一种哌嗪磺酰胺类化合物及其制备方法和应用与流程
[0001]
本发明属于药化领域,具体涉及一种哌嗪磺酰胺类化合物及其制备方法和应用,尤其涉及一种活性高的哌嗪磺酰胺类化合物及其制备方法和应用。
背景技术:
[0002]
阿尔茨海默症是一种进行性发展的致死性神经退行性疾病,临床表现为认知和记忆功能不断恶化,日常生活能力进行性减退,并有各种神经精神症状和行为障碍。阿尔茨海默症是老年期最为常见的一种痴呆类型,也是老年期最常见的慢性疾病之一。
[0003]
目前发现用于抗老年痴呆药物的作用机理主要包括:通过抑制乙酰胆碱酯酶的活性,减少乙酰胆碱的降解,提高脑内胆碱能水平而建立的乙酰胆碱酯酶抑制剂,和通过抑制nmda受体表面或离子通道内部的各个作用位点,降低nmda受体的兴奋效应,抑制其活性而建立起来的nmda受体拮抗剂。近年来出现一种比较流行和公认的作用机制为“β淀粉样蛋白学说(amyloid-βhypothesis,aβ)”,该机制认为app蛋白前体被水解断裂后形成不溶性的β淀粉样蛋白积聚构成了老年斑(amyloid plaques),由此引发了一系列的病理变化,包括神经炎症,神经细胞缺失和死亡等,最终导致阿尔茨海默病。根据该机制的流程,通过阻断循环中各个步骤以改变疾病的进程,就可以达到减轻或治疗的目的。目前根据此种作用机制所开发的各种新药如beta-水解酶抑制剂、gama-水解酶抑制剂、疫苗、tau蛋白磷酸化抑制剂、抗神经炎症等,绝大多数是基于β淀粉样蛋白学说。
[0004]
cn106187891b公开了一系列具有抗阿尔茨海默症作用的化合物,其以邻氨基苯甲酸为原料,通过与环己酮反应得到9-氯-1,2,3,4-四氢吖啶,然后和乙二胺反应得到9-(β-氨基乙二胺)-1,2,3,4-四氢吖啶;然后再和肉桂酸及其衍生物反应制备得到一系列具有抗阿尔茨海默症作用的化合物;整个工艺设计合理,产率高,副产物少,成本低,制备得到的化合物纯度高,具有很好的抗阿尔茨海默症功效,可实现工业化大生产。但是其反应路线长,合成不方便。其结构如下:
[0005][0006]
cn105535131a公开了熟大黄微粉在制备阿尔茨海默症药物中的用途。其还提供了一种药物组合物,含有熟大黄微粉为活性成分,加上药学上可接受的辅料或辅佐性成分制备而成的中药制剂。其有针对性地治疗阿尔茨海默症、疗效确切、无成瘾性和依赖性、无明显的毒副作用。但其采用中药制剂可能对病人产生潜在影响。
[0007]
cn103788058b公开了一种由金山五味子提取的木脂素类化合物,该化合物对神经细胞具有保护作用,对阿尔茨海默症具有预防和治疗作用,可用于制备神经细胞保护剂和防治阿尔茨海默症的药物。其结构如下:
[0008][0009]
目前阿尔茨海默症严重威胁人类生命健康,但是目前尚无有效治疗药物,因此,如何提供一种制备简单、治疗效果好的阿尔茨海默症治疗药物,成为了亟待解决的问题。
技术实现要素:
[0010]
针对现有技术的不足,本发明的目的在于提供一种哌嗪磺酰胺类化合物及其制备方法和应用,尤其提供一种活性高的哌嗪磺酰胺类化合物及其制备方法和应用。本发明提供的产品制备工艺简单,原料易得,适合工业化大规模生产,活性高,安全性高。
[0011]
为达到此发明目的,本发明采用以下技术方案:
[0012]
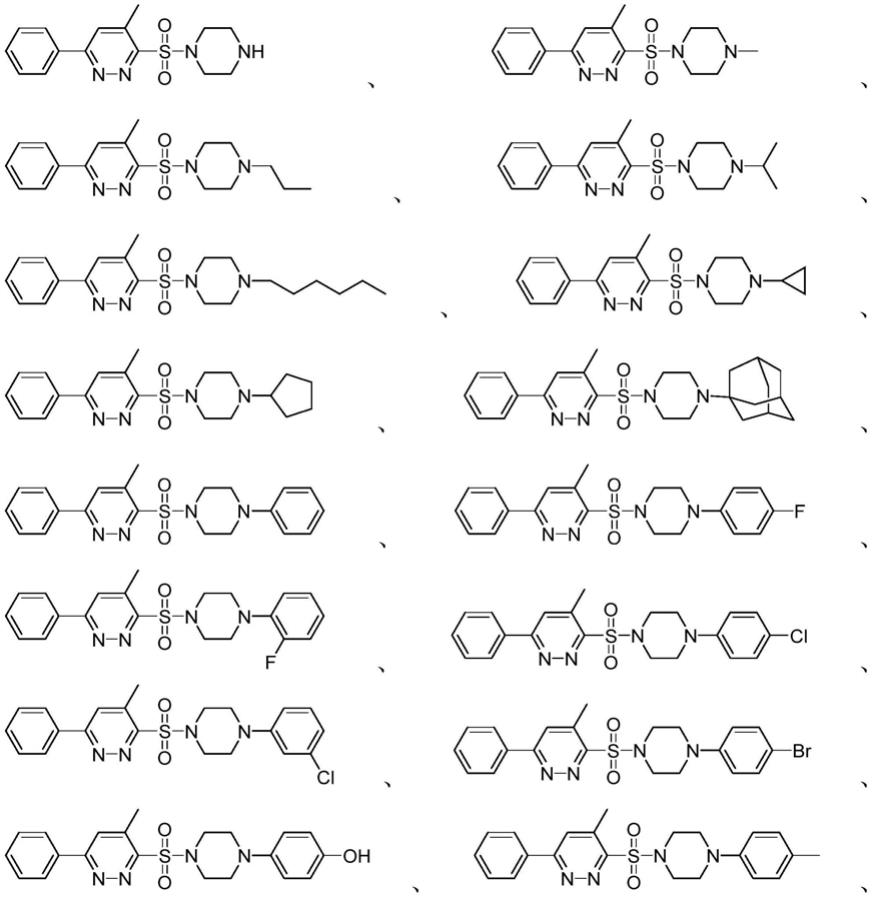
第一方面,本发明提供了一种哌嗪磺酰胺类化合物,所述哌嗪磺酰胺类化合物的结构如式i所示。
[0013][0014]
所述式i中r选自氢、取代或未取代的烷基、取代或未取代的环烷基、取代或未取代的芳基、取代或未取代的杂芳基或取代或未取代的杂环烷基中任意一种。
[0015]
上述化合物能有效抑制小胶质细胞(bv2)分泌白介素-1β(il-1β)的作用,对阿尔茨海默病具有良好的治疗效果。
[0016]
优选地,所述烷基、环烷基、芳基、杂芳基和杂环烷基中的取代基独立地选自芳基、芳基烷基、c1-c6烷基、c1-c6烷氧基、卤烷基、卤素、羟基、氨基或氰基中任意一种,其中c1-c6分别代表基团结构中含有1个碳原子、2个碳原子、3个碳原子、4个碳原子、5个碳原子和6个碳原子。
[0017]
所述取代基中,芳基例如可以是苯基、萘基、蒽基或菲基中任意一种,芳基烷基例如可以是苯甲基、苯乙基、苯异丙基、对苯二甲基、邻苯二甲基或间苯二甲基中任意一种,c1-c6烷基例如可以是甲基、乙基、丙基、异丙基、丁基、仲丁基、异丁基或叔丁基中任意一种,c1-c6烷氧基例如可以是甲氧基、乙氧基、丙氧基、异丙氧基、丁氧基、仲丁氧基、异丁氧基或叔丁氧基中任意一种,卤烷基例如可以是氯甲基、二氯甲基、二氟甲基、三氟甲基、全氟乙基或2,2,2-三氟乙基中任意一种,卤素例如可以是氟、氯、溴或碘中任意一种。
[0018]
优选地,所述烷基选自c1-c6烷基,例如可以是甲基、乙基、丙基、异丙基、丁基、仲丁基、异丁基或叔丁基中任意一种。
[0019]
优选地,所述环烷基选自c3-c6环烷基,例如可以是环丙基、环丁基、环戊基或环己基中任意一种。
[0020]
优选地,所述杂芳基选自哒嗪基、吲哚基、喹唑啉基、吡咯基、噻吩基、吲唑基、吡唑基、喹啉基、吡啶基、呋喃基、咪唑基、吡嗪基、嘧啶基、噻唑基、异喹啉基、苯并噻唑基或二氮杂萘基中任意一种。
[0021]
优选地,所述杂环烷基选自四氢呋喃基、四氢吡喃基、四氢吡咯基、哌啶基、四氢噻吩基、哌嗪基、六氢嘧啶基、吗啉基、1,3-恶嗪烷基或1,3-噻嗪烷基中任意一种。
[0022]
优选地,r选自氢、甲基、丙基、异丙基、己基、环丙基、环戊基、金刚烷基、苯基、4-氟苯基、2-氟苯基、4-氯苯基、3-氯苯基、4-溴苯基、4-羟基苯基、4-甲基苯基、2-甲氧基苯基、4-甲氧基苯基、3-三氟甲基苯基、2,4-二氟苯基、2,3-二氯苯基、2-氯-6-氟苯基、3,4-二甲基苯基、3,4-二甲氧基苯基、嘧啶-2-基、5-氟嘧啶-2-基、5-溴嘧啶-2-基、5-三氟甲基嘧啶-2-基、4,6-二甲基嘧啶-2-基、吡啶-2-基、吡啶-4-基、吡啶-3-基、吡嗪-2-基、噻吩-2-基或噻唑-2-基中任意一种。
[0023]
优选地,所述哌嗪磺酰胺类化合物的结构选自
[0024]
中任意一种。
[0025]
第二方面,本发明提供了如上所述的哌嗪磺酰胺类化合物在药学上可接受的盐、水合物。
[0026]
具体的,所述哌嗪磺酰胺类化合物在药学上可接受的盐、水合物例如可以是上述哌嗪磺酰胺类化合物的盐酸盐、氢溴酸盐、氢碘酸盐、磷酸盐、硫酸盐、硝酸盐、乙磺酸盐、甲苯磺酸盐、苯磺酸盐、乙酸盐、马来酸盐、酒石酸盐、琥珀酸盐、柠檬酸盐、苯甲酸盐、抗坏血酸盐和水杨酸盐、丙二酸盐、己二酸盐、己酸盐、精氨酸盐、富马酸盐、烟酸盐、邻苯二甲酸盐、草酸盐、锂盐、钠盐、钾盐、钡盐、钙盐、镁盐、铝盐、铁盐、亚铁盐、铜盐、锌盐、吗啉盐、二乙胺盐、三乙胺盐、异丙胺盐、三甲胺盐、赖氨酸盐或组胺酸盐及其水合物中任意一种。
[0027]
第三方面,本发明提供了如上所述的哌嗪磺酰胺类化合物的制备方法,所述制备方法包括以下步骤:
[0028]
(1)将化合物a与化合物b混合,加入溶剂和缩合剂反应,得到化合物c;
[0029]
(2)将步骤(1)得到的化合物c与氧化剂和溶剂混合反应,得到所述哌嗪磺酰胺类化合物。
[0030]
上述步骤的反应式如下:
[0031][0032]
所述r具有与如上所述相同的限定范围。
[0033]
上述制备方法制备工艺简单,原料易得,适合工业化大规模生产。
[0034]
优选地,步骤(1)中所述化合物a与化合物b的摩尔比为1:1-1:2。
[0035]
优选地,步骤(1)中化合物a与缩合剂的摩尔比为1:8-1:12。
[0036]
优选地,所述缩合剂包括次氯酸钠和/或磺酰氯。
[0037]
优选地,步骤(1)中所述反应的温度为20-30℃。
[0038]
优选地,步骤(1)中所述反应的时间为8-14h。
[0039]
优选地,步骤(2)中所述化合物c与氧化剂的摩尔比为1:2-1:6。
[0040]
优选地,步骤(2)中所述氧化剂包括间氯过氧苯甲酸、双氧水、过氧乙酸或过一硫酸氢钾复合盐中任意一种。
[0041]
优选地,步骤(2)中所述反应的温度为20-30℃。
[0042]
优选地,步骤(2)中所述反应的时间为8-14h。
[0043]
其中,化合物a与化合物b的摩尔比可以是1:1、1:1.1、1:1.2、1:1.3、1:1.4、1;1.5、1:1.6、1:1.7、1:1.8、1:1.9或1:2等,化合物a与缩合剂的摩尔比可以是1:8、1:8.5、1:9、1:9.5、1:10、1:10.5、1:11、1:11.5或1:12等,步骤(1)中所述反应的温度可以是20℃、21℃、22℃、23℃、24℃、25℃、26℃、27℃、28℃、29℃或30℃等,步骤(1)中所述反应的时间可以是8h、9h、10h、11h、12h、13h或14h等,化合物c与氧化剂的摩尔比可以是1:2、1:2.5、1:3、1;3.5、1:4、1:4.5、1:5、1:5.5或1:6等,步骤(2)中所述反应的温度可以是20℃、21℃、22℃、23℃、24℃、25℃、26℃、27℃、28℃、29℃或30℃等,步骤(2)中所述反应的时间可以是8h、9h、10h、11h、12h、13h或14h等,但并不仅限于所列举的数值,上述各数值范围内其他未列举的数值同样适用。
[0044]
优选地,步骤(1)和步骤(2)中所述溶剂选自二氯甲烷、四氢呋喃或乙腈中任意一种。
[0045]
作为本发明优选的技术方案,所述制备方法包括以下步骤:
[0046]
(1)将化合物a与化合物b以摩尔比1:1-1:2混合,加入溶剂和缩合剂在20-30℃下反应8-14h,得到化合物c;
[0047]
(2)将步骤(1)得到的化合物c与氧化剂以摩尔比1:2-1:6和溶剂混合,在20-30℃下反应8-14h,得到所述哌嗪磺酰胺类化合物。
[0048]
第四方面,本发明还提供了如上所述的哌嗪磺酰胺类化合物或哌嗪磺酰胺类化合物在药学上可接受的盐、水合物在制备预防或治疗神经退行性疾病、精神病、癫痫、惊厥或中风药物中的应用。
[0049]
优选地,所述药物包括如上所述的哌嗪磺酰胺类化合物和/或哌嗪磺酰胺类化合物在药学上可接受的盐、水合物以及药用辅料。
[0050]
优选地,所述药物的剂型包括液体剂型、半液体剂型或固体剂型中任意一种。
[0051]
与现有技术相比,本发明具有如下有益效果:
[0052]
本发明提供的哌嗪磺酰胺类化合物制备工艺简单,原料易得,适合工业化大规模生产;能够高效抑制bv2分泌il-1β的作用,半数有效抑制浓度(ic
50
)达到46.28μmol,显示了优秀的治疗效果;对于胶质瘤细胞(c6)分泌il-1β没有抑制作用,显示了高安全性。
具体实施方式
[0053]
为更进一步阐述本发明所采取的技术手段及其效果,以下结合本发明的优选实施例来进一步说明本发明的技术方案,但本发明并非局限在实施例范围内。
[0054]
以下实施例中3-巯基-4-甲基-6-苯基哒嗪根据journal of medicinal chemistry,44(17),2707-2718;2001中公开的方法制备,其他初始原料、反应试剂等若无特殊说明均为市售产品;
[0055]
bv2细胞系购自于美国sciencell;
[0056]
c6细胞系购自于通派(上海)生物科技有限公司;
[0057]
km小鼠购自于南方医科大学。
[0058]
实施例1 4-甲基-6-苯基-3-((4-(嘧啶-2-基)哌嗪-1-基)磺酰基)哒嗪的制备,其结构如下:
[0059][0060]
(1)将3-巯基-4-甲基-6-苯基哒嗪(610mg,3mmol)和2-嘧啶哌嗪(740mg,4.5mmol)混合,加入二氯甲烷30ml,混合液冷却至0℃,缓慢滴加15%次氯酸钠溶液10ml,滴加完毕。缓慢升至25℃,搅拌12h。待反应完全后,加入水50ml,萃取分出有机层,干燥,浓缩硅胶柱层析,分离得到4-甲基-6-苯基-3-((4-(嘧啶-2-基)哌嗪-1-基)硫基)哒嗪(340mg,收率31%);
[0061]
(2)将步骤(1)得到的4-甲基-6-苯基-3-((4-(嘧啶-2-基)哌嗪-1-基)硫基)哒嗪(300mg,0.82mmol)溶于二氯甲烷25ml,加入85%间氯过氧苯甲酸(666mg,3.28mmol)。25℃搅拌12h,待反应完全后,加入饱和的硫代硫酸钠搅拌20min,分出有机层,饱和碳酸氢钠洗,饱和食盐水洗,干燥,过滤,浓缩硅胶柱层析,得到4-甲基-6-苯基-3-((4-(嘧啶-2-基)哌嗪-1-基)磺酰基)哒嗪(81mg,收率25%)。
[0062]
对产物4-甲基-6-苯基-3-((4-(嘧啶-2-基)哌嗪-1-基)磺酰基)哒嗪进行表征,结果如下:1h-nmr(400mh,cdcl3)δ8.33(d,j=4.8hz,2h),8.03-8.05(m,2h),7.82(s,1h),
7.53-7.55(m,3h),6.54(t,j=4.8hz,1h),4.07(bs,4h),3.76(m,4h),2.76(s,3h);ms:m/e 397.1(m+h
+
),证明成功合成4-甲基-6-苯基-3-((4-(嘧啶-2-基)哌嗪-1-基)磺酰基)哒嗪。
[0063]
实施例2 4-甲基-6-苯基-3-((4-甲基哌嗪-1-基)磺酰基)哒嗪的制备,其结构如下:
[0064][0065]
(1)将3-巯基-4-甲基-6-苯基哒嗪(610mg,3mmol)和n-甲基哌嗪(450mg,4.5mmol)混合,加入二氯甲烷30ml,混合液冷却至0℃,缓慢滴加15%次氯酸钠溶液10ml,滴加完毕。缓慢升至30℃,搅拌8h。待反应完全后,加入水50ml,萃取分出有机层,干燥,浓缩硅胶柱层析,分离得到4-甲基-3-((4-甲基哌嗪-1-基)硫基)-6-苯基哒嗪(315mg,收率35%);
[0066]
(2)将步骤(1)得到的4-甲基-3-((4-甲基哌嗪-1-基)硫基)-6-苯基哒嗪(300mg,0.998mmol)溶于二氯甲烷25ml,加入85%间氯过氧苯甲酸(810mg,3.992mmol)。20℃搅拌14h,待反应完全后,加入饱和的硫代硫酸钠搅拌20min,分出有机层,饱和碳酸氢钠洗,饱和食盐水洗,干燥,过滤,浓缩硅胶柱层析,得到4-甲基-6-苯基-3-((4-甲基哌嗪-1-基)磺酰基)哒嗪(93mg,28%)。
[0067]
对产物4-甲基-6-苯基-3-((4-甲基哌嗪-1-基)磺酰基)哒嗪进行表征,结果如下:ms:m/e 333.2(m+h
+
),证明成功合成4-甲基-6-苯基-3-((4-甲基哌嗪-1-基)磺酰基)哒嗪。
[0068]
实施例3 4-甲基-6-苯基-3-((4-环丙基哌嗪-1-基)磺酰基)哒嗪的制备,其结构如下:
[0069][0070]
(1)将3-巯基-4-甲基-6-苯基哒嗪(610mg,3mmol)和n-环丙基哌嗪(567mg,4.5mmol)混合,加入二氯甲烷30ml,混合液冷却至0℃,缓慢滴加15%次氯酸钠溶液10ml,滴加完毕。缓慢升至20℃,搅拌14h。待反应完全后,加入水50ml,萃取分出有机层,干燥,浓缩硅胶柱层析,分离得到3-((4-环丙基哌嗪-1-基)硫基)-4-甲基-6-苯基哒嗪(323mg,收率33%);
[0071]
(2)将步骤(1)得到的3-((4-环丙基哌嗪-1-基)硫基)-4-甲基-6-苯基哒嗪(300mg,0.919mmol)溶于二氯甲烷25ml,加入85%间氯过氧苯甲酸(746mg,3.68mmol)。30℃搅拌8h,待反应完全后,加入饱和的硫代硫酸钠搅拌20min,分出有机层,饱和碳酸氢钠洗,饱和食盐水洗,干燥,过滤,浓缩硅胶柱层析,得到4-甲基-6-苯基-3-((4-环丙基哌嗪-1-基)磺酰基)哒嗪(102mg,收率31%)。
[0072]
对产物4-甲基-6-苯基-3-((4-环丙基哌嗪-1-基)磺酰基)哒嗪进行表征,结果如下:ms:m/e 359.1(m+h
+
),证明成功合成4-甲基-6-苯基-3-((4-环丙基哌嗪-1-基)磺酰基)哒嗪。
[0073]
实施例4 3-((4-(4-氟苯基)哌嗪-1-基)磺酰基)-4-甲基-6-苯基哒嗪的制备,其结构如下:
[0074][0075]
(1)将3-巯基-4-甲基-6-苯基哒嗪(610mg,3mmol)和n-对氟苯基哌嗪(811mg,4.5mmol)混合,加入二氯甲烷30ml,混合液冷却至0℃,缓慢滴加15%次氯酸钠溶液10ml,滴加完毕。缓慢升至25℃,搅拌12h。待反应完全后,加入水50ml,萃取分出有机层,干燥,浓缩硅胶柱层析,分离得到3-((4-(4-氟苯基)哌嗪-1-基)硫基)-4-甲基-6-苯基哒嗪(410mg,收率36%);
[0076]
(2)将步骤(1)得到的3-((4-(4-氟苯基)哌嗪-1-基)硫基)-4-甲基-6-苯基哒嗪(300mg,0.788mmol)溶于二氯甲烷25ml,加入85%间氯过氧苯甲酸(640mg,3.152mmol)。25℃搅拌12h,待反应完全后,加入饱和的硫代硫酸钠搅拌20min,分出有机层,饱和碳酸氢钠洗,饱和食盐水洗,干燥,过滤,浓缩硅胶柱层析,得到3-((4-(4-氟苯基)哌嗪-1-基)磺酰基)-4-甲基-6-苯基哒嗪(94mg,29%)。
[0077]
对产物3-((4-(4-氟苯基)哌嗪-1-基)磺酰基)-4-甲基-6-苯基哒嗪进行表征,结果如下:ms:m/e 413.2(m+h
+
),证明成功合成3-((4-(4-氟苯基)哌嗪-1-基)磺酰基)-4-甲基-6-苯基哒嗪。
[0078]
实施例5 3-((4-(吡啶-2-基)哌嗪-1-基)磺酰基)-4-甲基-6-苯基哒嗪的制备,其结构如下:
[0079][0080]
(1)将3-巯基-4-甲基-6-苯基哒嗪(610mg,3mmol)和n-(吡啶-2-基)哌嗪(735mg,4.5mmol)混合,加入二氯甲烷30ml,混合液冷却至0℃,缓慢滴加15%次氯酸钠溶液10ml,滴加完毕。缓慢升至25℃,搅拌12h。待反应完全后,加入水50ml,萃取分出有机层,干燥,浓缩硅胶柱层析,分离得到4-甲基-6-苯基-3-((4-(吡啶-2-基)哌嗪-1-基)硫基)哒嗪(349mg,收率32%);
[0081]
(2)将步骤(1)得到的4-甲基-6-苯基-3-((4-(吡啶-2-基)哌嗪-1-基)硫基)哒嗪(300mg,0.825mmol)溶于二氯甲烷25ml,加入85%间氯过氧苯甲酸(670mg,3.3mmol)。25℃搅拌12h,待反应完全后,加入饱和的硫代硫酸钠搅拌20min,分出有机层,饱和碳酸氢钠洗,饱和食盐水洗,干燥,过滤,浓缩硅胶柱层析,得到3-((4-(吡啶-2-基)哌嗪-1-基)磺酰基)-4-甲基-6-苯基哒嗪(88mg,收率27%)。
[0082]
对产物3-((4-(吡啶-2-基)哌嗪-1-基)磺酰基)-4-甲基-6-苯基哒嗪进行表征,结果如下:ms:m/e 396.1(m+h
+
),证明成功合成3-((4-(吡啶-2-基)哌嗪-1-基)磺酰基)-4-甲基-6-苯基哒嗪。
[0083]
体外活性测定:
[0084]
检测方法:取鼠源小胶质细胞系bv2和胶质瘤细胞系c6分别培养在含有10%fbs的dmem培养基中,培养第6代至第15代的细胞用于候选化合物活性筛选。培养细胞按50000个细胞/孔接种于24孔细胞培养板,在培养1天后更换为低血清培养基(添加2%fbs)继续培养16h。在培养基中加入300ng/ml(用于诱导bv2细胞)或1mg/ml(用于诱导c6细胞)脂多糖(鼠
伤寒沙门氏菌)诱导培养细胞分泌il-1β。并同时按终浓度200pm,20nm,2μm和200μm加入待测化合物(dmso≤0.1%);向空白加入0.1%dmso作为溶剂对照组。脂多糖诱导/药物处理24h后,收集培养液定量检测其中il-1β水平。培养液样品于4℃离心(8000g)10min,除去培养液中的悬浮颗粒杂质。将上清液稀释1倍,取150μl样品用于elisa(biosoμrce)检测。培养细胞处理和elisa检测采用双盲法进行。待测化合物抑制效率根据公式
①
计算,并计算每种候选化合物的ic
50
。以化合物对bv2分泌il-1β的抑制率阳性为判断化合物有效的标准;并以对c6分泌il-1β的抑制率为阴性作为判断化合物安全的指标。
[0085]
公式
①
:抑制率(%)=([il-1β]
lps诱导-[il-1β]
药物处理
)/[il-1β]
lps诱导
×
100%
[0086]
按上述方法对实施例1-5提供的化合物和阳性对照gibh-130进行测试,结果如下:
[0087]
表1实施例1-5提供的化合物抑制bv2分泌il-1β的ic
50
[0088]
组别ic
50
(μmol)组别ic
50
(μmol)gibh-13067.63实施例342.32实施例141.99实施例438.76实施例243.25实施例546.28
[0089]
表2实施例1-5提供的化合物对c6细胞分泌il-1β抑制的抑制率
[0090][0091][0092]
从上述数据可得,本发明提供的化合物在抑制bv2分泌il-1β中具有良好的活性,显示了优秀的治疗效果;同时对c6细胞分泌il-1β的抑制效果不明显,显示了高安全性。
[0093]
药代动力学实验:
[0094]
选取km小鼠8只,雄性,体重30-40g之间。将实施例1提供的化合物以2%dmso/4%乙醇/4%蓖麻油/90%去离子水配制。受试化合物分别给予灌胃5mg/kg和尾静脉注射1mg/kg。km小鼠被分成2组,每组4只。试验前禁食12h,自由饮水。于灌胃给药后,分别于5min、15min、30min、1h、3h、5h、8h由眼眶后静脉丛采血0.1ml;于尾静脉注射给药后,分别于5min、15min、30min、1h、3h、5h、8h由眼眶后静脉丛采血0.1ml,置肝素管中,4℃8000rpm离心6min分离血浆,于-20℃保存待测。然后采用lc-ms分析药物浓度,结果见下表:
[0095]
表3药代动力学分析
[0096][0097]
从上表可以看出本发明提供的化合物具有优良的药代动力学性质,能够用于制备安全有效的治疗阿尔茨海默病的新药。
[0098]
申请人声明,本发明通过上述实施例来说明本发明的哌嗪磺酰胺类化合物及其制备方法和应用,但本发明并不局限于上述实施例,即不意味着本发明必须依赖上述实施例才能实施。所属技术领域的技术人员应该明了,对本发明的任何改进,对本发明产品各原料的等效替换及辅助成分的添加、具体方式的选择等,均落在本发明的保护范围和公开范围之内。
[0099]
以上详细描述了本发明的优选实施方式,但是,本发明并不限于上述实施方式中的具体细节,在本发明的技术构思范围内,可以对本发明的技术方案进行多种简单变型,这些简单变型均属于本发明的保护范围。
[0100]
另外需要说明的是,在上述具体实施方式中所描述的各个具体技术特征,在不矛盾的情况下,可以通过任何合适的方式进行组合,为了避免不必要的重复,本发明对各种可能的组合方式不再另行说明。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1