40%之间的甘油,更优选包含20-30%之间的甘油,最优选包含约25%的甘油。在优选的实施方式中,组合物包含10-40mm之间的柠檬酸盐,更优选包含15-30mm之间的柠檬酸盐,最优选包含约20mm的柠檬酸盐。在另一优选的实施方式中,组合物包含5-40mm之间的组氨酸,更优选包含10-30mm之间的组氨酸,更优选包含15-25mm之间的组氨酸,最优选包含约20mm的组氨酸。因此,本发明提供了用于产生包含分离的碱性磷酸酶(ap)的物理稳定的组合物的方法,该方法包括将ap在缓冲液中溶解或稀释,得到ph在6.5至7.5之间且优选包含200-300mm之间的山梨糖醇和/或10-40%之间的甘油的组合物。优选地,所述组合物包含5-40mm之间的组氨酸和/或10-40mm之间的柠檬酸盐。在优选的实施方式中,包含ap的组合物包含少于100ppm的hcp。优选地,所述组合物包含由用于产生包含分离的碱性磷酸酶且包含减少含量的hcp的根据本发明的方法获得的洗脱的ap。因此,在优选的实施方式中,本发明了提供了根据本发明的方法,其用于产生包含分离的或重组碱性磷酸酶且包含减少含量的hcp的组合物,所述方法进一步包括在缓冲液中溶解或稀释其中所获得的ap的步骤,得到的组合物ph为6.5至7.5之间,优选为6.8至7.2之间,更优选为约7.0,并且优选包含200-300mm的山梨糖醇,更优选包含225-275mm之间的山梨糖醇,最优选包含约250mm的山梨糖醇和/或包含10-40%之间的甘油,更优选包含20-30%之间的甘油,最优选包含约25%的甘油。在优选的实施方式中,组合物包含10-40mm之间的柠檬酸盐,更优选包含15-30mm之间的柠檬酸盐,更优选包含约20mm的柠檬酸盐,和/或5-40mm之间的组氨酸,更优选包含10-30mm之间的组氨酸,更优选包含15-25mm之间的组氨酸,最优选包含约20mm的组氨酸。优选地,将ap溶解或稀释于其中以获得该组合物的缓冲液包含镁(mg)盐,例如mgcl2和/或锌(zn)盐,例如zncl2。优选地,mg以大于0.1mm的浓度存在于组合物中。优选地,mg浓度不超过100mm。mg浓度优选在0.1-100mm之间,更优选在0.5-20mm之间,更优选在1-5mm之间,最优选为约2mm。优选地,zn以大于10μm的浓度存在。优选地,zn浓度不超过1mm。zn浓度优选在10-1000μm之间,更优选在20-100μm之间,更优选在40-60μm之间,最优选为约50μm。如前文中所讨论,从形成可见颗粒角度,mg和zn盐不一定影响组合物的物理稳定性,但是作为金属络合酶,当存在zn和mg时,ap通常更加稳定。组合物可以进一步包含nacl,优选以生理学浓度,即约0.9%w/v的nacl。当组合物用做药物时,尤其是用于静脉给药时这是尤其有用的。尽管根据本发明的方法也可用于其他重组蛋白,但优选利用包含与seqidno:1的氨基酸序列具有至少90%序列同一性,优选具有至少95%序列同一性,更优选具有至少98%的序列同一性,更优选具有至少99%序列同一性,最优选具有100%序列同一性的氨基酸序列的碱性磷酸酶的组合物来实施根据本发明的方法。在优选的实施方式中,宿主细胞蛋白为哺乳动物宿主细胞蛋白,优选为组织蛋白酶样蛋白(cata),更优选为组织蛋白酶a(cathepsina)的仓鼠同源物,和/或重组碱性磷酸酶在基于细胞的表达系统中表达,优选包含哺乳动物宿主细胞,更优选包含cho宿主细胞。基于本发明提供了用于产生包含分离的碱性磷酸酶和减少含量的宿主细胞蛋白的组合物的方法,本发明将进一步提供通过根据本发明的方法获得的这样的组合物。因此,在一个实施方式中,本发明提供了包含分离的碱性磷酸酶和药学上可接受的赋形剂的组合物,其特征在于,该组合物包含少于100ppm,更优选少于50ppm,更优选少于20ppm,更优选少于10ppm,更优选少于5ppm,更优选少于2ppm,最优选少于1ppm或更少的宿主细胞蛋白。优选地,在2-8℃,优选在约5℃下持续进行2个月,更优选3个月的稳定性测试期间,该组合物不显示大量可见的颗粒形成。没有大量可见的颗粒形成意思是在1ml组合物中形成<20个可见的颗粒。优选地,在储存组合物的专用条件下进行稳定性测试期间,每1ml组合物形成15个或更少,更优选10个或更少,优选为5个或更少的颗粒,最优选少于1个颗粒。优选地,根据本发明的组合物是通过根据本发明的方法获得的,或者可根据本发明的方法获得的。优选地,组合物的ph在6.5-7.5之间,优选为约7并且优选包含10-40mm之间,优选15-30mm之间,更优选为约20mm的柠檬酸盐和/或10-40%之间,优选20-30,更优选约25%的甘油,和/或200-300mm之间,优选225-275mm之间,更优选约250mm的山梨糖醇,和/或5-40mm之间的组氨酸,优选10-30mm之间,最优选约20mm的组氨酸。在优选的实施方式中,组合物的ph在6.5-7.5之间,优选为约7并且包含10-40mm之间的柠檬酸盐,优选约20mm或5-40mm之间,优选约20mm的组氨酸。发明人已观察到优异的物理稳定性,即,利用这样配制的组合物在2-8℃下持续2个月进行的稳定性测试期间没有大量可见的颗粒形成。如上文所述,本文中所使用的术语“可见的颗粒”代表直径为50μm或更大,优选100μm或更大,更优选500μm或更大,最优选1mm或更大的颗粒。该颗粒能够通过肉眼,可选地利用放大装置或通过自动化处理,例如之前所述的胶片摄影机以及用于分析膜材料的适合装置观察到。在优选的实施方式中,根据本发明的组合物包含已在基于细胞的表达系统中表达的分离的碱性磷酸酶,所述基于细胞的表达系统优选包含哺乳动物宿主细胞,更优选包含cho宿主细胞。在优选的实施方式中,hcp是一种组织蛋白酶样蛋白,更优选为cathepsina的仓鼠同源物。优选地,碱性磷酸酶的序列与seqidno:1的氨基酸序列具有至少90%的序列同一性,更优选至少95%的序列同一性,更优选至少98%的序列同一性,更优选至少99%的序列同一性,最优选100%的序列同一性。现本发明提供了减少hcp的方法和包含分离的碱性磷酸酶且包含少于100ppmhcp的组合物,本发明将进一步提供根据本发明的组合物作为药物的用途。根据本发明的组合物作为药物的用途优选用于治疗碱性磷酸酶相关的疾病。碱性磷酸酶相关的疾病意思是与碱性磷酸酶缺陷相关的疾病或病况,或者可通过外源性施用碱性磷酸酶得到改善的疾病或病况。尤其,碱性磷酸酶相关的疾病可以是以下疾病中的任一种:败血症或感染性休克、炎性肠病或胃肠道的其他炎性疾病、(急性)肾损伤或其他肾脏疾病、缺血性再灌注病况、(外科)创伤和低磷酸酯酶症。在以下的非限制性实施例中将更加详细地阐释本发明。附图说明图1.在第0-7天在指示的缓冲液中配制的碱性磷酸酶组合物中的颗粒形成。y-轴表示定性的颗粒形成,其中1=~10个颗粒/ml;2=10-15个颗粒/ml和/或更大的颗粒;3=~15个颗粒/ml和/或相当更大的颗粒;4=15-20个颗粒/ml和/或相当更大的颗粒;5=~20个颗粒/ml和/或相当更大的颗粒;6=~30个颗粒/ml和/或相当更大的颗粒;7=30-40个颗粒/ml和/或相当更大的颗粒;8=40-50个颗粒/ml和/或相当更大的颗粒。如图中所示,在每一组条中(在a、在b、在c等中的每一组),顺序(从左至右),即第一个条=第0天、第二个条=第1天等。a、b、c等表示不同的赋形剂,如在表2中所解释。图2.在第7天在指示的缓冲液中配制的碱性磷酸酶组合物中的颗粒形成。y-轴表示定性的颗粒形成,其中1=~10个颗粒/ml;2=10-15个颗粒/ml和/或较大的颗粒;3=~15个颗粒/ml和/或相当更大的颗粒;4=15-20个颗粒/ml和/或相当更大的颗粒;5=~20个颗粒/ml和/或相当更大的颗粒;6=~30个颗粒/ml和/或相当更大的颗粒;7=30-40个颗粒/ml和/或相当更大的颗粒;8=40-50个颗粒/ml和/或相当更大的颗粒。图3.在指示的缓冲液中配制的碱性磷酸酶组合物中的颗粒形成。y-轴表示定性的颗粒形成,其中1=无可见的颗粒;2=1-5个小颗粒/ml;3=6-15个小颗粒/ml;以及4=16-25个小颗粒/ml。图4.物理应力条件对在不同制剂中recap的高分子量(hmw)物质形成的影响。通过尺寸排阻色谱确定的recap蛋白的主峰百分比和百分比hmw的图示。图5.captoadhere中间洗涤对用于初筛的hcp清除和产物产率的影响。(a)对于每一种洗脱液,归一化的hcp(归一化至基线洗脱液)与产物产率的图示。在每次试验中测试的中间洗涤用对应于在图中呈现的那些有色圆圈来标记。(b)每个完成的实验中残留hcp水平和产率的表格。为每个试验列举了每个测试的中间洗涤的配方以及原始残留hcp数据、归一化的hcp以及产物产率。表格中呈现的彩色圆圈对应(a)中的标记。所有缓冲液配方含有2mm的mgcl2和50μm的zncl2。图6.用于检测处理样品中cata的蛋白质印迹分析。(a)对于约10μg的每一个处理中的样品,在非还原条件下,使用cata抗体检测进行蛋白质印记分析。(b)列举了对于每一个分析样品的鉴定,包括残留hcp和产率。基线对照实验洗脱液(泳道5)相比于包括中间洗涤的洗脱液(泳道2和4),含有更多的cata。泳道3表示在含有0.5mamso4,ph8.0的中间洗涤步骤期间除去的cata。所有的缓冲液配方含有20mm的tris、2mm的mgcl2和50μm的zncl2。图7.来自二次筛选的captoadhere中间洗涤对hcp清除和产物产率的影响。(a)对于每一种洗脱液,归一化的hcp与产物产率的图示。使用相同的上样材料将hcp值归一化至基线洗脱液。在每次试验中测试的中间洗涤用对应图中表示的那些的彩色圆圈来标记。(b)概述了图中示出的数据的表。列出了用于每一个试验的中间体测试洗涤条件和上样材料。也列出了对于每一种洗脱液的原始hcp数据、归一化的hcp值以及产物产率。表格中的彩色圆圈对应(a)中的标记。所有的中间体缓冲液配方也含有20mm的tris、2mm的mgcl2和50μm的zncl2。图8.比较来自初始筛选和二次筛选研究的有希望的候选物的抗-cata蛋白质印迹分析。(a)用抗catahcp抗体的蛋白质印记。(b)处理样品的银染色。(c)在蛋白质印记分析中每条泳道的样品鉴定。指示了每条泳道上样产物的量。也列出了每种洗脱液所得的hcp值。基线对照实验洗脱液(泳道3)比其中含有5%甘油(泳道7)或没有5%甘油(泳道5)的0.1mnacl、0.2ml-arg,ph8.0的中间洗涤的洗脱液含有更多的cata。泳道4和泳道6显示了存在5%甘油(泳道6)或不存在5%甘油(泳道4)的、含有0.1mnacl、0.2ml-arg,ph8.0的不同的洗涤步骤期间除去的cata。所有缓冲液配方也含有20mm的tris、2mm的mgcl2和50μm的zncl2。图9.mbap中间洗涤对于初筛的hcp清除和产物产率的影响。(a)对于每种洗脱液,hcp浓度与产物产率的图示。(b)每个完成的实验中,hcp水平和产率的表格。列举了每次试验每种中间洗涤缓冲液的配方、原始hcp数据和产物产率。表格中示出的彩色圆圈对应(a)中的标记。只有1m尿素洗涤在维持高产率的同时显著降低了hcp。图10:sds-page和蛋白质印迹分析;来自初筛的mbap试验1(基线)和试验4(1m尿素四次)。(a)用gelcodeblue染色的sds-page分析。试验1和试验4洗脱液显示相同的条带图案。(b)用cata抗体检测的蛋白质印记分析。基线对照洗脱液(泳道4)比试验4洗脱液(泳道6)含有更多的cata。泳道5描绘了在含有1m尿素,ph8.0的中间洗涤步骤期间除去的cata。图11:mbap中间洗涤对初筛的hcp清除和产物产率的影响。(a)对于每种洗脱液的hcp浓度与产物产率的图示。(b)每个完成的实验中,残留hcp水平和产率的表格。列出了每次试验所测试的每一中间洗涤的配方、洗脱缓冲液nacl浓度、hcp结果、归一化的hcp以及产物产率。表格中示出的彩色圆圈对应(a)中的标记。所有制备的中间缓冲液配方含有20mm的tris、2mm的mgcl2和50μm的zncl2。图12:多重hcp去除洗涤筛选试验的sds-page和蛋白质印迹分析(a)具有样品浓度和hcpelisa数据的凝胶上样表。(b)用cata抗体检测的蛋白质印记。(c)用银染色的sds-page分析。图13:mbap中间洗涤对最终洗涤步骤优化的试验的hcp清除和产物产率的影响。(a)对于每种洗脱液,hcp浓度与产物产率的图示。红色圆圈强调了试验28的结果。(b)每个完成的实验的hcp水平和产率的表格。对于每次试验,列出了每一种中间洗涤缓冲液的配方,以及原始hcp数据、归一化的hcp以及产物产率的结果。表格中示出的彩色圆圈对应(a)中的标记。在上样3grecap/l树脂的情况下,40mm精氨酸洗涤缓冲液显示出最大的hcp清除,以及高的recap回收率(试验28,以黄色强调)。图14.验证试验过程中间体的抗-cata蛋白质印迹分析。(a)用抗catahcp抗体的蛋白质印记分析。(b)处理样品的银染色。(c)蛋白质印记分析中每条泳道的样品鉴定。包括用于蛋白质印记和银染色的样品上样。图15:hcp减少验证试验过程中间体和bds的分析结果。图16.针对演示试验的对每种下游过程中间体的分析测试的总结。来自该演示试验的过程中间体的分析结果。图17.在组氨酸缓冲液和柠檬酸盐缓冲液中配制的实施例3中获得的配方的长达3个月的稳定性测试。图18.改进的重组碱性磷酸酶的氨基酸序列。实施例实施例中使用的定义amso4:硫酸铵;ap:碱性磷酸酶;au:吸光度单位;bds:散装药物;bh:床高;bpc:生物过程容器;cata:组织蛋白酶样蛋白;cch:细胞培养物收获;cho:中华仓鼠卵巢;cv:柱体积;elisa:酶联免疫吸附测定;eq:平衡;hcp:宿主细胞蛋白;hic:疏水相互作用色谱;hmw:高分子量物质;id:内径;l-arg:l-精氨酸;lod:检测限;loq:定量限;mbap:mimeticblue碱性磷酸酶树脂;nascn:硫氰酸钠;pd:工艺开发;pes:聚醚砜;ppm:百万分之一;psi:磅/平方英寸;qpcr:定量聚合酶链反应;recap:重组碱性磷酸酶;rp-hplc:反相高效液相色谱;sec-hplc:尺寸排阻色谱法hplc;sds-page:十二烷基磺酸钠聚丙烯酰胺凝胶电泳;tmp:跨膜压力;ufdf:超滤/渗滤;v/v:体积/体积;wfi:注射用水;实施例中使用的设备aktaavant;aktabioprocessskid(gehealthcare);aktaprocess;akta净化器;biophotometerplusuv-vis分光光度计(eppendorf);bpg10cm和14cm直径色谱柱(gehealthcare);eppendorfplusuv-vis分光光度计;fisherscientificmini离心机;地磅(ohaus);invitrogennovexminicell;mettlertoledosevenmultiph和电导率测量仪;milliporevantage-l柱;蠕动泵(millipore);摇摆式吊桶离心机;实施例中使用的材料10lbpc(hyclone);20lbpc(hyclone);100lbpc(hyclone)invitrogen4-12%bis-trissds凝胶;15nplanova中空纤维过滤器(asahikasei)5lbpc(hyclone);乙酸;butyl650m树脂(tosohbioscience);captoadhere树脂(gehealthcare);d-山梨糖醇;乙醇;l-精氨酸;盐酸l-精氨酸;l-组氨酸;六水氯化镁;mimeticblueap树脂(prometicbiosciences,ltd.);pellicon2超滤biomax10kda0.1m2(millipore);pelliconbiomax10kda50cm2uf/df盒(millipore);planova15n病毒过滤器(asahikasei);poros50hq树脂(appliedbiosystems);氯化钠;氢氧化钠,50%;磷酸钠;tris盐酸盐;氨丁三醇;tris碱;tris盐酸盐;wfi(hyclone);氯化锌实施例1配方稳定性研究am药物签约的cmo,美国kbibiopharma已经针对工艺开发和制造进行了大量的对于recap的配方研究。初始配方是基于用于biap的配方,其是之前由am药物开发的一种ap形式。biap显示在含有高水平甘油(25-40%)的tris缓冲液中是非常稳定的。初始缓冲液的组成如下:5mmtris-hcl、2mmmgcl2、50μmzncl2、25%(w/v)甘油,ph8。recap在该缓冲液中被配制为10mg/ml。还未确定recap在该缓冲液中的最大溶解度。然而,在5mmtris-hcl、2mmmgcl2、50μmzncl2,ph8.2中,已经实现了高达35mg/ml的recap的浓度。稳健性研究进行的第一配方稳定性,关注:·ph稳健性(7.5-8.5)和5对50mm的tris缓冲液浓度·赋形剂类型和浓度(甘油25%、2%对250mm山梨糖醇)·可注射性通过ph、dsc、dls、克分子渗透压重量浓度、粘度分析样品。在25%甘油配方中,通过降低ph的dls观察到recap的物理稳定性稍有下降。z-平均直径从10增加至12nm。样品均一性随ph(pdi从0.12至0.33)下降并且粒径分布的峰宽度增加。在2%甘油或250mm山梨糖醇配方中未观察到趋势。未充分考虑5mm配方的ph稳健性,但并未观察到明显的问题。在这些研究之后,将recap的配方缓冲液变为:50mmtris-hcl、2mmmgcl2、50μmzncl2、25%(w/v)甘油,ph8强力降解研究·热应力:在50℃下孵育1周。·冷冻/解冻应力:ο2.5ml直接在-75℃冰箱中冷冻24小时,接着在室温下解冻。ο2.5ml在冻干机中以0.05℃/分钟冷冻至-65℃,接着以0.05℃/分钟解冻至25℃。ο暴露于1、3或5个冷冻/解冻循环·搅拌应力:通过旋转支架和轨道振荡在室温下5天。·脱酰胺作用/碱水解:用1mtris碱将样品至ph≥10,在37℃下孵育五天。·脱酰胺作用/酸水解:用1nhcl将样品至ph≤4,在37℃下孵育五天。·氧化:在37℃下暴露于0.04%(v/v)过氧化氢4小时。·光稳定性:暴露于8.00klux的冷光持续150小时,之后暴露于10.0瓦特小时/平方米的uv光持续20小时。所有样品通过如下来分析:·sec·rp-hplc·活性(动力学)·cief·a280·sds-cge·pepmapw/lc/ms.加速稳定性研究利用来自10l/5-10克规模生产试验的散装药物进行加速稳定性研究(参见表1)。将recap样品在-75℃、2-8℃和25℃下储存并且每月对样品进行测试。在t=2个月时,没有观察到对ph和活性的影响。从蛋白质含量增加以及体积活性的增加看,25℃时的一些蒸发是明显的。来自t=2个月样品测试的数据集尚不完整。然而,显然在这些样品中颗粒形成持续发生。表1稳定性数据t=1个月颗粒形成和表征的研究在以10克规模产生的recap批次中观察到最新颗粒形成。在0.22μm过滤后数天内再次出现颗粒。外观上,颗粒看上去本质上含有蛋白质。尺寸分布分析表明较宽的尺寸分布。过滤使得较大颗粒减少100倍,但较小颗粒仅减少10倍。已经进行了第一次重新配方研究,其中测试了ph(7.0-8.5)、缓冲液类型(柠檬酸盐与tris)、缓冲液强度(5与50mmtris)和添加剂(山梨糖醇、蔗糖、甘油、nacl和精氨酸)(参见表2)。所有26个配方均不能防止过滤后的颗粒形成。然而,这些似乎倾向于更快地形成颗粒和较高的ph。表2颗粒评价更新-完整配方的视觉评估:基于颗粒含量将在7天的过程中配方筛选样品的外观分成1至8级(表3),并且将结果描绘在图1和图2中。表3可见颗粒的评分排序~10个颗粒/ml110-15个颗粒/ml和/或较大的颗粒2~15个颗粒/ml和/或相当更大的颗粒315-20个颗粒/ml和/或相当更大的颗粒4~20个颗粒/ml和/或相当更大的颗粒5~30个颗粒/ml和/或相当更大的颗粒630-40个颗粒/ml和/或相当更大的颗粒740-50个颗粒/ml和/或相当更大的颗粒8实施例2recap配方开发评价了三个配方来评估在冷冻-解冻和热应力期间recap的颗粒形成和稳定性/聚集。如下列举了所评价的配方:20mm组氨酸、250mm山梨糖醇、50μmzncl2、2mmmgcl2,ph7.020mm柠檬酸盐、250mm山梨糖醇、50μmzncl2、2mmmgcl2,ph7.050mmtris、25%甘油、50μmzncl2、2mmmgcl2,ph8.0该研究的目标蛋白质浓度为10mg/ml。利用未过滤的和过滤的(通过0.2μmpes膜)演示4ds作为两个独立的样品组来进行该研究。利用amiconultra15,10kmwco再生纤维素过滤器将蛋白质样品缓冲液交换到配方缓冲液中。用适合的缓冲液漂洗该过滤器,之后加入蛋白质。向每个uf/df中加入总共2ml的recap。向每个样品中加入体积为10ml的指定缓冲液,并将总体积减小至~2ml。将该过程重复进行总共4个循环。将配制的样品分成三个等分试样。将1ml的等分试样置于2-8℃并在时间0、3天和1周时监测颗粒形成。使两个0.5ml等分试样承受应力-将1个等分试样置于50℃持续1周,将1个等分试样暴露于5次冷冻-解冻循环。在研究结束时,2-8℃样品和应力样品一起通过sec、rp-hplc和活性对其进行分析。外观结果在所有的时间点,所有样品均呈现清澈且无色。在缓冲液交换之前,在时间0时以及研究结束时,未过滤的演示4ds呈现出清澈且无色,每ml具有~50个小颗粒。在缓冲液交换之前,过滤的演示4ds呈现出清澈且无色,在时间0时没有可见的颗粒,而在研究结束时每ml具有4个小颗粒。在配制的样品中,颗粒存在的范围为1-5个小可见颗粒至~20个可见的颗粒,从过滤和未过滤的开始材料,在tris/甘油配方中均观察到具有较大数目的颗粒(图3)。表4通过尺寸排阻色谱法确定的recap蛋白的主峰百分比和hmw百分比。sec结果多轮冷冻解冻并未诱导hmw形成。在基于柠檬酸盐/山梨糖醇和组氨酸/山梨糖醇的配方中,热应力造成有限的hmw形成。在基于tris/甘油的配方中,热应力诱导了显著hmw形成(表4和图4)。实施例3中间洗涤筛选策略选择两个柱步骤以用于开发具体的中间洗涤,从而提供宿主细胞蛋白清除。该洗涤主要聚焦保持产物产率同时显著柱洗脱液中的降低hcp水平。该中间洗涤最初是基于目前针对混合模式captoadhere树脂或亲和mimeticblue树脂的洗脱机制来选择的。在确定初筛中有希望的条件时,检查了含有特定流动相调节剂的一系列洗涤以确定能够破坏组织蛋白酶-样hcp和树脂配体之间的相互作用、hcp和产物之间的相互作用,或破坏二者的洗涤配方。单独、顺序或组合研究表5中示出的流动相调节剂。表5.调节剂及它们对hcp和树脂配体之间的相互作用、hcp与产物之间的相互作用、或两种相互作用的潜在影响的列表。调节剂调节剂影响nascn减少疏水相互作用尿素弱化氢键,离液剂甘油弱化疏水相互作用乙二醇弱化疏水相互作用和氢键l-精氨酸弱化疏水相互作用,离液剂nacl减少静电相互作用进一步优化每个柱步骤的前三种洗涤条件,随后连续测试该该柱步骤以表明添加剂作用。然后,以实验室规模实验进行整个纯化过程,以证明优化的过程步骤产生的散装药物满足hcp规格要求而不降低recap比活性或严重影响总体工艺产率。在整个研究中,常规使用具有cata检测的hcpelisa和蛋白质印记来监测hcp清除水平,并结合各种中间洗涤,以用于与基线试验进行比较。captoadhere色谱优化初级中间洗涤筛选该研究的目的在于确定用于captoadhere柱步骤,能够破坏产物和已知hcp杂质之间的相互作用、hcp杂质和树脂配体之间的相互作用或这两种相互作用,而不影响产物:配体相互作用的中间洗涤。破坏与hcp杂质的相互作用最终导致产物纯度增加。因此,采用了最初筛选来测试用于captoadhere捕获步骤的各种中间洗涤,并检查它们提高hcp清除率的能力。在初级筛选中使用的中间洗涤是基于针对混合模式色谱的现有洗脱机制来选择的,其包括破坏静电相互作用、疏水相互作用或破坏两种相互作用的组合。包括含有调节剂(即,离液剂、疏水改性剂、盐或烷基醇)的中间洗涤步骤能够减少任何与hcp杂质之间存在的相互作用,同时不损害产物:配体的相互作用。除了筛选调节剂影响之外,还评估了ph7.0和8.0。用于初级筛选的上样材料是清澈的细胞培养收集物b02-14oct2012(即,1×15lcchload)。将该细胞培收集物结合在2012年10月采用的校订的进料策略/补充(1)。在确定的过程之后进行基线对照实验,其中用7.5cv的高盐缓冲液(20mmtris、0.25mnacl、2mmmgcl2、50μmzncl2,ph8.0)洗涤柱,之后用3.0cv平衡缓冲液(20mmtris、0.1mnacl、2mmmgcl2、50μmzncl2,ph8.0)洗涤(2)。将来自对照实验的残留hcp水平与结合了中间洗涤缓冲液的一系列实验进行对比,图3中列出了对于总共5.0cv之后立即用7.5cv高盐上样后洗涤,并且之后用3.0cv平衡缓冲液洗涤。当uv280吸光度达到≥1.75au/cm的初始条件时,以升流方式洗脱每一筛选实验的产物,之后针对2.0cv进行收集。图5示出了所测试的中间洗涤的配方以及hcp水平和产物产率。将图中存在的残留hcp值归一化为基线洗脱液。在整个筛选研究中,产物产率(利用rp-滴定确定)和hcp清除水平(利用hcpelisa确定)是所分析的关键反应。选择相比于基线产生具有较高hcp清除率的洗脱液的中间洗涤作为有希望的洗涤候选物。基于这种策略,初筛的结果揭示了三种最可能的中间洗涤候选物:(1)0.1mnacl、0.2ml-arg,ph8.0;(2)1m尿素,ph8.0和(3)0.5mamso4,ph8.0。与基线对照相比,所测试的所有其他中间洗涤都不能降低残留hcp水平超过20%。并入该中间洗涤步骤,在许多情况下不利影响了产物产率,包括具有最高hcp清除率的实验(图5)。然而,产物产率的稍微下降能够由对hcp清除率的显著影响得到平衡。计划另外的缓冲液配方的二次筛选,用于鉴定对于该柱步骤能够平衡产物产率和hcp清除的中间洗涤。通过在非还原条件下的蛋白质印记分析来监测具体的残留hcp,组织蛋白酶样蛋白的量。图6示出了并入含有0.1mnacl、0.2ml-arg,ph8.0或0.5mamso4,ph8.0两种有希望的中间洗涤的影响。来自对照实验(泳道5)的洗脱液与包括中间洗涤的实验的洗脱液(泳道2&4)的比较证实了cata杂质的减少。泳道3代表在用0.5mamso4,ph8.0的中间洗涤期间从柱洗脱的组织蛋白酶样蛋白杂质。0.1mnacl、0.2ml-arg,ph8.0中间洗涤也证明了cata去除,然而洗涤样品并不包括在该具体的蛋白质印记分析中。当与基线洗脱液比较时,对于并入0.5mamso4,ph8.0洗涤的洗脱液,组织蛋白酶样蛋白减少了大约30%(泳道4),而对于接收了0.1mnacl、0.2ml-arg,ph8.0中间洗涤洗涤的洗脱液,则降低了大约50%(泳道2)。蛋白质印记分析的结果与图5中所示出的hcpelisa数据一致,证实了这两个测试指导筛选研究的可靠性。基于产物产率和hcp清除(如通过蛋白质印记和hcpelisa确定的)的编辑结果,从初级筛选鉴定的排名靠前的中间洗涤候选为0.1mnacl、0.2ml-arg,ph8.0。二次中间洗涤筛选初级筛选揭示了一种能够使得残留hcp减少至少50%的中间洗涤(0.1mnacl、0.2ml-arg,ph8.0)。产物产率受到这种特定洗涤的不利影响,因此具有能够潜在加强蛋白质:配体相互作用的调节剂的中间洗涤缓冲液被并入二次筛选中。还顺序和组合测试了来自初级筛选最有效的中间洗涤缓冲液并结合评估它们的累加效应以及变化ph值(图7)。利用上述工艺条件,进行基线对照实验并且如图7所示与结合了中间洗涤缓冲液的一系列实验比较hcp水平。评估中间洗涤效力的所有实验包括四个洗涤模块(block):(1)具有高盐缓冲液(20mmtris,0.25mnacl、2mmmgcl2、50μmzncl2,ph8.0)的7.5cv洗涤,(2)具有具有平衡缓冲液(20mmtris、0.1mnacl、2mmmgcl2、50μmzncl2,ph8.0)的1.5cv洗涤,(3)利用图7中列出的缓冲液配方之一进行的5.0cv中间洗涤,以及(4)利用平衡缓冲液(20mmtris、0.1mnacl、2mmmgcl2、50μmzncl2,ph8.0)的3.0cv洗涤,之后进行产物洗脱。对于进行顺序中间洗涤步骤的实验,柱洗涤模块变化为包括两个5.0cv中间洗涤相,其通过用平衡缓冲液进行1.5cv洗涤来间隔以防止中间洗涤缓冲液的混合。还完成了在收获上样材料中包括低浓度调节剂的实验,以鉴定增加的产物:配体结合的潜力。当uv280吸光度达到≥1.75au/cm的初始条件时,以升流方式洗脱来自每一筛选实验的产物,随后收集用于2.0cv(2)。图7中示出了所测试的中间洗涤的配方以及归一化的hcp水平和产率值。图示中(图7a)包括归一化的hcp值,其作为两种不同的许多收获上样材料被用于整个二次筛选。利用相同的收获上样材料(即,1×15l或200lpdcch上样)将残留hcp水平归一化为基线对照试验(1)。降低了残留hcp水平,同时保持了产物回收率最好结果的数个中间洗涤条件来自于含有0.1mnacl、0.2ml-arg、5%甘油,ph8.0的中间洗涤。当与基线对照实验相比较时,并入这种中间洗涤使残留hcp浓度降低了2.9倍。并入0.1mnacl、0.2ml-arg,ph8.0,接着0.5mamso4,ph8.0的顺序洗涤的洗脱液与基线洗脱液相比,也表现出显著的hcp清除率,高达2.5倍。如同初级筛选一样,通过在非还原条件下的蛋白质印记分析来监测组织蛋白酶样蛋白残留hcp的量。图8示出了并入有或没有5%甘油的0.1mnacl、0.2ml-arg,ph8.0中间洗涤的效果。该0.1mnacl、0.2ml-arg,ph8.0中间洗涤在初级筛选中已被鉴定。来自对照实验的洗脱液(泳道3)与包括中间洗涤的实验的洗脱液(泳道5&7)的比较证实了cata杂质的减少。泳道4和6分别代表在存在或不存在5%甘油的情况下在用0.1mnacl、0.2ml-arg,ph8.0的中间洗涤期间从柱洗脱的cata杂质。当与基线洗脱液比较时,包括甘油的0.1mnacl、0.2ml-arg,ph8.0中间洗涤中显示出更高的cata去除,大约65%,而并入无甘油的中间洗涤的洗脱液是大约50%的cata去除。这些结果表明了将各种调节剂(即,甘油、盐和精氨酸)结合到单个中间洗涤中的累加效应。captoadhere中间洗涤的选择当完成captoadhere中间洗涤筛选研究时,编辑的分析数据,尤其是hcpelisa和蛋白质印记分析结果用于选择用于captoadhere捕获步骤的中间洗涤(图5、图7和图8)。在整个筛选研究中,与基线对照试验相比,两个中间洗涤明确显示出hcp的显著减少。两个中间洗涤为:(1)0.1mnacl、0.2ml-arg,ph8.0和(2)0.1mnacl、0.2ml-arg、5%甘油,ph8.0。图8中的蛋白质印记分析提供了来自并入了这些中间洗涤的洗脱液的直接比较。与基线试验相比,两种中间洗涤均提供了具体的hcp,组织蛋白酶样蛋白显著更高的降低,然而,降低的水平在接受了0.1mnacl、0.2ml-arg、5%甘油,ph8.0中间洗涤的洗脱液中稍高。基于hcpelisa数据,当与基线洗脱液相比时,接受了0.1mnacl、0.2ml-arg、5%甘油,ph8.0中间洗涤的洗脱液降低了大约65%,相比之下,用0.1mnacl、0.2ml-arg,ph8.0洗涤降低了大约46%。除了较高的hcp清除之外,接受了0.1mnacl、0.2ml-arg、5%甘油,ph8.0中间洗涤的洗脱液的产物产率较高。考虑到较高的hcp清除和产物产率,选择用于captoadhere柱步骤的中间洗涤为0.1mnacl、0.2ml-arg、5%甘油,ph8.0。针对captoadhere柱步骤的筛选研究成功鉴定了能够获得高于基线过程的hcp水平可观减少的中间洗涤。选择用于在捕获步骤期间去除组织蛋白酶样蛋白杂质的洗涤为20mmtris、0.1mnacl、0.2ml-arg、5%甘油、2mmmgcl2、50μmzncl2,ph8.0并且将其并入在总柱洗涤体积中含有17.0cv的优化过程中。将该优化的的captoadhere条件整合到小规模工艺试验中,以证实该最佳的条件在工序中实现了显著降低的hcp水平。mimeticblueap色谱优化初级中间洗涤筛选本研究的目的在于确定用于mimeticblueap柱步骤的中间洗涤,其能够破坏产物和已知hcp杂质之间的相互作用、hcp杂质和树脂配体之间的相互作用或二者,而不会影响产物:配体相互作用。mimeticblueap(mbap)是一种尤其针对碱性磷酸酶的纯化而开发的合成亲和吸附剂。配体由连接至功能性磷酸基团的蓝色生色团构成。recap和配体之间的结合在不存在磷酸盐的情况下发生。当将磷酸盐引入到流动相中时实现洗脱。该结合机制的特异性暗示了洗涤缓冲液可配制为破坏recap-hcp相互作用和/或树脂-hcp相互作用并除去流动相中的hcp,同时保持recap在树脂上固定直至用磷酸盐洗脱。最初洗涤筛选实验由8个色谱试验组成,其中每一个包括不同的中间洗涤步骤。在开发过程中发现了该方法的一个局限性,即在进料流中存在高nacl浓度(130mm)并且洗涤缓冲液降低了树脂容量。这种观察表明在mbap结合机制中也涉及静电吸引,这暗示了其为伪亲和树脂。为了保持高产率,通过在该色谱模式中发生的静电相互作用来限制中间洗涤缓冲液的离子强度。mimeticblueap柱上样材料由清澈的细胞培养收集物b02-14oct2012产生。将该细胞培收集物结合在2012年10月采用的校订的进料策略/补充(1)。通过captoadhere捕获和poroshq色谱步骤根据确定的纯化方法来纯化该收集物(2)。分析captoadhere和poroshq洗脱液的滴定度、活性和残留hcp。所得的poroshq洗脱液作为该过程的代表来进行评估并且将其用作本研究最初十二次mimeticblueap试验的上样材料。根据确定的mbap纯化方法来进行基线对照实验。将mimeticblueap树脂货号fa0345填充在2.2cm直径的柱中至床高达到16.2cm,产生61.6ml柱。在进行基线mbap试验时,未确定上样(2x稀释的poroshq洗脱液)的滴定度。因此,uv280分析用于估计上样的浓度。利用该估计的值,计算108ml的上样提供的recap上样密度为2至3grecap/l树脂。在0.5mnaoh中净化该柱并使其平衡。将108ml的2x稀释的poroshq洗脱液上样至该柱。对于初级筛选测试试验而言,对于每个试验在上样后eq洗涤步骤之后是利用图9中所列出的测试缓冲液进行≥3cv的中间洗涤步骤。监测该试验在洗涤步骤期间的的蛋白质洗脱。在中间洗涤之后,再次用eq缓冲液洗涤该柱以除去测试洗涤缓冲液组分。用20mmtris、25mm磷酸盐、130mmnacl、2mmmgcl2、50μmzncl2,ph8.0来洗脱产物。通过uv设门来收集洗脱液,随着峰值升高至25mau,直至其下降至50mau。两次试验之间清洗柱并净化。分析洗脱液的滴定度(rp-hplc),活性和残留hcp(hcpelisa和蛋白质印记)。降低洗脱液中残留hcp浓度而不显著影响产率的的任何洗涤条件,继续进行本研究的二次筛选阶段。初级筛选试验的结果示于图9和图10中。该基线试验条件实现了recap的100%产率,并且残留hcp浓度为2047ppm。四个中间洗涤条件(100mmnacl、200mmnacl、0.2mnascn和200mm精氨酸)在洗涤步骤期间造成显著的产物损失。与基线试验相比,5%甘油和10%乙二醇洗涤未不利影响产率,但也没有显示出显著的hcp清除。满足高产率(90%)和显著的hcp减少(1066ppm)标准的惟一条件是1m尿素洗涤步骤(试验4)。用组织蛋白酶样蛋白检测的蛋白质印记分析(图10b)表明,来自试验4的洗脱液与基线试验相比,显示出减少的cata条带。该印记分析也检测了以1m尿素洗涤的cata,表明洗涤有效破坏recap和该hcp物质之间,和/或树脂和hcp之间的关联。该结果证实了hcpelisa分析的发现。二次中间洗涤筛选二次筛选研究引入了新的洗涤条件以尝试改进初级筛选试验中看到的hcp清除。再次测试氯化钠和l-精氨酸,但以低于初级筛选试验的浓度进行。在captoadhere初级筛选试验中精氨酸显示出选择性hcp清除,因此我们想要进一步研究精氨酸在这种模式色谱中的作用。离液剂尿素在mbap初级筛选试验中表现出中等hcp清除,因此组合测试尿素与氯化钠或连续测试。10%的乙二醇也显示出hcp的少量下降,因此也将其与nacl组合测试以尝试增加hcp和recap之间或hcp和柱基质之间的分离。最后,在ph7.0和8.0条件下对下洗涤缓冲液进行测试以检测ph依赖性相互作用。除了测试中间洗涤步骤对hcp清除的影响之外,我们还测试了消除洗脱缓冲液中的nacl的影响。在初级筛选实验中,我们观察到在中间洗涤期间,100mm的nacl导致大约60%的产物穿透,以及200mm的nacl导致全部产物穿透,而不存在磷酸盐。理论上来说,在高离子强度下,静电机制主导结合动力学并且使得配体将蛋白质释放到流动相中。可通过用低磷酸盐浓度且无nacl的洗脱提高树脂的选择性,仅促使recap和配体的膦酸盐基团解离,并在保留在树脂上静电结合的hcp直至1mnacl+0.5m磷酸盐清洗。在第12次mbap测试试验之后制备了新批次的上样材料。同时的captoadhere纯化方法的研究表明了0.1mnacl、0.2ml-精氨酸,ph8.0中间洗涤步骤使得capto洗脱液中的hcp减少50%。因此,在captoadhere纯化步骤中包括0.1mnacl、0.2ml-arg,ph8.0中间洗涤步骤,制备了用于mbap二次筛选试验的新批次的上样材料。poroshq50纯化方法仍保持不变。图11中示出了二次筛选试验的总结。在试验10中,唯一从基线方法偏离的是在洗脱缓冲液中不包括nacl;上样材料和具有eq缓冲液的上样后洗涤是等同的。与基线试验相比,仅仅洗脱缓冲液配方的这种改变就使mbap洗脱液的hcp浓度下降大约40%。因此,其余的试验使用不含有nacl的洗脱缓冲液。使用新批次的mbap上样材料进行试验14至试验20,所述mbap上样材料由并入了含有0.1mnacl、0.2ml-精氨酸,ph8.0的中间hcp去除洗涤的captoadhere洗脱液产生。试验14显示capto步骤中实现的hcp减少结合mbap洗脱缓冲液中除去nacl,使得mbap洗脱液的hcp水平减少至667ppm,并且在mbap纯化方法中不包括含有调节剂的洗涤缓冲液。许多试验将hcp浓度减少至少于500ppm,但并不产生可接受的产物产率。10%的乙二醇和nacl,ph8.0的洗涤组合仅仅是增强hcp清除(128ppm)而不显著不利影响产率(77%)的中间洗涤配方。尽管这种洗涤有利于hcp清除,但是中间洗涤缓冲液中包括nacl是不可接受的风险,因为许多利用了含有nacl的洗涤缓冲液的试验都显示出大量的recap产物损失。利用了不含nacl的mbap洗脱缓冲液配方的试验10流分的蛋白质印迹分析得到的结论是洗脱液中hcp浓度的减小有可能是由于在用磷酸盐(非nacl)的洗脱步骤期间仍与树脂结合的hcp,而之前的含有130mmnacl的洗脱缓冲液将hcp与recap一起洗脱。hcpelisa结果显示,试验10清洗的残留hcp浓度与洗脱液相比升高(图12a)。蛋白质印记分析确认了elisa结果(图12b)。在上样(泳道2)、试验1洗脱液(泳道3)和试验10清洗(泳道6)中观察到大约50kda的cata条带。在试验10洗脱液中没有cata条带(泳道5),表明在该样品中的catahcp已被下降低于试验的检测限(估计<1000ppm)。相同的条带在试验10清洗峰样品中更加明显。并入20、40、60和80mml-精氨酸洗涤步骤的试验12显示出hcp的最大减少。洗脱液hcp浓度为<40ppm,但产率较差,为37%。在该试验中,分别收集精氨酸步骤以确定在recap从树脂解离的精氨酸浓度。蛋白质印记对洗涤馏分的分析(图12b)显示在所有的精氨酸洗涤中均存在catahcp杂质,而直到60和80mm精氨酸步骤才不会发生产物的明显损失。该发现表明40mm的精氨酸洗涤步骤能够从recap和/或树脂选择性除去大量的hcp,而不影响recap:配体结合相互作用。选择40mm的精氨酸洗涤以用于在第三轮mbap中间洗涤筛选实验中进行进一步研究。mbap中间洗涤步骤优化从之前的实验中选择三种中间洗涤调节剂以用于进一步的优化。在初级洗涤筛选研究中,1m尿素已被证实降低洗脱液中的hcp而不影响产物回收。在二次筛选研究中,乙二醇+nacl降低hcp而不导致严重的产物损失。精氨酸已显示选择性hcp去除,但高于40mm的浓度不利地影响产率。研究的下一阶段阐述了对这些调节剂的研究。也继续使用无nacl的洗脱缓冲液。最终的实验以3grecap/l树脂柱上样测试了前三种候选物,以测试由于调节剂洗涤造成的柱容量的减小。图13中示出了具体的测试洗涤缓冲液配方和优化的实验结果的总结。1m尿素洗涤与仅含磷酸盐的洗脱缓冲液组合之前没有进行过测试。试验21和22分别利用具有1m和2m尿素的洗涤缓冲液,并且用新的仅含磷酸盐的缓冲液洗脱。试验27也用1m尿素洗涤来进行,但是上样到柱上的recap的量增加至3grecap/l树脂的目标上样密度。试验21显示了上样1.8grecap/l树脂的柱与1m尿素洗涤缓冲液的组合产生具有59ppm的hcp和83%的recap回收的洗脱液。洗涤缓冲液中的尿素浓度或recap上样密度的增加导致产物洗脱液中更高的残留hcp浓度(分别为299和371ppm的hcp)。也评估了组合1m的尿素与10%的乙二醇或40mm的精氨酸的中间洗涤缓冲液的hcp清除率和产物产率。10%的乙二醇/1m尿素组合洗涤产生了具有高残留hcp浓度(777ppm)的产物。在以2.3和3.0grecap/l树脂上样的柱上进行了40mm精氨酸/1m尿素洗涤组合。以更低的产物上样,精氨酸/尿素洗涤产生了具有低残留hcp浓度(6ppm)的洗脱液。所得71%的recap产物产率低于期望的回收率。当以更高的recap上样密度进行相同的纯化时,hcp清除仍可接受(68ppm),但回收率进一步降低至66%。在洗涤步骤中利用40mm精氨酸,ph8.0进行试验23和试验29。对于试验23和试验29,分别以1.8和3grecap/l树脂对柱上样。两个试验均产生具有低残留hcp浓度(54和59ppm)的洗脱液并且实现了可接受的产率(83和88%)。该中间洗涤是以最大柱上样容量保持hcp清除能力和高产物产率的唯一候选。针对mimeticblueap柱步骤的筛选研究鉴定了两个工艺改变,其导致产物池中残留hcp的浓度下降至大约60ppm。第一工艺改进是在色谱方法中加入中间洗涤步骤以用于选择性清除hcp。为去除组织蛋白酶样蛋白杂质而选择的洗涤为20mmtris、40mml-arg、2mmmgcl2、50μmzncl2,ph8.0。这种选择的中间洗涤缓冲液包括3.0cv将使mbap处理时间增加20至30分钟。第二工艺改进为从洗脱缓冲液去除nacl。这些工艺修订的组合使得残留hcp的降低超过30倍,如通过hcpelisa所测量。将优化的mbap条件并入小规模工艺试验中以确认这些发现。工艺确认试验在hcp减少实验完成后,并入修订的工艺条件来进行小规模确认试验。除了病毒过滤之外,通过所有的单元操作来处理细胞培养收集物(200lpd16oct2012)。对过滤的uf/df截留物质进行最终的产物分析。每一色谱步骤进行一次循环。通过rp-hplc(针对滴定度和%异二聚体a)、elisa(活性和hcp)以及sds-page和蛋白质印记分析来分析工艺中间步骤。图15中示出了分析结果的总结。新的工艺回收了53mg纯化的recap,其总体工艺产率为34%。captoadhere捕获色谱利用0.1mnacl、0.2ml-arg、5%甘油,ph8.0中间洗涤(参见vi.c节)来进行hcp减少实验的captoadhere试验#32。该洗涤配方显示出产生最高的hcp清除,并且被选择用于包括在captoadhere方法中。因此,通过剩余的纯化过程来纯化来自试验#32的洗脱液。在captoadhere试验#32中处理总共297ml的200lpd16oct2012细胞培养收集物。通过rp-hplc,收集物的滴定度为0.63g/lrecap。实际的上样系数为7.8grecap/l树脂。在洗脱期间,收集到187mg的产物。色谱图显示出与在确立新的洗涤步骤之前看到的不同的洗脱峰图谱。在该试验中,uv280洗脱液峰值是分开的,而在之前的试验中,洗脱液表现为单个峰(2)。峰形状出现这种变化的原因不确定,但最有可能是由于额外的精氨酸洗涤步骤。尽管色谱图看起来不同,但是总体recap产率(93%)和活性产率(86%)与之前的演示试验相当。通过0.2μmpes过滤器过滤该洗脱液并储存于2-8℃下。病毒灭活和poros50hq纯化将captoadhere洗脱液平衡至室温。为了获得1.0%的tritonx-100浓度,将4.65ml的10%tritonx-100(在wfi中)加入到41.9ml的captoadhere洗脱液中。将该洗脱液充分混合,然后静态孵育60分钟以实现完全病毒灭活。在孵育后,用514ml的wfi将该材料稀释12x。电导率目标为≤4.5ms/cm;溶液的最终电导率为4.13ms/cm且ph为7.74。将poroshq50树脂上样在1.1cm柱中至床高为19.6cm,以达到最终柱体积为18.6ml。柱不对称度为1.16且板高度为0.025cm。该试验的实际上样系数为9.1grecap/l树脂。色谱方法与演示试验5操作参数相同(2)。收集56ml的洗脱液。分析显示出61%的recap质量产率和66%的活性恢复。通过0.2μmpes过滤器过滤该洗脱液并储存在2-8℃下。如期望的进行poroshq50色谱。mimeticblueap纯化mimeticblueap色谱步骤并入两种方法改进。添加含有40mm精氨酸的3.0cv中间洗涤缓冲液。而且,通过除去缓冲液中的nacl而改变洗脱缓冲液。实施这两个修订以减少工艺中间步骤中的hcp。将mimeticblueap(mbap)树脂上样到2.2cm柱中至床高为16.0cm,以使得最终柱体积为60.8ml。不测试柱的het和不对称度。将上样的柱在0.5m中净化并用20mmtris、2mmmgcl2、50μmzncl2,ph8.0平衡。该试验的实际上样系数为1.6grecap/l树脂。用wfi将poros洗脱液1:1稀释以降结合的电导率。洗脱液体积为41ml。分析显示出84%的recap质量产率和80%的活性恢复,其稍低于在演示试验5中观察到的恢复(2)。mbap洗脱液的hcp浓度为34ppm,其相比于上样材料下降了2500倍。通过0.2μmpes过滤器过滤洗脱液并储存在2-8℃下。butyl650m纯化工艺的下一步骤包括利用疏水相互作用下色谱进行的精加工步骤。将该butyl650m树脂上样到1.1cm柱中至床高为8.7cm,以达到最终最终柱体积为8.3ml。用于butyl纯化步骤的目标床高为19.5cm,但在该试验中降低了高度以接近容量上样树脂。柱不对称度为1.17且板高度为0.034cm。取样后的mbap洗脱液为37.8ml。通过添加31.9g的2.1mamso4将mbap洗脱液调节至1.0mamso4。上样的电导率为135ms/cm且ph为7.67。该试验中的实际上样系数为9.2grecap/l树脂。色谱方法与演示试验5操作参数相同(2)。收集48ml的洗脱液体积。为了避免形成沉淀,立即用96ml的wfi稀释洗脱液以使amso4浓度降低至最终稀释体积为144.5ml。分析显示出86%的recap质量产率和81%的活性恢复。通过0.2μmpes过滤器过滤洗脱液并储存在2-8℃下。如期望进行butyl650m色谱。超滤/渗滤50cm2pellicon(millipore)biomax10kdapes超滤盒用于进行uf/df步骤,提供了13g/m2的质量面积比。用wfi冲洗该盒,然后用配方缓冲液(20mm组氨酸、250mmd-山梨糖醇、2mmmgcl2、50μmzncl2,ph7.0)平衡。将145ml稀释的butyl洗脱液浓缩大约3倍至40ml。由于产物的体积较小,截留物质不再进行浓缩。在浓缩期间,通过膜的tmp保持在≤15psi。在浓缩之后,用配方缓冲液将产物渗滤10x。收集材料并通过0.2μmpes瓶顶过滤器进行过滤。产物回收之后,将30ml配方缓冲液再循环通过该盒以从膜表面回收产物。不将缓冲液流加入到截留物质中,而是分别储存。这样做是为了防止uf/df截留物质稀释。uf/df步骤在截留物质中的产率为67%recap质量产率且在缓冲液流中为15%,其等于82%的总recap回收率。hcp浓度低于2ppm的loq。将产物储存在2-8℃下。hcp减少确认试验的总结该纯化试验的目的在于实施从捕获步骤至uf/df和最终配方的完全纯化过程,结合了对captoadhere和mimeticblueap色谱步骤的工艺改变。我们想要证明这些变化能够将bds中的hcp浓度减少至显著低于≤100ppm的目标规格,而不会不利影响recap分子的活性或总体产率。该工艺中间步骤的蛋白质印迹分析显示出在mimeticblue中间洗涤步骤之后不可检出cata(图14)。在本试验中产生的bds中的残留hcp浓度,如通过hcpelisa确定的<2ppm(低于loq)。543u/mg的具体活性在规格之内,并且总体产率为34%。uf/df步骤中的产物回收低于演示试验4和5,但已知uf/df中的低截留物质体积导致较差的产物回收率。除了uf/df步骤之外,步骤产率与演示试验4和5相当,这表明captoadhere和mbap过程变化未显著降低该过程的总体产率。实施例4纯化过程recap的纯化过程包括六个单元操作。captoadhere色谱捕获步骤之后是用具有1%tritonx-100的洗脱液进行病毒灭活。用poroshq50的阴离子交换色谱除去了去垢剂并部分纯化了进料流。进行两个另外的色谱步骤(mimeticblueap和butyl650m),以除去杂质(即,宿主细胞蛋白、dna)至达到规格。浓缩该材料并利用10kdamwcopes膜将缓冲液交换到配方缓冲液中。最后,利用15nplanova中空纤维过滤器对纯化的recap进行病毒过滤。captoadhere捕获色谱捕获步骤的目的在于将目标分子与细胞培养基分离,以浓缩产物并部分纯化recap。选择混合模式树脂captoadhere作为纯化过程中的捕获步骤。captoadhere柱的容量之前已确定为8grecap/l树脂(3)。将清澈的细胞培养物收集物14oct2012b01和b03合并以提供大约20l的收集物以用于下游纯化。将captoadhere树脂上样到净化的bpg100柱(gehealthcare)至床高为23.7cm,最终柱体积为1.9l。柱不对称度为1.44且板高度为0.049cm。上样的柱在0.5mnaoh中净化并平衡。通过rp-hplc确定的合并的收集物的滴定度为0.7g/lrecap,因此处理了全部20l的收集物。实际上样系数为7.3g/l树脂。在将收集物上样到柱上之后,进行nacl盐洗涤步骤,之后是eq缓冲液洗涤以降低nacl浓度。用0.55ml-精氨酸+0.1mnacl实现洗脱。在该方法的洗脱阶段期间,逆转流动方向以使洗脱液体积最小。uv280升高至1.75au/cm时,开始洗脱液峰收集。通过0.2μmpes过滤器将洗脱液的两个cv收集到无菌50lbpc中;洗脱液体积为3.9l。电导率为38.2ms/cm且ph为7.79。选择大体积bpc以使得能够在下一色谱步骤中进行13x水稀释。与之前的演示试验一样进行该试验。病毒灭活和poros50hq纯化将captoadhere洗脱液平衡至室温。将533ml的10%tritonx-100(在wfi中)加入到洗脱液中,使得最终浓度为1.2%。(因为重量测量误差,超过了1.0%的目标浓度)。将洗脱液充分混合,然后静态孵育60分钟。孵育之后,用63.9l的wfi将该材料稀释13x。电导率目标为≤4.5ms/cm;溶液的最终电导率为3.21ms/cm且ph为7.72。将poroshq50树脂上样到净化的bpg100柱(gehealthcare)中至床高为20.0cm,最终柱体积为1.6l。柱不对称度为1.63且板高度为0.020cm。下表10中列出了poroshq50步骤的纯化参数。在0.5mnaoh中上样的柱被净化并平衡。poroshq50柱的容量之前已被确定为10grecap/l树脂(3)。本试验的实际上样系数为8.0grecap/l树脂。在柱上样之后,进行50mmnacl盐洗涤步骤以除去杂质。用130mmnacl在降流中进行洗脱。uv280升高至0.5au/cm时,开始洗脱液峰收集。通过0.2μmpes过滤器将洗脱液的三个cv收集到无菌10lbpc中;体积为4.7l。洗脱液的电导率为14.6ms/cm且ph为8.22。除了recap产率(61%)低于演示试验4(85%),该试验与之前的演示试验同样相同。产率下降的原因不明。检查洗脱缓冲液的电导率以确定是否由于不正确的缓冲液配方导致了较低的产率。缓冲液为14ms/cm,其为缓冲液的目标电导率,因此排除了较低nacl浓度的原因。洗涤缓冲液的电导率也很好地在规格之内,这暗示了低产率并不是由于洗涤缓冲液中的产物损失造成的。通过rp-hplc(滴定度和%异二聚体a)、sec-hplc(纯度)和elisa(活性和hcp)分析对poroshq50洗脱液进行了分析。分析示出了61%的recap质量产率和60%的活性恢复。将poros洗脱液与上样材料进行比较,异二聚体a的百分比从82%增加至93%并且通过sec的纯度从36%增加至87%。hcp降低三倍。将洗脱液储存在2-8℃下。mimeticblueap纯化将mimeticblueap(mbap)树脂上样到净化的quickscale14cm直径柱(millipore)中至床高为17.0cm,最终柱体积为2.6l。柱不对称度为1.37且板高度为0.039cm。该上样的柱在0.5mnaoh中被净化并平衡。mbap柱的容量之前已经确定为3grecap/l树脂(3)。该试验的实际上样系数为2.9grecap/l树脂。用wfi将poros洗脱液1:1稀释,以降低电导率。在将稀释的poros洗脱液上样到柱之后,用eq缓冲液洗涤除去未结合的材料。用25mmnapo4、130mmnacl在降流中进行洗脱。当uv280升高超过0.125au/cm时开始洗脱液,直至其在峰的尾端降至0.25au/cm。通过0.2μmpes过滤器将洗脱液收集到无菌10lbpc中;体积为2.4l或0.9cv。该试验与之前的演示试验同样相同。通过uv280(滴定度)、rp-hplc(%异二聚体a)、sec-hplc(纯度)和elisa(活性和hcp)来分析洗脱液。分析显示出92%的recap质量产率和96%的活性恢复。通过sec确定的产物纯度从87%增加至99.6%。hcp下降大约50倍。洗脱液储存在2-8℃下。butyl650m纯化将butyl650m树脂上样到净化的bpg100柱(gehealthcare)至床高为19.0cm,最终柱体积为1.5l。柱不对称度为1.37且板高度为0.039cm。在0.5mnaoh中净化上样的柱,并用1.0mamso4平衡。通过添加2.2l的2.1mamso4将2.4lmbap洗脱液调节至1.0mamso4。上样材料的电导率为125.7ms/cm且ph为7.77。butyl650m柱的容量之前已被设定为10grecap/l树脂(4),但没有对当前处理条件下柱的真实容量进行严格的研究。该试验的实际上样为4.6grecap/l树脂,大约为目标上样系数的一半。在柱上样之后,用eq缓冲液洗涤除去未结合的材料。用6mamso4实现洗脱。在0.25au/cm直至uv在峰尾上降至0.25au/cm之间收集洗脱液峰值。通过0.2μmpes过滤器将洗脱液收集到20lbpc中;体积为4.4l。为了避免形成沉淀,立即用8.8lwfi稀释该洗脱液以降低amso4浓度。该试验与之前的演示试验相同进行。超滤/渗滤在演示试验5中,使用新配方缓冲液以增强易形成颗粒的物质的溶解度。该新配方为20mm组氨酸、250mmd-山梨糖醇、2mmmgcl2、50μmzncl2,ph7.0。从过程中除去vf后加入25%甘油,因此uf/df最终浓度目标从≥13.5g/l变为≥11.5g/l。对于演示试验5,使用0.1m2pelliconxl(millipore)biomax10kdapes超滤盒,其提供的质量面积比为57g/m2。用wfi冲洗该盒,然后用配方缓冲液平衡。将稀释的hic洗脱液浓缩至15g/l。在浓缩过程中,穿过膜的tmp保持在8-15psi。浓缩之后,用配方缓冲液将产物渗滤10x。将材料收集到1lpetg瓶中。产物回收之后,将100ml配方缓冲液再循环通过该盒以从膜表面回收产物。将缓冲液流加入到截留物质中并通过0.2μmpes瓶顶过滤器过滤溶液。最终体积为460ml。通过uv280(滴定度)、rp-hplc(%异二聚体a)、sec-hplc(纯度)和elisa(活性和hcp)来分析产物。表6:演示试验5uf/df的分析结果。tuf/df步骤产率为105%的recap质量产率和85%的活性恢复。hcp浓度为120ppm,其高于100ppm的目标。洗脱液储存在2-8℃下。表6:演示试验5uf/df的分析结果。病毒过滤和散装充填0.01m2planova15n中空纤维病毒过滤器(asahi)用于病毒过滤步骤。将柱筒排空储存缓冲液,并利用蠕动泵用配方缓冲液平衡。将流速调节至5ml/min以使跨膜的压力达到15psi。从冷藏中取出uf/df截留物质平衡至室温。注意到,在截留物质过夜时形成了5-10个小的白色颗粒。以5ml/min泵送该截留物质通过过滤器并将其收集到无菌petg瓶中。在180分钟内完成过滤,并在整个过程中将压力保持在15psi。将10ml的配方缓冲液泵送通过该柱筒、收集并加入产物至最大回收率。添加96ml配方缓冲液将浓度调节至9.9g/l。病毒滤液的最终体积为490ml,产生4.8g产物。取样用于通过uv280(滴定度)、rp-hplc(%异二聚体a)、sec-hplc(纯度)和elisa(活性和hcp)进行分析。相比于uf/df截留物质,vf的步骤产率为95%的recap质量产率和94%的活性恢复。该过程中的最终步骤是通过0.2μmpes瓶顶过滤器过滤vf产物。最终bds是清澈的并且在过滤后立即观察无颗粒。相比于病毒滤液,bds的分析(表7)示出了97%的recap质量产率和98%的活性恢复。hcp浓度在123ppm处没有变化,其高于100ppm的目标。bds储存在2-8℃下。表7:vf的分析结果和配方。演示试验5的总结表8中示出了演示试验5中间工艺步骤和最终bds的分析的总结。表8:每一步骤的recap和活性产率以及累积过程产率演示试验5扩大规模总体累积过程质量产率为40%且活性产率为43%(表9)。该产率稍低于45-50%的预期产率。满足了浓度、活性、纯度和残留dna的规格。bds中的hcp浓度为123ppm,其高于100ppm的目标。表9:演示试验5的产物规格和结果。对所有参数进行满足规格的测试,hcp除外。实施例5演示试验的结果和讨论过程描述利用不同的上游细胞培养收集物在一系列小规模纯化过程中最初形成整个过程的参数。在来自之前的演示试验的许多bds中,进一步优化用于captoadhere和mimeticblue色谱步骤的工艺参数,以在发现颗粒后增加宿主细胞蛋白清除率水平,其被鉴定为组织蛋白酶-样宿主细胞蛋白(更具体地,如用质谱所证实的,其为cathepsina的仓鼠同源物)。一旦下游纯化工艺被修订,就进行小规模确认试验以确认最终工艺条件(1)。本报告中演示试验的结果利用清澈的细胞培养收集物(200lpd生产),其是最终上游工艺的代表(5)。对于该演示试验,上游工艺开发团队提供了大约70-升的清澈的细胞培养收集物,其反相(rp)-滴定度为大约0.6g/lrecap。一部分收集物材料(大约24-升)用于演示试验6。通过四个色谱步骤利用所列出的柱尺寸、柱上样、流速、保留时间和洗脱方式(表10)来纯化大约14克的产物。一旦通过四个色谱步骤纯化产物之后,就进行超滤/渗滤(uf/df)以浓缩产物并在butyl650m工艺中间步骤中除去硫酸铵。然后通过planova15n病毒过滤器过滤该tuf/df截留物质。最后,将产物通过0.2微米pes过滤到适当的容器中,以获得其散装药物。应该注意,该演示试验包括使用测量的recap消光系数,其针对依赖于uv280的所有浓度计算为1.01ml/mg*cm。表10.演示试验6柱色谱条件的总结captoadhere捕获步骤captoadhere柱步骤用于在清澈的收集物中捕获recap产物并浓缩进料流。对于captoadhere树脂而言,收集物上样材料的调整不是必须的,因为这种树脂能够耐受具有高电导率的进料流。将上样材料平衡至环境温度并上样到1.86l上样的captoadhere柱上,并利用表11中列出的条件进行纯化。上样的产物的最大量为14.0g,因此对于上样的captoadhere柱而言仅需要以8grecap/l树脂上样的一次循环。用最初观察uv命令进行产物洗脱,之后进行2.0柱体积(cv)的收集。洗脱之后,使中间体经过0.2微米过滤至100lbpc(hyclone)中,以适应病毒灭活,并且之后进行12倍稀释。收集之后,立即将captoadhere中间体转移至病毒灭活和稀释步骤。表11.captoadhere捕获色谱试验条件实际上样到captoadhere柱上的为7.5grecap/l树脂。采用高盐洗涤(250mmnacl)来去除离子结合于混合模式树脂的杂质(即,dna,hcp)。在高盐洗涤之后,用平衡缓冲液进行1.5cv洗涤以防止由于高盐洗涤缓冲液和中间洗涤液与盐、l-精氨酸和甘油的潜在混合而导致的产物损失。将该中间洗涤液(0.1mnacl、0.2ml-arg、5%甘油)结合到改进的下游工艺中,以增加宿主细胞蛋白的去除,尤其是组织蛋白酶样蛋白(cata),该下游工艺在这个那个基线下游工艺中进行。为了防止中间洗涤液和洗脱缓冲液的混合,该工艺中包括3.0cv平衡洗涤。利用l-精氨酸和氯化钠的组合以升流方式实现产物的洗脱,就像在初步开发步骤中确定的一样。一旦出现uv观察条件,则最大洗脱体积为2.0cv。在captoadhere柱步骤之后,进料流从23.7升减少至大约3.7升。结果提示,对于下游处理的捕获步骤,并入具有调节剂组合的中间洗涤(洗涤#3)对于增加hcp清除率是有益的。总之,针对captoadhere工艺中间步骤的分析结果与之前的演示试验的结果高度相当,但是具有显著增加的hcp清除率。病毒灭活和poros50hq纯化包括病毒灭活步骤以有效失活潜在的包膜病毒。在产物上样并向人和动物给药之前,必须从产物中除去用于失活的tritonx-100去垢剂,这能够通过后续的失活处理步骤来实现。纯化步骤之后的病毒灭活包括利用poroshq树脂进行的阴离子交换色谱。该工艺步骤将有助于从产物除去残留的tritonx-100以及另外的杂质(即,dna、hcp)。在捕获步骤之后,回收了大约13.8g的产物,其电导率为38.8ms/cm。poroshq树脂的最大上样不超过10grecap/l树脂,因此,对于1.53升poroshq柱仅需要一次循环。在同一天完成captoadhere色谱和病毒灭活。对于病毒灭活,将1:9v/v的10%tritonx-100储液加入到captoadhere洗脱液中,以制备1%tritonx-100最终浓度。将产物充分混合,然后在环境温度下保持静置1小时,以完成病毒灭活。在上样到上样的poroshq柱上之前,用wfi将病毒失活的capto洗脱液稀释12x(1+11),以使电导率下降至4.4ms/cm,在≤4.5ms/cm的规格之内。以大约9grecap/l树脂的上样量将该病毒失活的和稀释的captoahdere洗脱液上样到1.53lporoshq柱上。利用表12中列出的既定条件纯化该产物。在材料上样后进行低盐洗涤(50mmnacl)以除去杂质。一旦出现uv观察条件,对于3.0cv的最大洗脱液收集,利用高盐(130mmnacl)来实现产物洗脱。洗脱液经过0.2微米过滤到10lbpc(hyclone)中,之后储存在2-8℃下。表12.poroshq色谱试验条件poroshq步骤的色谱结果与之前的结果一致,由于上样材料中存在tritonx-100,因此在上样期间得到轻微uv吸光度。色谱图显示除了陡峭的洗脱液峰,其最大吸光度为3.2au。在2mnacl清洗期间存在强峰,这是之前鉴定过的非产物相关杂质。通过rp-hplc滴定方法、终点活性测试、hcpelisa和sec-hplc来分析poroshq洗脱液,并与captoadhere中间体进行比较。纯化步骤的recap回收和活性回复与演示试验5类似并且分别以63%和68%进行了小试规模验证试验(1、2)。相比于上样材料,残留hcp中的减少超过3倍,这提示最初的两个工艺步骤确实能够降低hcp,但是另外的纯化步骤也是必要的以满足≤100ppm的目标hcp规格。异二聚体a%在中间体中增加,通过sec-hplc保持98.6%的高recap纯度。mimeticblue纯化在poros50hq纯化完成之后,随后的纯化步骤包括利用mimeticblueap树脂的亲和色谱。通过mimeticblue柱步骤纯化大约9g的产物。使用2.6l上样的柱,仅需以大约3.3grecap/l上样的的一次循环。将poroshq洗脱液升温至环境温度,接着用wfi进行2x(1+1)稀释以在上样前降低电导率。将等分试样稀释后,将其上样到上样的2.6lmimeticblue柱上并利用表13中列出的修订条件进行纯化。工艺条件从包括具有l-精氨酸的中间洗涤的之前确定的条件稍微改变以除去残留hcp之后用平衡缓冲液洗涤以防止由于缓冲液混合造成的产物损失。除了中间洗涤之外,洗脱条件从仅使用磷酸钠的之前演示试验改变,而不是磷酸钠和盐的组合。针对mimeticblue柱步骤的优化的研究表明,用不包括盐的洗脱缓冲液的处理中间体中残留hcp显著下降,因此其被从工艺去除(1)。在洗脱过程中,预定义的uv门(≥0.125au/cm至≤0.25au/cm)被用于收集产物。产物经过0.2微米pes过滤到无菌5lbpc(hyclone)中,之后储存在2-8℃下。表13.mimeticblue纯化色谱试验条件色谱洗脱方式非常类似于之前的演示试验。对分析组的的rp-滴定度、残留dna(通过qpcr)、酶促活性、残留hcp和纯度(通过sec-hplc)进行分析来分析洗脱液。mimeticblue纯化步骤的结果揭示了recap的回收率为92%且活性恢复为91%。通过sec-hplc测量的纯度增加至大约100%,这证实了recap是中间体中存在的主要产物。残留dna的水平下降至<0.5pg/mg,明显低于≤35pg/mg的目标规格。与小试规模证实试验相一致的是,利用优化的的mimeticblue条件使mimeticblue洗脱液中存在的残留hcp显著下降了大约2000倍,这相对于演示试验中存在的超过50倍下降是很大的改善(1、2)。残留hcp显著低于针对药物物质的≤100ppm目标规格。来自mimeticblue纯化的结果证明了mimeticblue柱步骤提供了相比于先前柱步骤最大的hcp清除率。butyl650m精加工该过程中的最终色谱步骤包括利用butyl650m树脂的疏水相互作用下色谱(hic)。在hic色谱之前,将mimeticblue洗脱液升温至环境温度,然后加入硫酸铵。mimeticblue洗脱液的uv280用于计算butyl650m循环的最小数目,其不超过10grecap/l树脂。对于上样的1.48lbutyl650m柱,对于5.5grecap/l树脂的最大上样仅需一次循环。柱上样之后,立即用2.1mamso4ph8.0将mimeticblue洗脱液稀释至1.0mamso4的最终浓度。柱上样在1小时内完成,加入硫酸铵以降低潜在的沉淀形成。然后利用表14中列出的确定的条件来纯化产物。用0.6mamso4利用预先定义的uv设门(≥0.25au/cm至≤0.25au/cm的设门)洗脱产物。在从柱洗脱之后,立即用wfi将洗脱液稀释3x(1+2)并用0.2微米pes将其过滤到无菌20lbpc(hyclone)中,然后储存在2-8℃下。表14.butyl650m精加工色谱试验条件hic步骤的色谱图与之前的试验一致,得到1.6au的最大吸光度的洗脱液峰值。对butyl650m工艺中间体,通过uv280(分析浓度)、终点活试验、rp-滴定度、qpcr(分析残留dna)、elisa(分析hcp)和sec-hplc(分析纯度)进行分析,并与mimeticblue中间体进行比较。针对recap和活性将洗脱液保持在高比活性和大于80%的回收率。将通过sec-hplc确定的recap的纯度保持在99.9%。通过sec-hplc未观察到高分子量(hmw)聚集体,这提示添加硫酸铵不改变产物质量或者导致产物发生聚集。残留hcp浓度低于hcpelisa测定的检测限(≤3ppm)。因此,在hic色谱步骤之后,残留hcp浓度下降了至少12倍,这提示hic步骤对于通过该工艺进一步清除hcp是必需的。超滤/渗滤该工艺的下一步骤包括超滤/渗滤至缓冲液交换以及浓缩产物。分子量截留为10kda的0.1m2pellicon2超滤biomax盒用于浓缩产物,之后进行渗滤以除去硫酸铵。首先用0.5mnaoh净化膜1小时,用wfi冲洗,并用配方缓冲液(20mml-组氨酸、250mmd-山梨糖醇、2mmmgcl2、50μmzncl2,ph7.0)平衡。将稀释的hic洗脱液升温至环境温度,之后将其上样到膜上,上样在膜上的产物为70g/m2。对于0.1m2的膜,在整个试验中利用0.5l/min的漫流流速(crossflowrate)将tmp保持在≤15psi。最初将稀释的butyl650m洗脱液浓缩至14g/l,之后用配方缓冲液进行10x渗滤。然后用大约31ml的配方缓冲液清洗膜以达到11.5g/l的目标浓度(通过uv280确定)。一旦达到目标浓度,利用0.2微米pes过滤器单元过滤截留物质并立即通过病毒过滤进行处理。分析上样和uf/df截留物质(经过清洗)的活性、浓度(通过uv280)、残留dna(通过qpcr)、纯度(通过sec-hplc)和残留hcp。recap的纯度保持在100%,这表明没有聚集物形成。uf/df步骤的回收率为大约70%,其低于演示试验5的100%回收率(2)。在演示试验5处理中使用的相同的膜被用于该演示试验中并且其重复利用可能造成产物损失。uf/df截留物质中的残留hcp浓度低于hcpelisa定量分析的检测限。病毒过滤和散装充填在散装充填之前,该工艺中的最终步骤为病毒过滤。用50l/m2配方缓冲液(20mml-组氨酸、250mmd-山梨糖醇、2mmmgcl2、50μmzncl2,ph7.0)平衡0.001m2planova15n过滤器。以430l/m2上样将uf/df截留物质施加至平衡的病毒过滤器,保持分压为≤15psi。一旦所有材料施加之后,用10l/m2的配方缓冲液清洗过滤器以回收过滤器外壳中的蛋白质。然后向滤液中加入清洗液以达到10.4g/l的终浓度。散装药物的目标规格为9.0至11.0mg/ml,因此该最终浓度在pes之前的规格范围内。为了制备bds,然后通过0.2微米pes过滤系统将该病毒滤液过滤到1lpetg容器中并储存在2-8℃下。对上样、经过清洗的病毒滤液和散装药物的酶促活性、残留dna(通过qpcr)、rp-滴定度、纯度(通过sec-hplc)、浓度(通过uv280)和残留hcp(通过elisa)进行分析。在病毒过滤步骤之后回收了大约98%的产物。在过滤期间均保持比活性和纯度。散装药物中的残留hcp明显低于≤100ppm的目标规格。这一结果证实了,优化的工艺能够获得比基线工艺更大的hcp清除率,其在之前的演示试验的整个过程中都接近大约100ppm。演示试验结果的总结在演示试验完成后,在图16中列表并示出了对于每一工艺中间体的分析结果。对于该演示试验而言最重要的因素在于,利用针对captoadhere和mimeticblue柱步骤已被进一步确立的优化条件,hcp显著清除率达到<0.5ppm,明显低于<100ppm的目标规格(1)。稳定性测试利用在实施例3中获得的配方进行稳定性试验。图17中总结了在所示条件下在组氨酸缓冲液和在柠檬酸盐缓冲液中的配方的结果。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1