一种由双荧光素酶报告基因检测鸡RIPK2启动子活性的方法
本发明涉及一种由双荧光素酶报告基因检测鸡ripk2启动子活性的方法,属于基因工程领域。
背景技术:
ripk2是ripk激酶家族的一个成员,主要通过nod/ripk2通路进行免疫炎症调节。抑制ripk2基因可减少各种炎症因子的释放和缓解炎症反应综合征。而高表达ripk2时可激活p38mapk通路,能更有效的清除大肠杆菌。这表明ripk2对胞内菌的识别、炎症反应的缓解以及其nod/ripk2通路对宿主免疫的调节都发挥着关键作用。因此,调控基因ripk2的表达对机体免疫炎症的调节具有重要实践意义。
基因表达是一个受时间和空间等多因素调控的复杂过程。基因是否表达通常由特定启动子是否启动来决定的。转录因子可以特异性结合启动子区的dna序列,并协助rna聚合酶在启动子区启动基因转录。启动子本身并不控制基因活动,而是通过转录因子结合位点与转录因子结合来控制基因活动。转录因子结合位点的改变可直接影响转录因子的结合,继而改变基因的转录效率并引起基因表达的差异。因此,筛选启动子核心区域即验证启动子最大活性区域对进一步探究基因的表达调控具有非常重要的现实意义。
双荧光素酶报告基因检测系统使实验报告基因测试的结果归一化,具有检测速度快、灵敏度高等特点,且在哺乳动物中已应用的十分纯熟。本方法将鸡ripk2基因启动子区系列缺失片段连接到荧光素酶报告载体pgl3-basic上,转染免疫细胞,检测荧光素酶活性,以确定鸡ripk2基因启动子核心区域,为筛选调控鸡ripk2表达的因素提供了一种快速便捷的方法。
技术实现要素:
本发明的目的在于针对上述问题,提供一种由双荧光素酶报告基因检测鸡ripk2启动子活性的方法,本发明利用双荧光素酶报告基因检测系统,将不同长度的鸡ripk2启动子插入荧光素酶报告质粒,检测荧光素酶表达量来确定启动子活性,为筛选调控鸡ripk2表达的因素提供了一种快速便捷的方法。
本发明的目的是这样实现的,一种由双荧光素酶报告基因检测鸡ripk2启动子活性的方法,灵敏度高、检测迅速且费用较低,包括以下步骤:
步骤1)、鸡ripk2基因启动子全长的克隆;
利用软件tfsearch和proscan分析鸡ripk2基因启动子区域,利用primer5.0设计启动子区引物,具体为固定3′端,从5′端开始设计,每次截短500bp设计启动子区系列缺失片段7对引物,由上海生物工程有限公司合成;提取鸡巨噬细胞系hd11基因组dna;pcr扩增出鸡ripk2启动子区系列缺失片段;琼脂糖凝胶电泳检测pcr扩增产物;对产物进行纯化;送上海生物工程有限公司进行测序分析;
步骤2)、鸡ripk2启动子区系列缺失片段报告质粒的构建和鉴定;
将提纯后的不同长度的启动子片段和pgl3-basic连接,进行单酶切和双酶切,构建含有ripk2启动子不同长度的不同重组质粒,并对其进行筛选和鉴定。
步骤3)、鸡ripk2启动子活性定性分析;
将鸡ripk2启动子全长的重组质粒转染至鸡巨噬细胞系hd11,48h后在倒置荧光显微镜下观察荧光表达状态;
步骤4)、双荧光素酶报告基因检测鸡质粒ripk2启动子的活性;
将鸡ripk2启动子报告质粒和prl-tk共转染至鸡巨噬细胞系hd11中,24h后裂解细胞,并收集裂解液进行双荧光素酶检测;
步骤5)、鸡ripk2核心启动子区的确定;
(1)构建鸡ripk25′端缺失片段载体;
对鸡ripk25′端每隔500bp进行截短并设计特异性引物,构建重组报告质粒,分别命名为pgl3-basic-ripk2-wt、pgl3-basic-ripk2-1、pgl3-basic-ripk2-2、pgl3-basic-ripk2-3、pgl3-basic-ripk2-4、pgl3-basic-ripk2-5、pgl3-basic-ripk2-6并进行双荧光素酶活性检测;
(2)用脂质体转染试剂盒将不同长度的启动子荧光素酶报告质粒和内参质粒prl-tk共转染至细胞中,并检测荧光素酶报告基因的表达活性;
(3)通过对鸡ripk2缺失片段载体启动子活性统计分析,初步确定ripk2核心启动子位置;
步骤6)、鸡ripk2核心启动子区转录因子的筛选;
利用软件transfac和alibaba2.1对鸡ripk2启动子区转录因子结合位点进行预测分析。
步骤4)中,所述的双荧光素酶报告基因为renilla荧光素酶基因。
步骤1)中,所述的7对引物,引物pgl3-basic-ripk2-wt的上游引物序列为
5′-cgtgctagcccgggctcgagtgataaaccactgaaactaa-3′,
引物pgl3-basic-ripk2-wt的下游引物序列为
5′-aacagtaccggaatgccaagcttccggcagcttgctagaggg-3′,
引物pgl3-basic-ripk2-1的上游引物序列为
5′-cgtgctagcccgggctcgagggtctggagcccccagtacaa-3′,
引物pgl3-basic-ripk2-1的下游引物序列为
5′-aacagtaccggaatgccaagcttccggcagcttgctagaggg-3′,
引物pgl3-basic-ripk2-2的上游引物序列为
5′-cgtgctagcccgggctcgagggggcactggagattcatgat-3′,
引物pgl3-basic-ripk2-2的下游引物序列为
5′-aacagtaccggaatgccaagcttccggcagcttgctagaggg-3′,
引物pgl3-basic-ripk2-3的上游引物序列为
5′-cgtgctagcccgggctcgagacagtgctactgtcaggagtc-3′,
引物pgl3-basic-ripk2-3的下游引物序列为
5′-aacagtaccggaatgccaagcttccggcagcttgctagaggg-3′,
引物pgl3-basic-ripk2-4的上游引物序列为
5′-cgtgctagcccgggctcgaggaacaggctgcccagagaagc-3′,
引物pgl3-basic-ripk2-4的下游引物序列为
5′-aacagtaccggaatgccaagcttccggcagcttgctagaggg-3′,
引物pgl3-basic-ripk2-5的上游引物序列为
5′-cgtgctagcccgggctcgagttgtgccttcagtcgcatgcc-3′,
引物pgl3-basic-ripk2-5的下游引物序列为
5′-aacagtaccggaatgccaagcttccggcagcttgctagaggg-3′,
引物pgl3-basic-ripk2-6的上游引物序列为
5′-cgtgctagcccgggctcgagtgaaatgagcaaatgcagccg-3′,
引物pgl3-basic-ripk2-6的下游引物序列为
5′-aacagtaccggaatgccaagcttccggcagcttgctagaggg-3′。
步骤1)中,pcr反应的反应条件为:预变性95℃5min,扩增阶段(95℃变性15s,55℃退火30s,72℃延伸10s)循环30次,终延伸72℃10min。
步骤2中,不同长度的鸡ripk2启动子片段与载体质粒pgl3-basic的连接采用dna连接试剂盒,进行连接反应。
步骤2中,所述的筛选重组子的具体方法,取连接产物直接转化感受态大肠杆菌;1天后挑取单克隆进行测序检测,测序正确的单菌落进行保种和无内毒素质粒大提备用;最后经过酶切、pcr及测序鉴定。
步骤2)中,所述的单酶切具体用的是hindiii,双酶切用的是xhoi和hindiii。
本发明的有益效果,本发明相对于现有技术具有如下的优势和效果:
(1)本发明将包含鸡ripk2基因第一个外显子上游3000bp和下游200bp的片段连接至renilla荧光素酶报告质粒,构建了pgl3-basic-ripk2-wt重组质粒,转染细胞并检测了启动子活性,便于对鸡ripk2基因启动子的调控方式及调控元件进行研究。
(2)本发明具有灵敏度高、准确性强、检测迅速、费用较低等特点,避免了转染效率对结果的影响。
(3)本发明初步确定鸡ripk2启动子核心区域为-2300~-1839bp,在此核心区域预测有与ap-1、cebpa和nf-κb等转录因子结合的位点,这为后续筛选调控鸡ripk2基因表达的转录因子奠定了基础。
附图说明
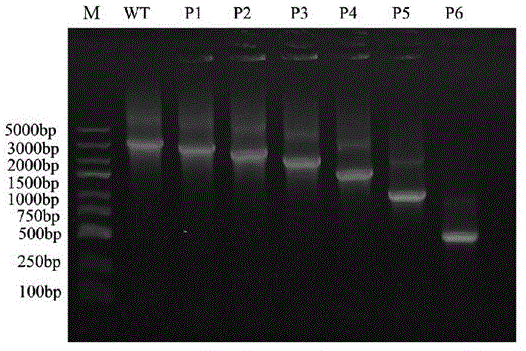
图1为本发明中不同长度的鸡ripk2启动子片段pcr产物的电泳图。
图2为本发明中不同长度鸡ripk2启动子片段重组质粒的单酶切检测的电泳图。
图3为本发明中不同长度鸡ripk2启动子片段重组质粒的双酶切检测的电泳图。
图4为本发明中鸡ripk2基因启动子-3216bp至+38bp片段的活性分析。
图5为本发明中鸡ripk2启动子系列缺失片段的活性检测。注:图中不同上标(a、b和c)代表不同组间存在显著性差异(p<0.05),相同上标差异不显著(p>0.05)。
具体实施方式
为了使本发明所解决的技术问题、技术方案及有益效果更加清楚明白,以下结合实施例,对本发明进行进一步详细说明。
以下例子在此用于示范本发明的优选实施方案,本领域内的技术人员会明白,下述例子中披露的技术代表发明人发现的可以用于实施本发明的技术,因此可以视为实施本发明的优选方案。但是本领域内的技术人员根据本说明书应该明白,这里所公开的特定实施例可以做很多修改,仍然能得到相同的或者类似的结果,而非背离本发明的精神或范围。
除非另有定义,所有在此使用的技术和科学的术语,和本发明所属领域内的技术人员所通常理解的意思相同,在此公开引用及他们引用的材料都将以引用的方式被并入。本领域内的技术人员将意识到或者通过常规试验就能了解许多这里所描述的发明的特定实施方案的许多等同技术。这些等同将被包含在权利要求书中。
下述实施例中的实验方法,如无特殊说明,均为常规方法。下述实施例中所用的仪器设备,如无特殊说明,均为实验室常规仪器设备;下述实施例中所用的试验材料,如无特殊说明,均为自常规生化试剂商店购买得到的。
本发明的实施例:利用双荧光素酶报告基因检测启动子活性的方法,具体如下:
一、菌株、质粒以及试剂的准备;
菌株:大肠杆菌感受态细胞dh5α购自大连takara公司;
细胞系:鸡巨噬细胞系hd11购自上海雅吉生物技术有限公司;
质粒:报告载体pgl3-basic、prl-tk、pegfp-n1购自promega公司;
试剂来源:2×pcrtaqmix购自天根公司;
t4dnaligase、无内毒素质粒提取试剂盒购自大连takara公司;
限制性内切酶hindiii、xhoi、dna提取试剂盒、质粒dna抽提试剂盒、琼脂糖凝胶回收试剂盒购自上海生物工程有限公司;
荧光素酶检测试剂盒购自碧云天公司;
lipofectamine2000transfectionreagent购自invitrogen公司;
胎牛血清购自杭州天杭生物科技有限公司;
dmem高糖培养基购自hyclone公司;
胰酶-edta和pbs购自吉诺生物医药技术有限公司;
其它试剂均为国产分析纯。
二、重组质粒的构建;
步骤1:不同长度鸡ripk2启动子片段重组质粒的构建;
(1)根据鸡ripk2基因5′调控区序列设计7组上下游引物,引物序列如表1:
表1鸡ripk2基因启动子系列缺失片段的引物
注:下划线为酶切位点,xhoi识别序列ctcgag,hindiii识别序列aagctt。
(2)以基因组dna为模板,利用常规pcr反应进行扩增;
(3)反应条件为:预变性95℃5min,扩增阶段(95℃变性15s,55℃退火30s,72℃延伸10s)循环30次,终延伸72℃10min。
(4)反应结束后,取5μl反应产物用1.5%的琼脂糖凝胶电泳检测;
(5)按试剂盒说明书凝胶回收pcr产物;
步骤2)、7组不同长度的鸡ripk2启动子片段的酶切和纯化;
琼脂糖凝胶纯化后的pcr产物进行hindiii单酶切以及hindiii和xhoi双酶切,单酶切体系为:hindiii0.5μl,rbuffer2μl,dna8μl,ddh2o9.5μl。双酶切体系为:xhoi和hindiii各0.5μl,rbuffer2μl,dna8μl,ddh2o9μl。反应条件为:混匀,37℃水浴2h-4h。
步骤3)、连接反应;
酶切产物用琼脂糖凝胶回收试剂盒纯化,通过t4dnaligase将鸡ripk2基因启动子目的片段连接到pgl3-basic载体。连接体系为:10×ligationbuffer2μl,t4dnaligase1μl,目的片段酶切回收产物16μl,pgl3-basic酶切回收产物1μl。混匀反应液,16℃连接过夜。
步骤4)、重组质粒的鉴定;
分别将构建的7组重组质粒转化感受态细胞dh5α,1天后挑取单克隆进行测序检测,测序正确的单菌落进行保种和无内毒素质粒大提。
三、鸡ripk2基因启动子定性分析;
将hd11细胞接种于24孔板,每孔细胞浓度为2×105,细胞汇合度为50%左右时按lipofectamine2000transfectionreagent说明书将重组质粒pegfp-n1/ripk2-wt转染hd11细胞,48h后在倒置荧光显微镜下观察荧光表达状态。pegfp-n1转染hd11细胞为阳性对照,plinker-egfp转染hd11细胞为阴性对照。
四、鸡ripk2启动子双荧光素酶活性检测
将报告基因细胞裂解液充分混匀,吸尽细胞培养液后可以直接加入报告基因细胞裂解液,加入100μl细胞裂解液,重悬细胞,充分裂解5min后,离心收集上清液。取100μl上清液加入到96孔板中,加入100μlrenilla荧光素酶,开启荧光测定仪进行检测。
五、鸡ripk2启动子区转录因子的预测;
利用软件transfac和alibaba2.1对鸡ripk2启动子区转录因子结合位点进行预测分析。
五、结果;
凝胶电泳结果显示扩增出鸡ripk2基因不同长度启动子片段,具体见图1,测序后与预期大小一致。将鸡ripk2基因启动子区系列缺失载体经转化、提取后用酶hindiii作单酶切、酶xhoi和hindiii作双酶切,琼脂糖凝胶电泳结果见图2和图3。单酶切后条带单一,双酶切后得到线性载体片段和ripk2启动子目的片段,均得到两条与预期结果大小相同的条带。说明载体构建正确,可用于后续试验。将pegfp-n1/ripk2-wt、pegfp-n1、plinker-egfp分别转染鸡hd11细胞48h。结果表明pegfp-n1/ripk2-wt转染的hd11细胞可以观察到绿色荧光,可表达gfp,但其荧光强度比阳性对照pegfp-n1弱;而缺失启动子的阴性对照plinker-egfp未检测到gfp的表达;具体结果见图4。因此,克隆片段-3162~+38bp具有启动子活性,可用于后续试验。将ripk2基因6个缺失载体和内参质粒prl-tk共转染鸡hd11细胞,检测结果见图5。双荧光素酶试验结果显示与pgl3-basic-ripk2-2(-2300~+38bp)相比,pgl3-basic-ripk2-3(-1839~+38bp)区域的转录活性明显下降且差异显著,表达出极低的转录活性。这表明-2300~-1839bp之间存在重要的正调控元件,同时也证明-2300~+38bp区域是鸡ripk2基因启动子的基本活性区域。转录因子结合位点分析软件预测显示鸡ripk2基因启动子核心区域存在转录因子ap-1、cebpa和nfκb等。
序列表
<110>扬州大学
<120>一种由双荧光素酶报告基因检测鸡ripk2启动子活性的方法
<160>14
<170>siposequencelisting1.0
<210>1
<211>40
<212>dna
<213>人工序列(artificialsequence)
<400>1
cgtgctagcccgggctcgagtgataaaccactgaaactaa40
<210>2
<211>42
<212>dna
<213>人工序列(artificialsequence)
<400>2
aacagtaccggaatgccaagcttccggcagcttgctagaggg42
<210>3
<211>41
<212>dna
<213>人工序列(artificialsequence)
<400>3
cgtgctagcccgggctcgagggtctggagcccccagtacaa41
<210>4
<211>42
<212>dna
<213>人工序列(artificialsequence)
<400>4
aacagtaccggaatgccaagcttccggcagcttgctagaggg42
<210>5
<211>41
<212>dna
<213>人工序列(artificialsequence)
<400>5
cgtgctagcccgggctcgagggggcactggagattcatgat41
<210>6
<211>42
<212>dna
<213>人工序列(artificialsequence)
<400>6
aacagtaccggaatgccaagcttccggcagcttgctagaggg42
<210>7
<211>41
<212>dna
<213>人工序列(artificialsequence)
<400>7
cgtgctagcccgggctcgagacagtgctactgtcaggagtc41
<210>8
<211>42
<212>dna
<213>人工序列(artificialsequence)
<400>8
aacagtaccggaatgccaagcttccggcagcttgctagaggg42
<210>9
<211>41
<212>dna
<213>人工序列(artificialsequence)
<400>9
cgtgctagcccgggctcgaggaacaggctgcccagagaagc41
<210>10
<211>42
<212>dna
<213>人工序列(artificialsequence)
<400>10
aacagtaccggaatgccaagcttccggcagcttgctagaggg42
<210>11
<211>41
<212>dna
<213>人工序列(artificialsequence)
<400>11
cgtgctagcccgggctcgagttgtgccttcagtcgcatgcc41
<210>12
<211>42
<212>dna
<213>人工序列(artificialsequence)
<400>12
aacagtaccggaatgccaagcttccggcagcttgctagaggg42
<210>13
<211>41
<212>dna
<213>人工序列(artificialsequence)
<400>13
cgtgctagcccgggctcgagtgaaatgagcaaatgcagccg41
<210>14
<211>42
<212>dna
<213>人工序列(artificialsequence)
<400>14
aacagtaccggaatgccaagcttccggcagcttgctagaggg42
- 还没有人留言评论。精彩留言会获得点赞!