一种两亲性壳聚糖衍生物及其制备方法
1.本发明涉及生物技术领域,特别是涉及一种两亲性壳聚糖衍生物及其制备方法。
背景技术:
2.随着生物技术的蓬勃发展和重大突破,生物医用材料已成为研究和开发的热点。目前,具有良好生物相容性的两亲性生物材料,由于具有亲水和疏水基团,表现出在水溶液中自发形成稳定胶束的特性,在医药领域受到广泛关注。一方面,利用胶束的疏水内核与药物的作用,可实现对疏水药物的增溶、输运。另一方面,通过对胶束的靶向修饰可实现胶束对病灶部位的特异性识别,大大提高药效、降低毒副作用,或实现病变组织的高分辨成像、提高疾病检测灵敏度。
3.18β
‑
甘草次酸为甘草中的活性成分,是一种重要的医药和高档化妆品原料,具有抗炎抗过敏、抑制细菌繁殖等作用。将其用于化妆品中可调节皮肤的免疫功能,增强皮肤免疫能力,消除炎症、预防过敏。解除化妆品中相关成分及其他外界因素对皮肤的毒副作用。它还能够有效抑制络氨酸酶活性,阻止黑色素的产生,具有美白功效。用于口腔产品可有效预防和消除牙龈炎症、口腔溃疡。
4.唾液酸为一种天然的碳水化合物,广泛存在于人体中,对细胞的代谢起重要作用。唾液酸可在细胞表面的位置保护大分子和细胞免受酶和免疫的攻击并促进内在免疫。在分子和细胞之间,细胞和细胞之间,细胞和外界之间,糖链末端的唾液酸既可作为识别位点,也可掩蔽识别位点。
5.壳聚糖(chitosan)是由甲壳素(chitin)在强碱条件下脱乙酰基得到的产物,是天然多糖中唯一的碱性多糖,而且壳聚糖无刺激性,具有良好的生物相容性和生物可降解性,其降解产物无毒,能被生物体完全吸收,因此是理想的纳米微胶囊壁材。相比于甲壳素,脱乙酰基后的壳聚糖溶解性能已有很大程度的改善,但也只能溶解在ph<6.0的酸性溶液中。由于壳聚糖溶解性差、粘度高,其应用范围受到限制,因此改善壳聚糖的溶解性能,是壳聚糖改性研究中最重要的方向之一。
6.通过对壳聚糖的化学改性,在壳聚糖分子中引入适量的亲水基团或疏水基团后,会破坏壳聚糖的结晶结构,使其溶解性极大提高,即利用壳聚糖主链上的氨基、羟基的反应活性,通过化学改性方法使壳聚糖主链上同时接上带有疏水性侧链和亲水性的侧基或侧链,从而使壳聚糖同时具有亲水性和疏水性,即两亲性,并且通过调节亲水性侧链与疏水性侧链的比例,对壳聚糖分别用亲水、疏水基团进行修饰,使壳聚糖的两亲性控制在合理的范围内,可得到两亲性壳聚糖衍生物,有利于扩大壳聚糖的应用范围。同时还可以包埋脂溶性药物的,实现靶向输运至病灶部位,最大程度提高药物的生物利用度,并减轻药物的毒副作用。
技术实现要素:
7.本发明的目的是提供一种两亲性壳聚糖衍生物及其制备方法,以解决上述现有技
术存在的问题,以丰富壳聚糖衍生物的种类,扩大壳聚糖在生物医药方面的应用范围,同时使得制备方法简单。
8.为实现上述目的,本发明提供了如下方案:
9.本发明提供一种两亲性壳聚糖衍生物,具有式ⅰ所示的结构:
[0010][0011]
本发明还提供一种上述两亲性壳聚糖衍生物的制备方法,包括以下步骤:
[0012]
(1)18β甘草次酸接枝壳聚糖:
[0013]
a.将壳聚糖、水与hobt混合搅拌,待壳聚糖溶解,备用;
[0014]
b.将18β甘草次酸加入步骤a中的体系中,搅拌均匀,再加入edac
·
hcl,搅拌混匀后调节体系ph值为4.8
‑
5.2,在室温下搅拌反应1
‑
12h;
[0015]
c.反应结束后,在水中用透析袋对步骤b所得的体系进行透析,将透析后的产物进行真空干燥,洗涤,再次真空干燥得到18β甘草次酸
‑
壳聚糖;
[0016]
(2)唾液酸接枝18β甘草次酸
‑
壳聚糖:
[0017]
d.将步骤(1)制备的18β甘草次酸
‑
壳聚糖与水及hobt混合搅拌,待18β甘草次酸
‑
壳聚糖溶解后,备用;
[0018]
e.将唾液酸加入步骤d的体系中,搅拌均匀,加入edac
·
hcl,搅拌混匀后调节体系ph值为4.8
‑
5.2,然后在室温下搅拌反应1
‑
12h;
[0019]
f.反应结束后,在水中用透析袋对步骤e所得的体系进行透析,将透析后的产物进行真空干燥,得到唾液酸
‑
壳聚糖18β甘草次酸接枝产物,即为两亲性壳聚糖衍生物。
[0020]
进一步地,步骤a中壳聚糖的浓度为5
‑
20mg/ml,壳聚糖氨基与hobt的摩尔比为2∶1
‑
1∶2。
[0021]
进一步地,步骤a中壳聚糖氨基与hobt的摩尔比为1:1。
[0022]
进一步地,步骤b中所述edac
·
hcl、18β甘草次酸与步骤a中hobt的摩尔比为(1
‑
6):(1
‑
3):1;步骤b反应时间为3h;利用稀盐酸或酸性pbs调节步骤b中体系ph值。
[0023]
进一步地,步骤b中所述edac
·
hcl、18β甘草次酸与步骤a中hobt的摩尔比为4∶2∶1。
[0024]
进一步地,步骤c中洗涤所用的溶剂为乙醇、甲醇或二氯甲烷中的一种;步骤c和步骤f中所述透析袋的截留分子量为2000
‑
3500da,透析时间为48
‑
72h。
[0025]
进一步地,步骤c中所述洗涤溶剂为乙醇;步骤c和步骤f中所述透析袋的截留分子
量为3500da,透析时间为72h。
[0026]
进一步地,步骤d中所述18β甘草次酸
‑
壳聚糖浓度为5
‑
20mg/ml,18β甘草次酸
‑
壳聚糖氨基与hobt的摩尔比为2∶1
‑
1∶2;
[0027]
步骤e中所述edac
·
hcl、唾液酸与步骤d中hobt的摩尔数比为(1
‑
6):(1
‑
3):1;利用稀盐酸或酸性pbs调节步骤e中体系ph值;步骤e的反应时间为3h。
[0028]
进一步地,步骤d中所述8β甘草次酸
‑
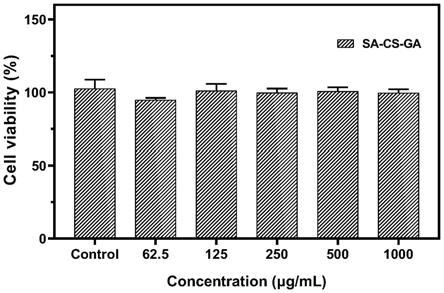
壳聚糖氨基与hobt的摩尔比为1:1;步骤e中所述edac
·
hcl、唾液酸与步骤d中hobt的摩尔数比为4∶2∶1。
[0029]
本发明公开了以下技术效果:
[0030]
本发明制备的sa
‑
cs
‑
ga所用原料之一的壳聚糖,是自然界唯一的阳离子天然多糖,来源于虾蟹壳等,资源十分丰富;本发明制备的sa
‑
cs
‑
ga水溶性好,对细胞没有毒副作用及血液相容性良好,该两亲性壳聚糖衍生物具有两亲性基团,可作为疏水药物的载体,在抗炎,抗菌抗病毒及药物载体方面具有良好的应用前景,在生物医药领域具有潜在的应用价值。
[0031]
本发明的反应条件温和,仅在hobt和edac
·
hcl的存在下即可将18β甘草次酸和唾液酸接枝到壳聚糖分子链上,制备方法简单、反应时间短、条件温和且反应高效,同时便于实施,不需要复杂设备,便于工业化批量生产。
附图说明
[0032]
为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
[0033]
图1为本发明实施例1合成的sa
‑
cs
‑
ga的1h
‑
nmr图谱;
[0034]
图2为本发明实施例1合成的sa
‑
cs
‑
ga的红外光谱图;
[0035]
图3为本发明实施例1合成的sa
‑
cs
‑
ga细胞毒性试验结果图;
[0036]
图4为本发明实施例1合成的sa
‑
cs
‑
ga血液相容性试验结果图,图(a)为溶血率,图(b)为直观图。
[0037]
图5为本发明实施例1合成的sa
‑
cs
‑
ga抗炎实验结果图,其中,图(a)为不同处理组的耳厚差,图(b)为不同处理组的耳重差。
具体实施方式
[0038]
现详细说明本发明的多种示例性实施方式,该详细说明不应认为是对本发明的限制,而应理解为是对本发明的某些方面、特性和实施方案的更详细的描述。
[0039]
应理解本发明中所述的术语仅仅是为描述特别的实施方式,并非用于限制本发明。另外,对于本发明中的数值范围,应理解为还具体公开了该范围的上限和下限之间的每个中间值。在任何陈述值或陈述范围内的中间值以及任何其他陈述值或在所述范围内的中间值之间的每个较小的范围也包括在本发明内。这些较小范围的上限和下限可独立地包括或排除在范围内。
[0040]
除非另有说明,否则本文使用的所有技术和科学术语具有本发明所述领域的常规
技术人员通常理解的相同含义。虽然本发明仅描述了优选的方法和材料,但是在本发明的实施或测试中也可以使用与本文所述相似或等同的任何方法和材料。本说明书中提到的所有文献通过引用并入,用以公开和描述与所述文献相关的方法和/或材料。在与任何并入的文献冲突时,以本说明书的内容为准。
[0041]
在不背离本发明的范围或精神的情况下,可对本发明说明书的具体实施方式做多种改进和变化,这对本领域技术人员而言是显而易见的。由本发明的说明书得到的其他实施方式对技术人员而言是显而易见的。本申请说明书和实施例仅是示例性的。
[0042]
关于本文中所使用的“包含”、“包括”、“具有”、“含有”等等,均为开放性的用语,即意指包含但不限于。
[0043]
本发明的两亲性壳聚糖衍生物,具有式ⅰ所示的结构:
[0044][0045]
实施例1
[0046]
式ⅰ所示两亲性壳聚糖衍生物的制备方法:
[0047]
(1)18β甘草次酸接枝壳聚糖的合成:
[0048]
a.将壳聚糖(浓度为20mg/ml)、去离子水与1
‑
羟基苯并三唑(hobt)混合搅拌,搅拌10min待壳聚糖溶解后,备用;其中,壳聚糖氨基与hobt的摩尔比为1∶1。
[0049]
b.将18β甘草次酸缓慢加入步骤a中的体系中,搅拌均匀,再缓慢加入1
‑
(3
‑
二甲氨基丙基)
‑3‑
乙基碳二亚胺盐酸盐(edac
·
hcl),搅拌混匀后利用稀盐酸调节体系ph值为4.8,然后在室温下搅拌反应3h;
[0050]
edac
·
hcl和18β甘草次酸的摩尔数与hobt的摩尔数比为4∶2∶1。
[0051]
c.反应结束后,在水中用透析袋(透析袋的截留分子量为3500da)透析产物,透析时间为72h;真空干燥,用乙醇洗涤,真空干燥得到18β甘草次酸
‑
壳聚糖。
[0052]
(2)唾液酸接枝18β甘草次酸
‑
壳聚糖
[0053]
d.将步骤1中制备的18β甘草次酸
‑
壳聚糖(浓度为5mg/ml)与水及hobt混合搅拌10min,待溶解后,备用;
[0054]
18β甘草次酸
‑
壳聚糖氨基与hobt的摩尔比为1∶1。
[0055]
e.将唾液酸缓慢加入步骤d中的体系中,搅拌均匀,再缓慢加入1
‑
(3
‑
二甲氨基丙基)
‑3‑
乙基碳二亚胺盐酸盐(edac
·
hcl),搅拌混匀后利用酸性pbs调节体系ph值为4.8,然后在室温下搅拌反应3h;
[0056]
加入edac
·
hcl和唾液酸的摩尔数与步骤d中hobt的摩尔数比为4∶2∶1。
[0057]
f.反应结束后,在水中用透析袋透析产物(截留分子量为3.5kda),透析72h,真空干燥,得到唾液酸
‑
壳聚糖18β甘草次酸接枝产物(sa
‑
cs
‑
ga)。
[0058]
图1是本发明实施例1合成的sa
‑
cs
‑
ga的1h
‑
nmr图谱;图2是本发明实施例1合成的sa
‑
cs
‑
ga的红外光谱图。
[0059]
实施例2
[0060]
式ⅰ所示两亲性壳聚糖衍生物的制备方法:
[0061]
(1)18β甘草次酸接枝壳聚糖的合成:
[0062]
a.将壳聚糖(浓度为5mg/ml)、去离子水与1
‑
羟基苯并三唑(hobt)混合搅拌10min,待壳聚糖溶解后,备用;
[0063]
壳聚糖氨基与hobt的摩尔比为1.2∶2。
[0064]
b.将18β甘草次酸缓慢加入步骤a中的体系中,搅拌均匀,再缓慢加入1
‑
(3
‑
二甲氨基丙基)
‑3‑
乙基碳二亚胺盐酸盐(edac
·
hcl),搅拌混匀后利用稀盐酸调节体系ph值为5.0,然后在室温下搅拌反应12h;
[0065]
edac
·
hcl和18β甘草次酸的摩尔数与hobt的摩尔数比为1∶1∶1。
[0066]
c.反应结束后,在水中用透析袋(透析袋的截留分子量为3000da)透析产物,透析时间为48h;真空干燥,用甲醇洗涤,真空干燥得到18β甘草次酸
‑
壳聚糖;
[0067]
(2)唾液酸接枝18β甘草次酸
‑
壳聚糖
[0068]
d.将步骤1中制备的18β甘草次酸
‑
壳聚糖(浓度为20mg/ml)与水及hobt混合搅拌10min,待溶解后,备用;
[0069]
18β甘草次酸
‑
壳聚糖氨基与hobt的摩尔比为1.5∶1。
[0070]
e.将唾液酸缓慢加入步骤d中的体系中,搅拌均匀,再缓慢加入1
‑
(3
‑
二甲氨基丙基)
‑3‑
乙基碳二亚胺盐酸盐(edac
·
hcl),搅拌混匀后利用酸性pbs调节体系ph值为5.0,然后在室温下搅拌反应12h;
[0071]
所述加入edac
·
hcl和唾液酸的摩尔数与步骤d中hobt的摩尔数比为3∶2∶1。
[0072]
f.反应结束后,在水中用透析袋透析产物(截留分子量为3.0kda),透析时间为48h,真空干燥,得到唾液酸
‑
壳聚糖18β甘草次酸接枝产物。
[0073]
实施例3
[0074]
式ⅰ所示两亲性壳聚糖衍生物的制备方法:
[0075]
(1)18β甘草次酸接枝壳聚糖的合成:
[0076]
a.将壳聚糖(浓度为15mg/ml)、去离子水与1
‑
羟基苯并三唑(hobt)混合搅拌11min,待壳聚糖溶解后,备用;
[0077]
壳聚糖氨基与hobt的摩尔比为2∶1.5;
[0078]
b.将18β甘草次酸缓慢加入步骤a中的体系中,搅拌均匀,再缓慢加入1
‑
(3
‑
二甲氨基丙基)
‑3‑
乙基碳二亚胺盐酸盐(edac
·
hcl),搅拌混匀后利用稀盐酸调节体系ph值为5.2,然后在室温下搅拌反应1h;
[0079]
edac
·
hcl和18β甘草次酸的摩尔数与hobt的摩尔数比为5∶3∶1。
[0080]
c.反应结束后,在水中用透析袋(透析袋的截留分子量为2500da)透析产物,透析时间为50h;真空干燥,用二氯甲烷洗涤,真空干燥得到18β甘草次酸
‑
壳聚糖。
[0081]
(2)唾液酸接枝18β甘草次酸
‑
壳聚糖
[0082]
d.将步骤1中制备的18β甘草次酸
‑
壳聚糖(浓度为15mg/ml)与水及hobt混合搅拌12min,待溶解后,备用;
[0083]
18β甘草次酸
‑
壳聚糖氨基与hobt的摩尔比为1∶2;
[0084]
e.将唾液酸缓慢加入步骤d中的体系中,搅拌均匀,再缓慢加入1
‑
(3
‑
二甲氨基丙基)
‑3‑
乙基碳二亚胺盐酸盐(edac
·
hcl),搅拌混匀后利用稀盐酸调节体系ph值为5.2,然后在室温下搅拌反应10h;
[0085]
所述加入edac
·
hcl和唾液酸的摩尔数与步骤d中hobt的摩尔数比为5∶3∶1。
[0086]
f.反应结束后,在水中用透析袋透析产物(截留分子量为2000da),透析时间为55h,真空干燥,得到唾液酸
‑
壳聚糖18β甘草次酸接枝产物。
[0087]
实施例4
[0088]
式ⅰ所示两亲性壳聚糖衍生物的制备方法:
[0089]
(1)18β甘草次酸接枝壳聚糖的合成:
[0090]
a.将壳聚糖(浓度为10mg/ml)、去离子水与1
‑
羟基苯并三唑(hobt)混合搅拌10min,待壳聚糖溶解后,备用;壳聚糖氨基与hobt的摩尔比为1.5∶1。
[0091]
b.将18β甘草次酸缓慢加入步骤a中的体系中,搅拌均匀,再缓慢加入1
‑
(3
‑
二甲氨基丙基)
‑3‑
乙基碳二亚胺盐酸盐(edac
·
hcl),搅拌混匀后利用稀盐酸调节体系ph值为5.2,然后在室温下搅拌反应6h;
[0092]
edac
·
hcl和18β甘草次酸的摩尔数与hobt的摩尔数比为4∶1∶1。
[0093]
c.反应结束后,在水中用透析袋(透析袋的截留分子量为2700da)透析产物,透析时间为60h;真空干燥,用乙醇洗涤,真空干燥得到18β甘草次酸
‑
壳聚糖。
[0094]
(2)唾液酸接枝18β甘草次酸
‑
壳聚糖
[0095]
d.将步骤1中制备的18β甘草次酸
‑
壳聚糖(浓度为10mg/ml)与水及hobt混合搅拌10min,待溶解后,备用;
[0096]
18β甘草次酸
‑
壳聚糖氨基与hobt的摩尔比为2∶1;
[0097]
e.将唾液酸缓慢加入步骤d中的体系中,搅拌均匀,再缓慢加入1
‑
(3
‑
二甲氨基丙基)
‑3‑
乙基碳二亚胺盐酸盐(edac
·
hcl),搅拌混匀后利用酸性pbs调节体系ph值为5.2,然后在室温下搅拌反应6h;
[0098]
所述加入edac
·
hcl和唾液酸的摩尔数与步骤d中hobt的摩尔数比为4∶1∶1。
[0099]
f.反应结束后,在水中用透析袋透析产物(截留分子量为2800da),透析时间为65h;真空干燥,得到唾液酸
‑
壳聚糖18β甘草次酸接枝产物。
[0100]
效果验证:
[0101]
一、细胞毒性试验:
[0102]
对于实施例1制备的sa
‑
cs
‑
ga进行细胞毒性试验,试验过程如下:
[0103]
收集hacat细胞后调整细胞浓度,将其以2
×
104个/孔的密度接种于96孔培养板上(每孔100μl),每组设6个复孔,培养结束后,对细胞进行mtt检测。mtt检测具体操作步骤如下:细胞处理结束后,每孔加入10μlmtt溶液(5mg/ml),37℃孵育4h后,小心吸弃各孔内液体,每孔再加入150μl dmso,避光振荡10
‑
15min,全自动酶标仪测定570nm波长处各孔吸收值(od)。细胞存活率按以下公式计算:
[0104]
细胞存活率(%)=实验组(od)/对照组(od)
×
100%
[0105]
细胞毒性试验结果见图3,由图3可以看出与正常细胞比较,这种两亲性壳聚糖衍生物的存活率没有显著差异,说明合成的壳聚糖衍生物没有细胞毒性。
[0106]
二、血液相容性试验:
[0107]
对于实施例1制备的sa
‑
cs
‑
ga进行血液相容性试验,试验过程如下:
[0108]
1.新鲜抗凝小鼠血在2000rpm/min下离心15min,弃上清液,得到红细胞沉淀,用1
×
pbs缓冲液(0.1m,ph=7.2
‑
7.4)清洗红细胞沉淀多次,直至上清液呈清透状态,而后用pbs将红细胞沉淀稀释至体积浓度为2%(v/v);
[0109]
2.将500μl红细胞悬液与500μl不同浓度的样品(用pbs溶解过滤除菌后,紫外灭菌30min)在1.5ml离心管中混合均匀,密封、轻轻地摇匀试管后置于37℃水浴中孵育1h,同时以500μl去离子水和500μl pbs分别与500μl红细胞悬液共混培养作为阳性和空白对照,3次平行实验。
[0110]
图4为血液相容性试验的结果,图中(a)为溶血率,(b)为直观图,由图4可以看出,经sa
‑
cs
‑
ga处理的样品与水处理的样品相比,没有发现明显的红细胞破裂现象,与pbs处理的样品相似,在浓度范围内溶血率<5%。结果表明,sa
‑
cs
‑
ga在浓度<500μg/ml时对红细胞无损伤作用,具有良好的血液相容性。
[0111]
三、急性抗炎效果试验:
[0112]
对于实施例1制备的sa
‑
cs
‑
ga进行急性抗炎效果试验,试验过程如下:
[0113]
将6周龄的雄性icr小鼠在自由获取食物和水的情况下饲养1周使其适应实验环境后开始进行后续实验。小鼠分为4组,分别为正常组,模型组,cs组,sa
‑
cs
‑
ga组,每组8只。正常组不作任何处理,正常饲养;其他组小鼠的右耳给予tpa丙酮溶液(5μg/20μl)处理(连续3天),左耳用不含tpa的纯丙酮液体处理(20μl),如图5所示,在1小时后对小鼠右耳给予cs及sa
‑
cs
‑
ga的治疗处理。在第四天,小鼠耳拍照及处死小鼠进行后续的检测。用厚度仪测量小鼠右耳与左耳的厚度,随后用8mm的小鼠耳打孔器取得小鼠右耳和左耳组织,用分析天平称量耳重。
[0114]
图5为tpa诱导小鼠耳肿胀的耳厚差与耳重差图。图5(a)为不同处理组的耳厚差值,结果显示,tpa诱导小鼠右耳后,与正常组比较,模型组右耳与左耳的厚度差明显增大。与模型组比较,cs组的耳厚差没有显著差异,而sa
‑
cs
‑
ga组的耳厚差显著下降,表明sa
‑
cs
‑
ga对tpa诱导的炎症发展有抑制作用。图5(b)为不同处理组的耳重差值,与耳厚差类似,与正常组比较,模型组的耳重差增大,具有极显著性。与模型组比较,cs组的耳重差没有显著性差异,然而sa
‑
cs
‑
ga组的耳重差显著性下降。说明sa
‑
cs
‑
ga具有抗炎效果。
[0115]
以上所述的实施例仅是对本发明的优选方式进行描述,并非对本发明的范围进行限定,在不脱离本发明设计精神的前提下,本领域普通技术人员对本发明的技术方案做出的各种变形和改进,均应落入本发明权利要求书确定的保护范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1