单磷酸阿糖腺苷的合成方法及应用与流程
本发明属于药物合成领域,涉及一种抗病毒药物的合成制备,具体地说是一种单磷酸阿糖腺苷的合成方法及应用。
背景技术:
单磷酸阿糖腺苷【化学名为:9-(β-d-阿拉伯呋喃糖)腺嘌呤5’-单磷酸酯,i】为核苷酸类抗病毒药物,其药理作用是与病毒的脱氧核糖核苷酸聚合酶结合,使其活性降低而抑制dna合成。单磷酸阿糖腺苷进入细胞后,经过磷酸化生成阿糖腺苷二磷酸(ara-adp)和阿糖腺苷三磷酸(ara-atp)。抗病毒活性主要由阿糖腺苷三磷酸(ara-atp)所引起,ara-atp与脱氧腺苷三磷酸(datp)竞争地结合到病毒dnap上,从而抑制了酶的活性及病毒dna的合成,同时抑制病毒核苷酸还原酶的活性而抑制病毒dna的合成,还能抑制病毒dna末端脱氧核苷酰转移酶的活性,使ara-atp渗入到病毒的dna中并连接在dna链3’-oh位置的末端,抑制了病毒dna的继续合成,其化学式为:
单磷酸阿糖腺苷是阿糖腺苷的水溶性衍生物,对各种dna病毒如i型和ii型单纯性疱疹病毒、带状疱疹病毒、水痘病毒、牛痘病毒都有明显的抑制活性;在中国,单磷酸阿糖腺苷还广泛用于治疗病毒性乙型肝炎。
但是,单磷酸阿糖腺苷临床用药剂量较大,为满足临床用药需要必须具备合适工业生产的单磷酸阿糖腺苷的合成工艺。
文献报道的化学合成方法主要有两种。
第一条合成路线(m.ikebara,etal.tetrahedronlet1972;28:3695.)以单磷酸腺苷为原料,先制备阿糖腺苷,后者经磷酸化得到单磷酸阿糖腺苷:
反应过程包括脱磷酸和磷酸化两个过程,步骤较长,产率较低,总产量只有8%;特别是磷酸化副产物较多,给分离纯化带来困难。
第二条合成路线(masakatsukaneko,etal.chempharmbull1977;25:1892.)以单磷酸腺苷为原料,经保护后溴化,再经氨化、巯基化、氢化等反应,得到单磷酸阿糖腺苷:
此工艺整个过程无需脱磷酸和磷酸化,总产率可达到12%。但是大部分中间体及最终产品需用活性碳亲和层析和离子交换树脂层析分离,而且反应涉及多次保护和脱保护,不利于工业化生产。
技术实现要素:
为解决现有技术中存在的以上不足,本发明旨在提供一种单磷酸阿糖腺苷的合成方法,以达到缩短单磷酸阿糖腺苷的合成工艺,降低工业生产成本,提高总反应收率的目的。
为实现上述目的,本发明所采用的技术方案如下:
一种单磷酸阿糖腺苷的合成方法,所述合成方法包括以下步骤:
1)取5-碘-2-((磷羧基氧代)甲基)-4-(甲苯磺酰氧代)四氢呋喃-3-基乙酸酯(2)与叔-丁基(8-羟基-9h-嘌呤-6-基)氨基甲酸酯(3)混于有机溶剂a中,加入碱和钯催化剂进行缩合反应,经过滤浓缩后,得到化合物(4)粗品;
2)取化合物(4)粗品溶于thf,再加入碱性溶剂,进行环氧化反应,反应结束后,进行纯化处理i,得到化合物(5)粗品;
3)取化合物(5)粗品与硫氢化物在有机溶剂b中混合,进行开环反应,反应结束后,进行纯化处理ii,得到化合物(6)粗品;
4)取化合物(6)粗品溶于低级醇,加入雷尼镍进行脱硫反应,即得单磷酸阿糖腺苷(1),其总反应式为:
作为本发明的限定,所述有机溶剂a为四氢呋喃(thf)、甲苯、n,n-二甲基甲酰胺(dmf)或二甲基亚砜(dmso);所述碱为碳酸盐或有机碱;所述碳酸盐为碳酸钠、碳酸氢钠、碳酸钾或碳酸铯;所述有机碱为三乙胺(tea)或n,n-二异丙基乙胺(dipea);所述钯催化剂为四(三苯基膦)钯或[1,1'-双(二苯基膦基)二茂铁]二氯化钯;
作为本发明的另一种限定,所述缩合反应的反应温度为60~150℃、反应时间为2~10h;
作为本发明的第三种限定,所述碱性溶剂为氨水或浓度为40%的氢氧化钠溶液;所述环氧化反应的反应温度为90~120℃、反应时间为8~12h;
作为本发明的第四种限定,所述纯化处理i为用1n的盐酸调节ph至中性,再加入乙酸乙酯萃取,保留并浓缩有机相,直至再无溶剂蒸出,即得化合物(5)粗品;
作为本发明的第五种限定,所述硫氢化物为硫氢化钾、硫氢化钠和硫氢酸中的至少一种;所述有机溶剂b为四氢呋喃(thf)、二氯甲烷(dcm)或三氯甲烷;
作为本发明的第六种限定,所述开环反应的反应温度为40~70℃、反应时间为5~8h;所述纯化处理ii为加入二氯甲烷萃取,并加入硅胶,浓缩,进行柱层析,洗脱剂为体积比为13~19:1的石油醚:二氯甲烷,收集并浓缩洗脱剂,即得化合物(6)粗品;
作为本发明的第七种限定,所述低级醇为甲醇(meoh)或乙醇(etoh);所述脱硫反应的反应温度为50~90℃、反应时间为2~5h;
本发明还提供单磷酸阿糖腺苷的合成方法的一种应用,所述单磷酸阿糖腺苷的合成方法用于合成单磷酸阿糖腺苷;所得单磷酸阿糖腺苷用于制备注射用单磷酸阿糖腺苷。
由于采用了上述的技术方案,本发明与现有技术相比,所取得的有益效果是:
(1)本发明所提供的单磷酸阿糖腺苷的合成方法,采用小分子缩合制成单磷酸阿糖腺苷的中间体,缩短了工艺路线,简化了合成步骤,省去了对保护基的引入,避免其他副反应的生成;
(2)本发明所提供的单磷酸阿糖腺苷的合成方法,由于简化了合成步骤,提高了合成单磷酸阿糖腺苷的总反应收率,间接减少了工业生产成本。
(3)本发明所提供的单磷酸阿糖腺苷的合成方法所合成的单磷酸阿糖腺苷,不仅可以用于制备注射用单磷酸阿糖腺苷,还可以制备成多种剂型,如,喷雾剂、软膏剂等,可以避免了单磷酸阿糖腺苷注射后,所产生的局部疼痛等症状。
综上所述,本发明所提供的单磷酸阿糖腺苷的合成方法,进一步简化了工业生产步骤,提高了总的反应收率,减少了工业生产成本。
本发明适用于合成单磷酸阿糖腺苷,所合成的单磷酸阿糖腺苷用于制备注射用单磷酸阿糖腺苷。
附图说明
下面结合附图及具体实施例对本发明作更进一步详细说明。
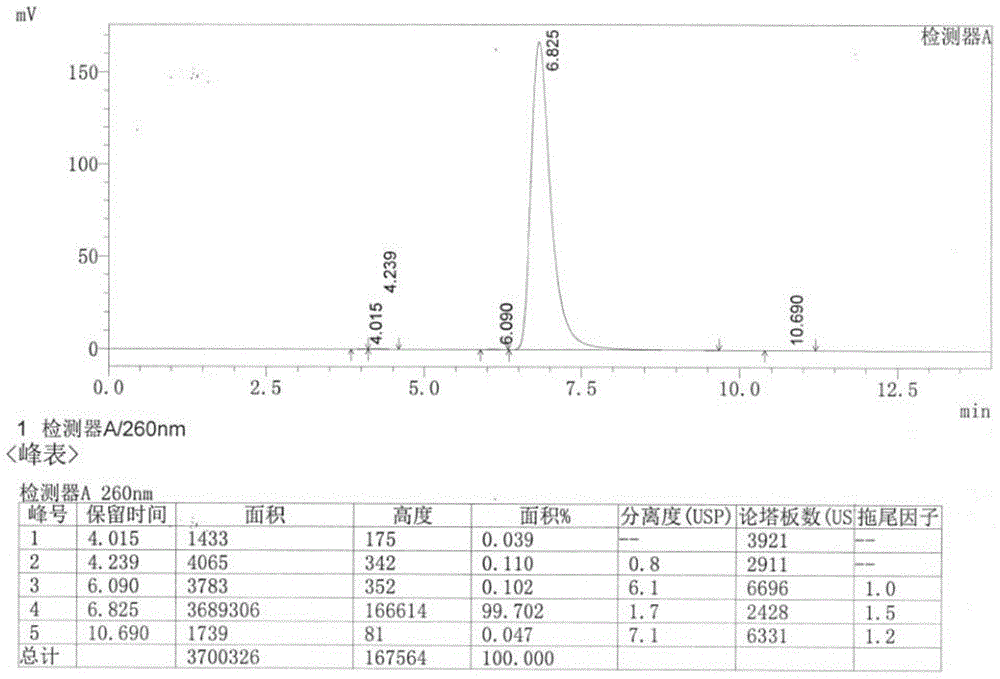
图1为本发明实施例1所合成的单磷酸阿糖腺苷的hplc图。
具体实施方式
以下结合附图对本发明的优选实施例进行说明。应当理解,此处所描述的优选实施例仅用于说明和理解本发明,并不用于限定本发明。
实施例1一种单磷酸阿糖腺苷m1的合成方法
本实施例提供一种单磷酸阿糖腺苷m1的合成方法,其合成方法包括以下步骤:
1)称取53.6g5-碘-2-((磷羧基氧代)甲基)-4-(甲苯磺酰氧代)四氢呋喃-3-基乙酸酯(2)与30g叔-丁基(8-羟基-9h-嘌呤-6-基)氨基甲酸酯(3)混于220mlthf中,加入31.8g碳酸钠和5.78g四(三苯基膦)钯在70℃,进行缩合反应8h,tlc监测反应进度,原料(2)的紫外显色点消失后,即为反应结束。将反应液过滤,滤饼用乙醇洗涤三次,收集滤液并减压浓缩至再无溶剂蒸出,得到39.54g化合物(4)粗品(收率为60%),直接用于下一步,其反应式为:
2)称取39.54g化合物(4)粗品溶于130mlthf,再加入15ml浓度为40%的氢氧化钠溶液,密封升温至90℃,混合搅拌12h,进行环氧化反应,取少量反应液,稀释后注入液相色谱仪中,化合物(4)的物料峰消失,即为反应终点,用1n的盐酸6ml调节ph至中性(广谱ph试纸测得ph=7),再加入300ml乙酸乙酯萃取三次,保留合并有机相,再浓缩所有溶剂,即得17.09g化合物(5)粗品(收率为64%),其反应式为:
3)称取17.09g化合物(5)粗品与2.79g硫氢化钠在75mldcm中溶解,加热到50℃并保温搅拌8h进行开环反应,抽取少量反应液,稀释后注入液相色谱仪,化合物(5)的物料峰消失,即为反应终点,加入50ml饱和硫代硫酸钠水溶液和200ml二氯甲烷萃取五次,收集有机相,浓缩出反应液总量五分之三的溶剂,再加入与反应物总量的重量比为1:1的硅胶,浓缩剩余溶剂,进行柱层析(200~300目硅胶装柱),使用体积比为13:1的石油醚:二氯甲烷的混合溶剂,收集并浓缩洗脱剂,得到10.33g化合物(6)粗品(收率为71%),其反应式为:
4)称取10.33g化合物(6)粗品溶于50mlmeoh,加入6g雷尼镍在65℃进行脱硫反应3h,抽取少量反应液稀释后,注入液相色谱仪,化合物(6)的物料峰消失,即为反应终点,浓缩所有溶剂,加入120ml饱和碳酸氢钠溶液搅拌至澄清,加入50g活性炭脱色,过滤并水洗滤饼,50℃减压浓缩掉一半水,加入体积比为5:8的丙酮:环己烷的混合溶液搅拌,逐滴加入10ml磷酸至大量固体析出,过滤、烘干,即得7.85g单磷酸阿糖腺苷m1(1)(收率为83%;总反应收率为22.6%;纯度为99.7%;),其反应式为:
取少量单磷酸阿糖腺苷m1,稀释后注入高效液相色谱仪根据《中国药典》四部通则0512进行检测,测定结果如图1所示。
实施例2~6单磷酸阿糖腺苷m2~m6的合成方法
实施例2~6提供了单磷酸阿糖腺苷m2~m6的合成方法,其合成方法与实施例1所提供的单磷酸阿糖腺苷的合成方法大致相同,区别仅在于部分合成工艺参数不同,具体合成工艺参数见表1。
表1:单磷酸阿糖腺苷m2~m6的合成工艺参数表
其余工艺参数均与实施例1相同。
对比例1单磷酸阿糖腺苷d1的合成方法
本对比例所提供的单磷酸阿糖腺苷d1的合成方法与实施例1基本相同,区别仅在于缩合反应温度为55℃,反应13h,tlc监测反应进度,原料(2)的紫外点仍有残留,继续保温搅拌3h,tlc监测仍有原料(2)残留,结束反应,按照实施例1中的处理方式处理,得到17.69g化合物(4)(收率为33%),继续按照实施例1步骤2)~4)中的方法合成单磷酸阿糖腺苷,所得单磷酸阿糖腺苷d14.55g(总收率为13.1%,纯度为98.7%)。
对比例2单磷酸阿糖腺苷d2的合成方法
本对比例所提供的单磷酸阿糖腺苷d2的合成方法与实施例1基本相同,区别仅在于缩合反应温度为155℃,反应6h,tlc监测反应进度,原料(2)的紫外点消失,取少量反应液做液相色谱仪检测,与实施例1中步骤1)所得化合物(4)对比,极性较化合物(4)大,分子量为559,所得分子式为:
对比例3单磷酸阿糖腺苷d3的合成方法
本对比例所提供的单磷酸阿糖腺苷d3的合成方法与实施例1基本相同,区别仅在于环氧化反应温度为80℃,反应15h,tlc监测反应进度,化合物(4)的紫外点存在,无新点产生,继续保温搅拌2h,tlc监测仍是没有新点产生,即合成失败。
对比例4单磷酸阿糖腺苷d4的合成方法
本对比例所提供的单磷酸阿糖腺苷d4的合成方法与实施例1基本相同,区别仅在于环氧化反应温度为130℃,反应15h,tlc监测反应进度,化合物(4)的紫外点消失,产生新点,取样注入液相色谱仪中,得到的物料峰极性较大且分子量为265,分子式为:
对比例5单磷酸阿糖腺苷d5的合成方法
本对比例所提供的单磷酸阿糖腺苷d5的合成方法与实施例1基本相同,区别仅在于开环反应温度为室温,反应10h,tlc监测反应进度,化合物(5)的紫外点存在,无新点产生,继续保温搅拌5h,tlc监测仍是没有新点产生,即合成失败。
对比例6单磷酸阿糖腺苷d6的合成方法
本对比例所提供的单磷酸阿糖腺苷d6的合成方法与实施例1基本相同,区别仅在于开环反应温度为80℃,反应5h,tlc监测反应进度,化合物(5)的紫外点消失,产生新点,与化合物(6)极性相差较大,取样送液相测得分子量为413,分子式为:
对比例7单磷酸阿糖腺苷d7的合成方法
本对比例所提供的单磷酸阿糖腺苷d7的合成方法与实施例1基本相同,区别仅在于步骤4)所用洗脱剂为体积比为10:1的石油醚:二氯甲烷的混合溶液洗脱,所有杂质混合单磷酸阿糖腺苷d7一起被洗脱出来,无法到达柱层析的分离目的,所得单磷酸阿糖腺苷d78.79g(收率为93%;纯度为90.6%)。
对比例8单磷酸阿糖腺苷d8的合成方法
本对比例所提供的单磷酸阿糖腺苷d8的合成方法与实施例1基本相同,区别仅在于步骤4)所用洗脱剂为体积比为22:1的石油醚:二氯甲烷的混合溶液洗脱48h,单磷酸阿糖腺苷d8仍无法洗脱出来,无法到达柱层析的分离目的,逐渐加大洗脱剂极性至19:1,洗脱出单磷酸阿糖腺苷,所得单磷酸阿糖腺苷d86.99g(收率为74%;总收率为20.17%;纯度为95.3%)。
对比例9单磷酸阿糖腺苷d9的合成方法
本对比例采用申请号为200410015563.4的中国发明专利,合成单磷酸阿糖腺苷d9,其方法步骤为:
(一)2-氧-对-甲苯磺酰酯基-5-磷酸腺苷(ii)的制备
在150毫升二氧六环和350毫升1当量氢氧化钠的混合溶液中加入34.7克5-amp;待溶解后,向此溶液中加入22.8克研细的对-甲苯磺酰氯,于0℃搅拌反应15小时后,加入6当量盐酸35毫升,调节ph至4.0。滤集析出的结晶,得46.4克结晶粉末ii。
(二)8-溴-2-氧-对-甲苯磺酰酯基-5-磷酸腺苷(iii)的制备
在240毫升2m醋酸钠(ph4)溶液中,加入46克ii,冷却至0~5℃。然后向此溶液中滴加19毫升溴,保持0-5℃。在此温度下搅拌18小时。在0~5℃,向反应液中加入28克亚硫酸氢钠。搅拌15分钟后,以5当量氢氧化钠调节ph至4.0(约90~100毫升)。减压蒸干,所得固体直接用于下步反应。
(三)8-羟基-n,3-二乙酰基-2-氧-对-甲苯磺酰酯基-5-磷酸腺苷(iv)的制备
在上步反应产物中加入80毫升乙酸和80毫升乙酸酐,搅拌回流反应2小时。冷至室温后,加入60毫升甲醇。蒸干。加入乙醇,蒸干;再加入乙醇,再蒸干。将残留物以尽可能少量的水加热溶解,搅拌下冷却至5℃,放置过夜。过滤,以少量水洗,真空干燥,得60.2克固体。
(四)5-磷酸8,2-环氧腺苷(v)的制备
在反应釜中,将60克iv悬浮于300毫升乙醇,在0~5℃通氨气至饱和,密封后于65~70℃搅拌反应20小时。冷至室温后,将钢釜置于干冰-甲醇浴(或冰盐浴)中2小时。滤集析出的固体,以冷甲醇洗,得35克v,可用于下步反应。
(五)单磷酸阿糖腺苷(i)的制备
将35克v以240毫升吡啶溶解,装于钢釜中,加入5克dowex50x4;通干燥的硫化氢至饱和。密封后于95-100℃加热15小时。通氮气赶去过量的硫化氢,将反应液减压浓缩至干。将残留物以600毫升水溶解,滤去不溶物。在滤液中加入20克瑞尼镍,回流反应2.5小时;再补加6克雷尼镍,回流反应0.5小时,再补加4克雷尼镍,回流反应0.5小时。滤去不溶物,将滤液减压浓缩至200毫升,用37%的盐酸调节ph2.5,加入晶种,搅拌下冷却至5℃,放置过夜。过滤,以少量水洗,真空干燥,得7.2克单磷酸阿糖腺苷(i)白色固体,总反应式为:
应用例注射用单磷酸阿糖腺苷的制备方法
单磷酸阿糖腺苷100g
甘露醇40g
5%氢氧化钠溶液适量
注射用水加至2000ml制成1000支。
制法:随机选取实施例5所合成的单磷酸阿糖腺苷m5100g,甘露醇40g加入1500ml注射用水溶解,调节ph=7,补足注射用水至2000ml,灌装,将灌装合格品置于冻干箱内,-50℃预冻2小时,将箱内抽真空至25pa,以2℃/小时缓慢升温进行升华至产品温度达-5℃,进一步升温进行解吸干燥,40℃平衡1小时,压全塞。将冻干品轧盖、灯检、贴签、包装。
实验例注射用单磷酸阿糖腺苷的稳定性测试
加速试验
以上述应用例中所制备的注射用单磷酸阿糖腺苷在温度40℃±2℃、相对湿度75%±5%的条件下放置6个月,经取样分析,各项指标分析结果与0月的比较,含量稍微下降、有关物质稍增加,但均在拟定的限度范围内,其他各项检测指标无明显变化,本品在温度40℃±2℃、相对湿度75%±5%的条件下质量基本稳定,测定结果见表2。
表2:注射用单磷酸阿糖腺苷的加速试验结果
长期试验
以上述应用例中所制备的注射用单磷酸阿糖腺苷在温度为25℃±2℃、相对湿度为60±10%的条件下放置24个月,经取样分析,各项指标分析结果与0月的比较,含量稍下降、有关物质稍增加,但均符合拟定的限度范围内,其他各项监测指标无明显变化,说明本品在长期试验的条件下质量基本稳定,测试结果见表3。
表3:注射用单磷酸阿糖腺苷的长期试验结果表
根据《药剂学》中有关药品有效期的规定,本品的有效期可暂定为24个月。
需要说明的是,以上所述仅为本发明的优选实施例而已,并不用于限制本发明,尽管参照上述实施例对本发明进行了详细的说明,对于本领域技术人员来说,其依然可以对上述各实施例所记载的技术方案进行修改,或者对其中部分技术特征进行等同替换。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!