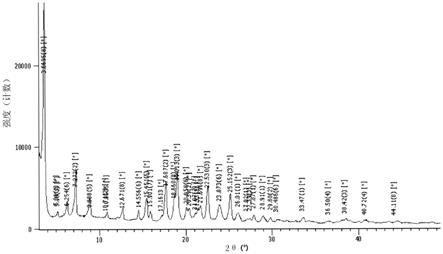
用于制备二苯基甲烷衍生物的方法与流程
用于制备二苯基甲烷衍生物的方法
1.本技术是申请号为201780036968.1的中国专利申请的分案申请,原申请是2017年6月15日提交的pct国际申请pct/kr2017/006271于2018年12月13日进入中国国家阶段的申请。
技术领域
2.本发明涉及用于制备二苯基甲烷衍生物的方法,更特别地,涉及用于制备二苯基甲烷衍生物的改进方法,所述二苯基甲烷衍生物可用作钠依赖性葡萄糖协同转运蛋白(sglt)的抑制剂。
背景技术:
3.钠依赖性葡萄糖协同转运蛋白(sglt)允许根据浓度梯度运输na
+
,同时允许逆着浓度梯度运输葡萄糖。目前,克隆了两种重要的sglt同工型,称为sglt1和sglt2。sglt1位于肠、肾和心脏中,并通过其表达调节心脏葡萄糖转运。由于是一种高亲和力、低容量转运蛋白,sglt1仅负责一部分肾葡萄糖重吸收。相反,sglt2是一种低亲和力、高容量转运蛋白,其主要位于早期近曲小管中的上皮细胞的顶端区域。在健康个体中,超过99%的在肾小球中过滤的血浆葡萄糖被重吸收,而不到1%的总过滤葡萄糖在尿液中排泄。据估计,90%的肾葡萄糖重吸收通过sglt2促进,而其余10%由晚期近端直管中的sglt1介导。sglt2的遗传突变对碳水化合物代谢没有任何具体的不利影响。然而,取决于突变,引起肾葡萄糖分泌增加约140g/天。根据人类突变研究,sglt2已经成为治疗研究的主题,因为据估计sglt2是大部分肾葡萄糖重吸收的原因。
4.美国特开专利公开号2015/0152075公开了一种对sglt2具有抑制活性之具有二苯基甲烷部分的化合物及其制备方法。该文献公开了二苯基甲烷衍生物化合物对于治疗糖尿病是有效的,其基于这样的事实:二苯基甲烷衍生物化合物对人sglt2活性表现出优异的抑制作用并且与公知的sglt2抑制剂达格列净(dapagliflozin)相比显著降低动物的尿糖排泄。另外,美国特开专利公开号2014/0274918公开了一种二苯基甲烷衍生物,其有效地作为钠依赖性葡萄糖协同转运蛋白1(sglt1)和钠依赖性葡萄糖协同转运蛋白2(sglt2)的双重抑制剂。
5.在美国特开专利公开号2015/0152075的实施例172等中,公开了以与以下反应方案1相同的方式制备二苯基甲烷化合物c28的方法。
6.[反应方案1]
[0007]
[0008][0009]
然而,根据制备化合物c28的常规方法,采用线性合成方法,例如在与葡萄糖基团偶联后在糖苷配基基团中形成五边形环。在这种线性合成的情况下,由于复杂的途径,获得了低的最终产率。此外,在葡萄糖基团的取代基或与二氢苯并呋喃键合的环丙基苄基的合成在中间出错,或者意在将取代基或环丙基苄基改变为另一种的情况下,不方便的是不得不通过从头开始再次该合成。此外,即使对于合成化合物c28的环丙基的过程,该方法也是通过在合成途径结束时通过simon
‑
smith反应使烯烃环化来进行的,因此产率根据试剂(二乙基锌,溶剂)等的状态(纯度、无水等)和反应浓度而变化很大。
[0010]
鉴于上述情况,本发明人发现可以通过汇聚合成方法有效地制备二苯基甲烷衍生物,其中各主要基团分别合成然后彼此偶联,而不是常规线性合成方法,并因此完成了本发明。
技术实现要素:
[0011]
技术问题
[0012]
因此,本发明的一个目的是提供用于制备可用作sglt的抑制剂的二苯基甲烷衍生物的改进方法。
[0013]
问题的解决方案
[0014]
根据本发明的一个方面,提供了用于制备下式1a的化合物的方法,所述方法包括以下步骤:
[0015]
(1)使下式2的化合物与下式3的化合物反应,并使生成物进行环化反应,得到下式4的化合物;
[0016]
(2)使式4化合物进行醛基化或酰胺化,然后使生成物与下式5的化合物反应并进行还原,得到下式6的化合物;以及
[0017]
(3)使式6化合物与下式7的化合物反应,并进行脱保护和还原,
[0018][0019]
在所述式中,
[0020]
a是氧(o)或硫(s);
[0021]
n是1或2;
[0022]
pg是保护基;
[0023]
x
′
是卤素或c1
‑
7烷基;
[0024]
x、y和hal各自独立地为卤素;
[0025]
b是(b
‑
1)或(b
‑
2)
[0026]
其中ra、rb、rc和rd各自独立地为氢、卤素、羟基、巯基、氰基、硝基、氨基、羧基、氧代、c1‑7烷基、c1‑7烷硫基、c2‑7烯基、c2‑7炔基、c1‑7烷氧基、c1‑7烷氧基
‑
c1‑7烷基、c2‑7烯基
‑
c1‑7烷氧基、c2‑7炔基
‑
c1‑7烷氧基、c3‑
10
环烷基、c3‑7环烷硫基、c5‑
10
环烯基、c3‑
10
环烷氧基、c3‑
10
环烷氧基
‑
c1‑7烷氧基、苯基
‑
c1‑7烷基、c1‑7烷硫基
‑
苯基、苯基
‑
c1‑7烷氧基、单c1‑7烷基氨基或二c1‑7烷基氨基、单c1‑7烷基氨基
‑
c1‑7烷基或二c1‑7烷基氨基
‑
c1‑7烷基、c1‑7烷酰基、c1‑7烷酰基氨基、c1‑7烷基羰基、c1‑7烷氧基羰基、氨甲酰基、单c1‑7烷基氨甲酰基或二c1‑7烷基氨甲酰基、
c1‑7烷基磺酰基氨基、苯基磺酰基氨基、c1‑7烷基亚磺酰基、c6‑
14
芳基硫烷基、c6‑
14
芳基磺酰基、c6‑
14
芳基、5至13元杂芳基、5至10元杂环烷基、5至10元杂环烷基
‑
c1‑7烷基、或5至10元杂环烷基
‑
c1‑7烷氧基;
[0027]
环c为c3‑
10
环烷基、c5‑
10
环烯基、c6‑
14
芳基、5至13元杂芳基、或5至10元杂环烷基;
[0028]
烷基、烯基、炔基和烷氧基各自独立地未经取代,或具有一个或更多个选自卤素、羟基、氰基、硝基、氨基、巯基、c1‑7烷基和c2‑7炔基的取代基;
[0029]
环烷基、环烯基、芳基、杂芳基和杂环烷基各自独立地未经取代,或具有一个或更多个选自卤素、羟基、氰基、硝基、氨基、巯基、c1‑4烷基和c1‑4烷氧基的取代基;并且
[0030]
杂芳基和杂环烷基各自独立地含有一个或更多个选自n、s和o的杂原子。
[0031]
根据本发明的另一个方面,提供了用于制备下式1b的化合物的方法,所述方法包括以下步骤:
[0032]
(1)使下式2的化合物与下式3的化合物反应,并使生成物进行环化反应,得到下式4的化合物;
[0033]
(2)使式4化合物进行醛基化或酰胺化,然后使生成物与式5化合物反应并进行还原,得到下式6的化合物;
[0034]
(3)使式6化合物与下式8的化合物反应然后进行还原,得到下式9的化合物;
[0035]
(4)在酸性条件下使式9化合物的呋喃糖环形成吡喃糖环然后向其中引入保护基,得到下式10的化合物;以及
[0036]
(5)用硫脲处理式10化合物,使生成物与c1‑7烷基卤化物反应,然后进行还原,
[0037][0038]
在所述式中,
[0039]
r是c1‑7烷硫基;
[0040]
b、n、pg、x
′
、x、y和hal如上式1中所限定。
[0041]
根据本发明的又一方面,提供了通过上述方法制备的化合物的晶形,特别是下式c28化合物的晶形。
[0042]
[式c28]
[0043][0044]
发明的有益效果
[0045]
制备本发明的二苯基甲烷衍生物的方法通过汇聚合成方法进行,其中主要基团被分别合成然后彼此偶联。因此,与现有技术文献中公开的线性合成方法相比,由于可以降低线性合成途径中固有的风险因素(例如,在合成中间失败时返回途径的最开始处并重新合成)的事实,可以实现简单的合成途径和高产率并提高了再现性。
[0046]
特别地,根据现有技术文献中公开的方法,即使在葡萄糖基团与糖苷配基基团偶联之后,也必须合成糖苷配基基团的残基。另一方面,根据本发明,糖苷配基基团的所有残基可在与葡萄糖基团偶联之前形成。另外,可以容易地合成与糖苷配基的末端基团键合的芳基,因此可以进行末端基团的多种设计。
[0047]
此外,通过上述方法制备的化合物的晶形具有优异的物理化学性质,因此可以有效地用于诸如药物生产的领域。
附图说明
[0048]
图1和2分别显示了在实验实施例4中获得的晶形a的xrd和dsc光谱。
[0049]
图3和4分别显示了在实验实施例4中获得的晶形b的xrd和dsc光谱。
[0050]
图5和6分别显示了在实验实施例4中获得的晶形c的xrd和dsc光谱。
[0051]
图7和8分别显示了在实验实施例4中获得的晶形d的xrd和dsc光谱。
具体实施方式
[0052]
本发明涉及用于制备式1化合物的方法:
[0053]
[式1]
[0054][0055]
在该式中,
[0056]
a是氧(o)或硫(s);
[0057]
r是羟甲基或c1‑7烷硫基;
[0058]
n是1或2;
[0059]
x
′
是卤素(例如,f、cl、br或i)或c1‑7烷基;
[0060]
b是(b
‑
1)或(b
‑
2)
[0061]
其中ra、rb、rc和rd各自独立地为氢、卤素、羟基、巯基、氰基、硝基、氨基、羧基、氧代、c1‑7烷基、c1‑7烷硫基、c2‑7烯基、c2‑7炔基、c1‑7烷氧基、c1‑7烷氧基
‑
c1‑7烷基、c2‑7烯基
‑
c1‑7烷氧基、c2‑7炔基
‑
c1‑7烷氧基、c3‑
10
环烷基、c3‑7环烷硫基、c5‑
10
环烯基、c3‑
10
环烷氧基、c3‑
10
环烷氧基
‑
c1‑7烷氧基、苯基
‑
c1‑7烷基、c1‑7烷硫基
‑
苯基、苯基
‑
c1‑7烷氧基、单c1‑7烷基氨基或二c1‑7烷基氨基、单c1‑7烷基氨基
‑
c1‑7烷基或二c1‑7烷基氨基
‑
c1‑7烷基、c1‑7烷酰基、c1‑7烷酰基氨基、c1‑7烷基羰基、c1‑7烷氧基羰基、氨甲酰基、单c1‑7烷基氨甲酰基或二c1‑7烷基氨甲酰基、c1‑7烷基磺酰基氨基、苯基磺酰基氨基、c1‑7烷基亚磺酰基、c6‑
14
芳基硫烷基、c6‑
14
芳基磺酰基、c6‑
14
芳基、5至13元杂芳基、5至10元杂环烷基、5至10元杂环烷基
‑
c1‑7烷基、或5至10元杂环烷基
‑
c1‑7烷氧基;
[0062]
环c为c3‑
10
环烷基、c5‑
10
环烯基、c6‑
14
芳基、5至13元杂芳基、或5至10元杂环烷基;
[0063]
烷基、烯基、炔基和烷氧基各自独立地未经取代,或具有一个或更多个选自卤素、羟基、氰基、硝基、氨基、巯基、c1‑7烷基和c2‑7炔基的取代基;
[0064]
环烷基、环烯基、芳基、杂芳基和杂环烷基各自独立地未经取代,或具有一个或更多个选自卤素、羟基、氰基、硝基、氨基、巯基、c1‑4烷基和c1‑4烷氧基的取代基;并且
[0065]
杂芳基和杂环烷基各自独立地含有一个或更多个选自n、s和o的杂原子。
[0066]
作为一个具体实例,环b
‑
1可选自以下:
[0067][0068]
在所述式中,r7为氢或c1‑7烷基;r
8a
和r
8b
各自独立地为c1‑7烷基或彼此连接以形成5至10元杂环烷基(其含有至少一个选自n、s和o的杂原子)。
[0069]
作为另一个具体实例,环b
‑
2可选自:
[0070][0071]
优选地,式1化合物可以是由下式1a表示的化合物或由下式1b表示的化合物:
[0072][0073]
在所述式中,a、b、r、x
′
和n如上式1中所限定。
[0074]
根据式1a化合物的一个优选实例,a可以是氧;n可以是1;x
′
可以是卤素;并且b可以是未经取代的或被一个或两个选自以下的取代基取代的苯基:卤素、羟基、氰基、硝基、氨基、巯基、c1‑7烷基、c3‑
10
环烷基和c1‑7烷氧基。
[0075]
此外,式1a和1b的化合物可以是其中葡萄糖为α形式、β形式或其外消旋形式的化合物。
[0076]
优选地,式1a和1b的化合物可以是其中葡萄糖为β形式的化合物。
[0077]
用于制备式1a(式1,其中r=羟甲基)化合物的方法
[0078]
根据本发明的一个方面,提供了用于制备式1a(式1,其中r=羟甲基)化合物的方法,该方法包括以下步骤:
[0079]
(1)使下式2的化合物与下式3的化合物反应,并使生成物进行环化反应,得到下式4的化合物;
[0080]
(2)使式4化合物进行醛基化或酰胺化,然后使生成物与式5化合物反应并进行还原,得到下式6的化合物;以及
[0081]
(3)使式6化合物与下式7的化合物反应,并进行脱保护和还原,
[0082][0083]
在所述式中,
[0084]
a是氧(o)或硫(s);
[0085]
n是1或2;
[0086]
pg是保护基;
[0087]
x
′
是卤素或c1‑7烷基;
[0088]
x、y和hal各自独立地为卤素;并且
[0089]
b如上式1中所限定。
[0090]
在上述制备方法中用作原料的式2化合物可以通过现有技术文献(美国特开专利公开号2015/0152075a1)中公开的合成途径制备。例如,式2化合物可以通过包括以下步骤来制备:
[0091]
(i)使以下式2a的甲酸化合物进行酯化反应,得到以下式2b的甲酯化合物;
[0092]
(ii)使式2b化合物进行氢化反应以使硝基还原,得到以下式2c的胺化合物;
[0093]
(iii)使式2c化合物与卤化试剂反应,得到以下式2d的卤化化合物;以及
[0094]
(iv)使式2d化合物进行sandmeyer反应。
[0095][0096]
在所述式中,y是卤素。
[0097]
如本文所用,术语“卤素”意指氟(f)、氯(cl)、溴(br)或碘(i)。
[0098]
步骤(1)
[0099]
在步骤(1)中,使式2化合物与式3化合物反应,并使生成物进行环化反应,得到式4化合物。
[0100]
因此,在与葡萄糖基团偶联之前,以及还在形成糖苷配基基团的末端残基(即,环b)之前,可以预先形成糖苷配基基团中含有氧的五边形或六边形环。
[0101]
作为一个具体实例,步骤(1)可以包括以下步骤:
[0102]
(i)使式2化合物与式3化合物反应,得到下式3a的化合物;
[0103]
(ii)使式3a化合物的烯丙基进行重排反应,并使生成物进行氧化或臭氧化反应,然后进行还原,得到下式3d的化合物;以及
[0104]
(iii)使式3d化合物进行环化反应,得到式4化合物:
[0105][0106]
在所述式中,n是1或2;x和y各自独立地为卤素。
[0107]
在步骤(i)中,在与式3化合物反应之前可以对式2化合物进行去甲基化步骤。例如,可以对式2化合物进行去甲基化以获得下式2e的化合物,并且式2e化合物可以与式3化合物反应:
[0108]
[式2e]
[0109][0110]
在该式中,x和y各自独立地为卤素。
[0111]
步骤(ii)中的重排反应可以例如在claisen重排反应中进行。
[0112]
重排反应可以通过添加路易斯酸来进行。路易斯酸可以是选自二异丁基氯化铝、二乙基氯化铝、氯化铝和三氯化硼中的至少一种。
[0113]
另外,重排反应可以在无溶剂反应中进行,或者在二乙胺中在高温(例如150℃至170℃)的加热条件下进行。
[0114]
在步骤(ii)中进行重排反应后,式3a化合物可以以下式3b的化合物获得:
[0115]
[式3b]
[0116][0117]
在该式中,n为1或2;x和y各自独立地为卤素。
[0118]
步骤(ii)中的氧化或臭氧化反应可以通过添加四氧化锇(oso4)、锇酸钾(vi)二水合物或臭氧(o3)来进行。
[0119]
在步骤(ii)中进行氧化或臭氧化反应后,式3b化合物可以以下式3c的化合物获得:
[0120]
[式3c]
[0121][0122]
在该式中,n为1或2;x和y各自独立地为卤素。
[0123]
然后,可以还原式3c化合物,得到式3d化合物。
[0124]
在步骤(iii)中,可以使式3d化合物进行环化反应,得到式4化合物。与现有技术文献(美国特开专利公开号2015/0152075a1)中公开的环化方法相比,根据这种方法可以提高产率。
[0125]
环化反应可以是使用vilsmeier试剂的环化反应,使用离去基团的环化反应,使用卤化物的环化反应,或使用mitsunobu反应的环化反应。
[0126]
根据一个实例,环化反应可以通过将vilsmeier试剂添加到式3d化合物来进行。此时的反应可在0℃至常温的温度下进行。就产率而言,优选在临合成时制备上述vilsmeier试剂并使用,例如,可以使用通过二甲基甲酰胺(dmf)与socl2或pocl3反应制备的vilsmeier试剂。
[0127]
根据另一个实例,可以通过引入甲苯磺酰基或甲磺酰基作为离去基团来进行环化反应。根据又一个实例,可以使用卤化物如i2和pbr3进行环化反应。根据又一个实例,也可以使用偶氮二甲酸二异丙酯(diad)等通过mitsunobu反应进行环化反应。
[0128]
这些反应是这样的反应,其中伯醇基团被能够充当离去基团的基团取代,并且经取代基团充当酚基团的亲核试剂,从而发生环化反应。
[0129]
步骤(2)
[0130]
在步骤(2)中,使式4化合物进行醛基化或酰胺化,然后与式5化合物反应。还原生成物,得到式6化合物。
[0131]
作为一个实例,步骤(2)可包括使式4化合物进行醛基化,以获得下式4a的化合物,然后使式4a化合物与式5化合物反应:
[0132]
[式4a]
[0133][0134]
在该式中,n为1或2;并且x和y各自独立地为卤素。
[0135]
具体地,醛基化反应可以通过以下进行:还原式4化合物以获得下式4c的化合物,然后与氯铬酸吡啶(pcc)、二氧化镁、三氧化硫吡啶络合物等反应。结果,可以获得式4a化合物:
[0136]
[式4c]
[0137]
[0138]
在该式中,n为1或2;x和y各自独立地为卤素。
[0139]
此时,在还原式4化合物时可以使用还原剂如nabh4和libh4。此外,在还原时,可以使用醇、四氢呋喃(thf)、或其混合物作为溶剂。作为一个优选的实例,在还原时,可以使用乙醇和thf的混合溶剂,并且其混合体积比可以是1∶1至1∶3。另外,在还原时,可以进一步使用路易斯酸,可以使用的路易斯酸的实例包括licl和cacl2。
[0140]
然后,式4a化合物可与式5化合物反应,得到下式6a的化合物:
[0141]
[式6a]
[0142][0143]
在该式中,n为1或2,x和y各自独立地为卤素,并且b如上式1中所限定。
[0144]
可以还原式6a化合物,得到式6化合物。
[0145]
作为另一个实例,步骤(2)可以包括使式4化合物进行酰胺化,以获得下式4b的化合物,然后使式4b化合物与式5化合物反应:
[0146]
[式4b]
[0147][0148]
在该式中,n为1或2;并且x和y各自独立地为卤素。
[0149]
具体地,酰胺化反应可以通过以下进行:使式4化合物水解,然后使生成物与n,o
‑
二甲基羟胺盐酸盐(meo(me)nh
·
hcl)等反应。结果,可以获得诸如式4b的weinreb酰胺形式。
[0150]
然后,式4b化合物可与式5化合物反应,得到下式6b的化合物:
[0151]
[式6b]
[0152][0153]
在该式中,n为1或2;x和y各自独立地为卤素,并且b如上式1中所限定。
[0154]
然后,可以还原式6b化合物,得到式6化合物。
[0155]
式5化合物可以是格氏试剂(grignard reagent)。
[0156]
根据制备格氏试剂的一般性方法,可使下式5a的化合物与金属镁(mg)反应,得到式5化合物。
[0157]
[式5a]
[0158][0159]
在该式中,b如上式1中所限定,并且hal是卤素。
[0160]
如上所述,根据本发明,在糖苷配基基团与葡萄糖基团偶联之前,可以根据制备格氏试剂的方法预先容易地引入最终化合物(式1a化合物)的基团b,其不仅允许多种衍生化,而且还允许最终产率得到提高。
[0161]
另一方面,根据现有技术文献(美国特开专利公开号2015/0152075 a1),为了完成最终化合物的基团b,在与葡萄糖基团偶联后,需要在合成途径结束时进行复杂的合成过程,因此存在反应产率和反应再现性由于长的过程而变化很大的问题。
[0162]
步骤(3)
[0163]
在步骤(3)中,使式6化合物与式7化合物反应,然后进行脱保护和还原。
[0164]
式6化合物与式7化合物的反应可在正丁基锂、仲丁基锂、叔丁基锂、异丙基氯化镁(i
‑
prmgcl)等存在下进行。
[0165]
式6化合物可与式7化合物反应,得到下式7a的化合物:
[0166]
[式7a]
[0167][0168]
在该式中,a是氧或硫;n是1或2;x是卤素;pg是保护基;并且b如上式1中所限定。
[0169]
保护基可以是例如三甲基甲硅烷基(tms)、苄基或乙酰基。
[0170]
然后,可以将式7a化合物脱保护,得到式1a化合物。例如,在保护基是三甲基甲硅烷基(tms)的情况下,通过向式7a化合物添加甲磺酸(ch3so3h)或三甲基甲硅烷基三氟甲磺酸盐(tmsotf)进行脱保护,从而可以获得式1a化合物。
[0171]
此外,在脱保护后,可进一步进行还原以获得式1a化合物。此时,二氯甲烷(ch2cl2)和乙腈(ch3cn)可以组合使用作为溶剂。
[0172]
通过上述步骤获得的式1a化合物可以是其中葡萄糖的α形式和β形式混合的化合物。
[0173]
因此,可以进行进一步的分离以仅获得所需的α形式或β形式。即,在脱保护和还原过程之后或期间,可以进一步仅分离其中葡萄糖为β形式的化合物。
[0174]
例如,将保护基引入通过脱保护和还原获得的化合物中。然后,将生成物在醇、乙酸乙酯或二氯甲烷中加热,分离所得沉淀物,然后脱保护,从而可以仅获得β形式。
[0175]
具体地,将通过脱保护和还原获得的化合物中葡萄糖的羟基用乙酰基等保护。然后,将生成物在c1‑6醇溶剂(乙醇、异丙醇等)中加热并搅拌,并分离所得沉淀物,使得可以仅获得其中葡萄糖为β型的下式7b的化合物:
[0176]
[式7b]
[0177][0178]
在该式中,a是氧或硫;n是1或2;x是卤素;pg是保护基;并且b如上式1中所限定。
[0179]
然后,可以将式7b化合物脱保护,最终仅获得可以由下式7c表示的β形式:
[0180]
[式7c]
[0181][0182]
在该式中,a、b、n和x
′
如上式1中所限定。
[0183]
根据一个优选的实例,步骤(3)可以通过包括以下步骤的方法进行:
[0184]
(3a
‑
1)使式6化合物与式7化合物在正丁基锂、仲丁基锂、叔丁基锂或异丙基氯化镁存在下进行反应,得到下式7a的化合物;
[0185]
(3a
‑
2)使式7a化合物在酸性条件下在甲醇存在下进行脱保护和甲基化反应,得到下式7d的化合物;
[0186]
(3b)还原式7d化合物,得到下式7e的化合物;以及
[0187]
(3c)向式7e化合物中引入保护基因,在醇、乙酸乙酯或二氯甲烷中加热生成物,分离所得沉淀物并脱保护,仅得到β形式:
[0188][0189]
在所述式中,pg是保护基;并且a、b、n和x如上式1a中所限定。
[0190]
在步骤(3a
‑
1)中的反应之后,优选通过进一步进行蒸发、萃取、干燥、过滤等获得式7a化合物,然后用于下一步骤(3a
‑
2)中。
[0191]
步骤(3a
‑
2)中使用的酸可以是盐酸、硫酸、乙酸、三氟乙酸、甲磺酸、三氟甲磺酸、对甲苯磺酸、氯化氢气体等。
[0192]
根据另一个优选实例,步骤(3)可以通过包括以下步骤的方法进行:
[0193]
(3a
′
)使式6化合物与式7化合物在正丁基锂、仲丁基锂、叔丁基锂或异丙基氯化镁存在下进行反应,并在酸性条件下在甲醇存在下对生成物进行脱保护和甲基化反应,而无需单独进行纯化,得到下式7d的化合物;
[0194]
(3b
′
)还原式7d化合物,得到下式7e的化合物;以及
[0195]
(3c
′
)向式7e化合物中引入保护基以仅分离β形式,并进行脱保护:
[0196][0197]
在所述式中,a、b、n和x如上式1a中所限定。
[0198]
在步骤(3a
′
)中,首先进行结合反应,此时,相对于1当量的式6化合物,可以以1.5至2.5当量,更优选1.7至2.3当量,特别是约2.0当量的量将式7化合物和反应试剂(即,正丁基锂、仲丁基锂、叔丁基锂或异丙基氯化镁)各自用于反应。此时的反应可以在
‑
80℃至
‑
10℃,更优选
‑
70℃至
‑
60℃的温度下进行1至12小时,或1至3小时。另外,作为反应溶剂,可以使用四氢呋喃或醚的单一溶剂,四氢呋喃/甲苯(1∶1)的混合溶剂等。
[0199]
另外,在步骤(3a
′
)中,脱保护和甲基化反应在酸性条件下进行。此时使用的酸的实例包括盐酸、硫酸、乙酸、三氟乙酸、甲磺酸、三氟甲磺酸、对甲苯磺酸和氯化氢气体。相对于1当量的式6化合物,可以以2至5当量,更优选3当量的量使用酸。此时的反应可以在0℃至40℃,更优选20℃至30℃的温度下进行6至24小时,或6至12小时。作为反应溶剂,可以使用甲醇等。
[0200]
然后,在步骤(3b
′
)中进行还原反应,此时,可以使用还原剂和酸。还原剂的实例可包括三乙基硅烷、三异丙基硅烷、叔丁基二甲基硅烷和硼氢化钠。酸的实例可包括三氟化硼二乙醚、三甲基甲硅烷基三氟甲磺酸盐、氯化铝、三氟乙酸和三氟甲磺酸。可以以2至5当量,更优选约3当量的量使用还原剂,可以以1.5至3当量,更优选约2当量的量使用酸。此时的反应可在
‑
50℃至0℃,更优选在
‑
20℃至
‑
10℃的温度下进行2至12小时,或2至5小时。此外,作为反应溶剂,可以使用单一溶剂(例如二氯甲烷、1,2
‑
二氯乙烷或乙腈),或混合溶剂(例如二氯甲烷/乙腈(1∶1)和1,2
‑
二氯甲烷/乙腈(1∶1))。
[0201]
接着,在步骤(3c
′
)中引入保护基,此时,可以进行使用乙酰化剂和碱的反应。乙酰化剂的实例包括乙酰氯、乙酰溴和乙酸酐,碱的实例包括氢氧化钠、碳酸钠、三乙胺、二异丙基乙胺、吡啶、二甲基吡啶和4
‑
二甲基氨基吡啶。可以以4至12当量,更优选约8当量的量使用乙酰化剂,可以以1至4当量,更优选约1.5当量的量使用碱。此时的反应可以在0℃至50℃,更优选20℃至30℃的温度下进行1至12小时,或1至3小时。作为反应溶剂,可以使用丙酮、乙酸乙酯、四氢呋喃、二甲基甲酰胺、二甲基乙酰胺、二氯甲烷、1,2
‑
二氯乙烷、氯仿等。
[0202]
最后,在步骤(3c
′
)中进行脱保护反应,并且此时,也可以以2至12当量,更优选约5当量的量使用试剂,例如氢氧化锂、氢氧化钠、氢氧化钾、甲醇钠和乙醇钠。此时的反应可以在0℃至50℃,更优选在20℃至30℃的温度下进行1至12小时,或1至3小时。作为反应溶剂,可以使用甲醇/水(1∶1至3∶1)、二氯甲烷/甲醇(1∶1至1∶2)、二氯甲烷/乙醇(1∶1至1∶2)、四氢呋喃/甲醇(1∶1至1∶2)、四氢呋喃/乙醇(1∶1至1∶2)、四氢呋喃/甲醇/水(1∶1∶3至2∶1∶3)、四氢呋喃/乙醇/水(1∶1∶3至2∶1∶3)等。
[0203]
根据另一个优选实例,步骤(3)可以通过包括以下步骤的方法进行:
[0204]
(3a
″
)使式6化合物与式7化合物在正丁基锂、仲丁基锂、叔丁基锂、或异丙基氯化镁存在下进行反应,并在酸性条件下在甲醇存在下对生成物进行脱保护和甲基化反应,而无需单独进行纯化,得到下式7d的化合物;
[0205]
(3b
″
)向式7d化合物中引入保护基,得到下式7f的化合物;以及
[0206]
(3c
″
)仅分离式7f化合物的β形式并进行还原,然后进行脱保护:
[0207][0208]
在所述式中,pg是保护基;并且a、b、n和x如上式1a中所限定。
[0209]
在步骤(3a
″
)中,进行结合反应、脱保护和甲基化。此时的优选条件如当量比、反应温度和溶剂如步骤(3a
′
)中所例示。
[0210]
然后,在步骤(3b
″
)中引入保护基,此时,可以进行使用乙酰化剂和碱的反应。优选的条件如乙酰化剂的类型、碱的类型、当量比、反应温度和溶剂如上面步骤(3c
′
)中所例示。
[0211]
接下来,在步骤(3c
″
)中进行还原反应。此时,可以使用还原剂和酸,并且优选的条件如还原剂的类型、酸的类型、当量比、反应温度和溶剂如上面步骤(3b
′
)中所例示。
[0212]
另外,在步骤(3c
″
)中进行脱保护反应,此时的优选条件如试剂的类型、当量比、反应温度、溶剂如上面步骤(3c
′
)中所例示。
[0213]
如上述优选示例所示,获得式7d化合物的方法可以在两个步骤中进行,或者可以在一个步骤中作为原位反应进行,以进一步提高最终产率。另外,在作为原位反应的一个步骤中进行的情况下,可以获得含有式7b化合物的粗浓缩残余物,或者可以通过结晶从其获得作为固体成分的式7d化合物,并用于下一步骤。在后一种情况下,通过除去反应副产物,可以容易地实现质量的提高和水分含量的控制。
[0214]
此外,式7d化合物可以在其合成后通过纯化用于下一步骤。例如,(i)在合成之后,可以使式7d化合物与诸如甲苯的有机溶剂形成共沸混合物,并且可以在下一步骤中使用通过重复浓缩过程以除去残留水分而获得的残余物,或(ii)在合成之后,可以使式7d化合物结晶,通过真空干燥除去残留水分得到的固体成分可以用于下一步骤。
[0215]
烷基化步骤
[0216]
另外,根据本发明,可以在步骤(3)之后进一步包括烷基化反应,结果,式1中的x
′
可以是c1‑7烷基。
[0217]
例如,步骤(4)后的产物可以与甲基硼酸反应,得到式1a化合物,其中x
′
被甲基取代。
[0218]
结晶步骤
[0219]
式1a化合物可以以晶形、无定形形式、或其混合物制备。然而,从稳定性和非吸湿性优异并因此具有促进配制的物理化学性质的观点来看,优选晶形的式1a化合物。
[0220]
因此,本发明的方法可以进一步包括在步骤(3)之后使式1a化合物结晶。可以使用多种溶剂进行结晶,因此可以获得多种晶形。
[0221]
例如,用于结晶的溶剂可选自甲苯;乙酸乙酯;二氯甲烷;丙酮;乙腈;2
‑
丙醇、四氢呋喃与二氯甲烷的混合物;以及四氢呋喃与正己烷的混合物,因此,可以制备晶形a。
[0222]
作为另一个实例,用于结晶的溶剂可以选自甲醇与蒸馏水的混合物;甲醇与正己烷的混合物;以及甲醇、二氯甲烷与正己烷的混合物,因此,可以制备晶形b。
[0223]
作为又一个实例,用于结晶的溶剂可以选自乙醇、蒸馏水与正己烷的混合物;以及四氢呋喃与甲苯的混合物,因此可以制备晶形c。
[0224]
作为又一个实例,用于结晶的溶剂可以是乙醇与正己烷的混合物,因此可以制备晶形d。
[0225]
作为一个优选的实例,用于结晶的溶剂可选自甲苯、乙酸乙酯、二氯甲烷,四氢呋喃与二氯甲烷的混合物,以及四氢呋喃与正己烷的混合物。
[0226]
用于制备式1b(式1,其中r=烷硫基,并且a=氧)化合物的方法
[0227]
根据本发明的另一方面,提供了制备式1b(式1,其中r=c1‑7烷硫基,并且a=氧)化合物的方法,该方法包括以下步骤:
[0228]
(1)使下式2的化合物与下式3的化合物反应,并使生成物进行环化反应,得到下式4的化合物;
[0229]
(2)使式4化合物进行醛基化或酰胺化,然后使生成物与式5化合物反应并进行还原,得到下式6的化合物;
[0230]
(3)使式6化合物与下式8的化合物反应,然后进行还原,得到下式9的化合物;
[0231]
(4)在酸性条件下,将式9化合物的呋喃糖环形成吡喃糖环,然后向其中引入保护基,得到下式10的化合物;以及
[0232]
(5)用硫脲处理式10化合物,使生成物与c1‑7烷基卤化物反应,然后进行还原。
[0233][0234]
在所述式中,
[0235]
r是c1‑7烷硫基;
[0236]
n是1或2;
[0237]
pg是保护基;
[0238]
x
′
是卤素或c1‑7烷基;
[0239]
x、y和hal各自独立地为卤素;并且
[0240]
b如上式1中所限定。
[0241]
在上述步骤中,步骤(1)和(2)可以与用于制备式1a(式1,其中r=羟甲基)化合物的方法的步骤(1)和(2)相同的方式进行。
[0242]
下面将详细描述步骤(3)至(5)。
[0243]
步骤(3)
[0244]
在步骤(3)中,使式6化合物与式8化合物反应,得到式9化合物。
[0245]
式8化合物可以根据已知方法制备,例如wo2009/014970中公开的方法。具体地,式8化合物可以根据wo2009/014970中公开的方法以l
‑
木糖开始制备。
[0246]
根据一个实例,式6化合物可与式8化合物反应,得到下式9a的化合物。
[0247]
[式9a]
[0248][0249]
在该式中,b、n和x如上式1中所限定。
[0250]
然后,可以还原式9a化合物,得到式9化合物。
[0251]
步骤(4)
[0252]
在步骤(4)中,使式9化合物的呋喃糖环在酸性条件下形成吡喃糖环,然后向其中引入保护基以获得式10化合物。通过该步骤,可以完成吡喃糖环构成葡萄糖基团。
[0253]
保护基可以是例如乙酰基。
[0254]
步骤(5)
[0255]
在步骤(5)中,将式10化合物用硫脲处理,与c1‑7烷基卤化物反应,然后还原。通过该步骤,可以将烷硫基引入最终化合物(式1b化合物)中。
[0256]
c1‑7烷基卤化物可以是例如c1‑7烷基碘化物。
[0257]
此外,在步骤(5)之后,可以进一步包括烷基化反应,结果,可以获得其中x
′
是c1‑7烷基的式1b化合物。
[0258]
晶形
[0259]
根据本发明的又一方面,提供了根据上述制备方法制备的化合物的晶形。
[0260]
例如,本发明提供式1a化合物的晶形。
[0261]
作为一个具体实例,本发明提供了式1a化合物的晶形,其中a是o,b是环丙基苯基,n是1,x
′
是cl,并且所述化合物为β形式。
[0262]
即,本发明提供下式c28化合物的晶形。
[0263]
[式c28]
[0264][0265]
式c28化合物可以通过上述式1a的制备方法制备。
[0266]
根据本发明,式c28化合物可以是多种晶形,下面将详细描述各晶形。
[0267]
在下文中,术语“约”可以表示在预定值或范围的5%内,优选在2%内。例如,“约10%”可以表示9.5%至10.5%,并且优选9.8%至10.2%。作为另一个实例,“约100℃”可以表示95℃至105℃,优选98℃至102℃。
[0268]
首先,本发明提供式c28化合物的晶形a。在使用cu
‑
k
α
光源照射的情况下,晶形a具有xrd光谱,其包括在6.2
°±
0.2
°
、7.2
°±
0.2
°
、8.8
°±
0.2
°
、17.6
°±
0.2
°
、19.0
°±
0.2
°
、22.5
°±
0.2
°
和25.1
°±
0.2
°
的衍射角(2θ)处的峰。这些峰可以是相对强度(i/i
o
)为约5%或更高,优选约10%或更高的峰。
[0269]
晶形a的xrd光谱可以还包括15.4
°±
0.2
°
、18.6
°±
0.2
°
、21.6
°±
0.2
°
和23.8
°±
0.2
°
的衍射角(2θ)处的峰。
[0270]
此外,该晶形在dsc(10℃/分钟)下可具有其中起始点为约157℃、最低点为约159℃的吸热峰。
[0271]
此外,本发明提供了式c28化合物的晶形b。在使用cu
‑
k
α
光源照射的情况下,晶形b具有xrd光谱,其包括在7.0
°±
0.2
°
、14.9
°±
0.2
°
、17.7
°±
0.2
°
、18.8
°±
0.2
°
、20.6
°±
0.2
°
、21.8
°±
0.2
°
和23.5
°±
0.2
°
的衍射角(2θ)处的峰。这些峰可以是相对强度(i/i
o
)为约5%或更高,优选约10%或更高的峰。
[0272]
晶形b的xrd光谱可以进一步包括5.6
°±
0.2
°
、9.4
°±
0.2
°
和11.0
°±
0.2
°
的衍射角(2θ)处的峰。
[0273]
此外,该晶形在dsc(10℃/分钟)下可具有其中起始点约为79℃、最低点约为88℃的吸热峰,以及起始点约为103℃、最低点约为111℃的吸热峰。
[0274]
此外,本发明提供了式c28化合物的晶形c。在使用cu
‑
k
α
光源照射的情况下,晶形c具有xrd光谱,其包括5.6
°±
0.2
°
、7.3
°±
0.2
°
、15.7
°±
0.2
°
、17.2
°±
0.2
°
、18.9
°±
0.2
°
、21.2
°±
0.2
°
和21.9
°±
0.2
°
的衍射角(2θ)处的峰。这些峰可以是相对强度(i/i
o
)为约5%或更高,优选约10%或更高的峰。
[0275]
晶形c的xrd光谱可以进一步包括19.9
°±
0.2
°
和23.1
°±
0.2
°
的衍射角(2θ)处的峰。
[0276]
此外,该晶形在dsc(10℃/分钟)下可具有其中起始点为约157℃、最低点为约159℃的吸热峰。
[0277]
此外,本发明提供了式c28化合物的晶形d。在使用cu
‑
k
α
光源照射的情况下,晶形d具有xrd光谱,其包括5.5
°±
0.2
°
、7.2
°±
0.2
°
、15.3
°±
0.2
°
、17.2
°±
0.2
°
、17.6
°±
0.2
°
、18.9
°±
0.2
°
和21.1
°±
0.2
°
的衍射角(2θ)处的峰。这些峰可以是相对强度(i/i
o
)为约5%或更高的峰。
[0278]
晶形d的xrd光谱可以进一步包括20.0
°±
0.2
°
、22.5
°±
0.2
°
和25.1
°±
0.2
°
的衍射角(2θ)处的峰。
[0279]
此外,该晶形在dsc(10℃/分钟)下可具有其中起始点为约157℃、最低点为约160℃的吸热峰。
[0280]
这种式c28化合物的晶形具有优异的物理化学性质(例如,吸湿性和化学稳定性),因此可以在多种领域(例如,药物的生产)中被容易地处理。
[0281]
实施发明的方式
[0282]
在下文中,将参考实施例更详细地描述本发明。然而,以下实施例仅旨在举例说明本发明,并且本发明的范围不限于这些实施例。
[0283]
以下实施例中指出的缩写的含义如下。
[0284]
‑
acoh:乙酸
[0285]
‑
acn:乙腈
[0286]
‑
ac2o:乙酸酐
[0287]
‑
bf3.oet2:三氟化硼醚合物
[0288]
‑
dipea:n,n
‑
二异丙基乙胺
[0289]
‑
dcm:二氯甲烷
[0290]
‑
dmap:4
‑
二甲基氨基吡啶
[0291]
‑
dmf:n,n
‑
二甲基甲酰胺
[0292]
‑
etoac:乙酸乙酯
[0293]
‑
etoh:乙醇
[0294]
‑
et3sih:三乙基硅烷
[0295]
‑
hbtu:2
‑
(1h
‑
苯并三唑
‑1‑
基)
‑
1,1,3,3
‑
四甲基脲六氟磷酸盐
[0296]
‑
hex:己烷
[0297]
‑
i
‑
proh:异丙醇
[0298]
‑
mei:碘甲烷
[0299]
‑
meoh:甲醇
[0300]
‑
mscl:甲磺酰氯
[0301]
‑
naome:甲醇钠
[0302]
‑
nbs:n
‑
溴代琥珀酰亚胺
[0303]
‑
pcc:氯铬酸吡啶
[0304]
‑
pd(pph3)4:四(三苯基膦)钯(0)
[0305]
‑
tea:三乙胺
[0306]
‑
tempo:(2,2,6,6
‑
四甲基哌啶
‑1‑
基)啶基
[0307]
‑
thf:四氢呋喃
[0308]
‑
tmsotf:三甲基甲硅烷基三氟甲磺酸盐
[0309]
‑
rt或rt:室温
[0310]
比较实施例1:(2s,3r,4r,5s,6r)
‑2‑
(7
‑
氯
‑6‑
(4
‑
环丙基苄基)
‑
2,3
‑
二氢苯并呋喃
‑4‑
基)
‑6‑
(羟甲基)四氢
‑
2h
‑
吡喃
‑
3,4,5
‑
三醇的合成
[0311]
标题化合物通过美国特开专利公开号2015/0152075中公开的方法制备。对于比较实施例1的具体合成步骤,参考上文背景技术部分中描述的方案1。
[0312]
步骤1:3
‑
甲氧基
‑2‑
硝基苯甲酸甲酯(化合物c2)
[0313]
在室温下向3
‑
甲氧基
‑2‑
硝基苯甲酸(25.0g,126mmol)和k2co3(35.0g,253mmol)在dmf(126ml)中的混合物添加mei(15.8ml,253mmol)。将混合物在室温下搅拌2小时。向混合物中倒入水(200ml),然后在5℃下搅拌30分钟。过滤收集沉淀的固体,用水和己烷洗涤。将固体减压干燥,得到作为白色固体的粗制形式的标题化合物(26.2g,98%)。
[0314]1h nmr(400mhz,cdcl3)δ7.60(dd,j=8.2,1.2hz,1h),7.50(t,j=8.2hz,1h),7.26(dd,j=8.2,1.2hz,1h),3.39(s,3h),3.99(s,3h);[m+na]
+
235.
[0315]
步骤2:2
‑
氨基
‑3‑
甲氧基苯甲酸甲酯(化合物c3)
[0316]
将3
‑
甲氧基
‑2‑
硝基苯甲酸甲酯(26.2g,124mmol)和pd/c(10wt.%,6.0g)在thf(400ml)和meoh(200ml)中的悬浮液在h2气氛下于室温下搅拌18小时。将etoac(300ml)添加到混合物并通过硅藻土垫过滤。将滤液真空浓缩,得到作为无色油状物的标题化合物(22.4g,99%)。
[0317]1h nmr(400mhz,cdcl3)δ7.47(dd,j=8.2,1,2hz,1h),6.85(dd,j=8.2,1.2hz,1h),6.85(t,j=8.2hz,1h),6.00(brs,2h),3.87(s,3h);[m+h]
+
182.
[0318]
步骤3:2
‑
氨基
‑5‑
溴
‑3‑
甲氧基苯甲酸甲酯(化合物c4)
[0319]
在0℃下向2
‑
氨基
‑3‑
甲氧基苯甲酸甲酯(22.4g,123mmol)在dmf(250ml)中的溶液分批添加n
‑
溴代琥珀酰亚胺(21.9g,123mmol)。将混合物在0℃下搅拌0.5小时。向混合物添加水并用etoac(500ml
×
2)萃取。将合并的有机层用mgso4干燥,过滤,并真空浓缩。将残余物通过硅胶柱色谱法纯化,得到作为白色固体的标题化合物(27.5g,86%)。
[0320]1h nmr(400mhz,cdcl3)δ7.60(d,j=2.2hz,1h),6.90(d,j=2.2hz,1h),6.03(brs,2h),3.87(s,3h);[m+h]
+
260.
[0321]
步骤4:5
‑
溴
‑2‑
氯
‑3‑
甲氧基苯甲酸甲酯(化合物c5)
[0322]
在0℃下向2
‑
氨基
‑5‑
溴
‑3‑
甲氧基苯甲酸甲酯(27.0g,103mmol)在h2o(70ml)和浓hcl(70ml)中的溶液滴加nano2(21.5g,311mmol)在h2o(50ml)中的溶液。搅拌1小时后,在0℃下将cu(i)cl在浓hcl(80ml)中的溶液滴加到反应混合物。将混合物在室温下搅拌18小时。向混合物添加水(300ml)并用etoac(500ml)萃取。将有机层用mgso4干燥,过滤,并真空浓缩。将粗制标题化合物在高真空下干燥,并作为白色固体不经进一步纯化(29.0g,100%)用于下一步骤。
[0323]1h nmr(400mhz,cdcl3)δ7.49(d,j=2.4hz,1h),7.16(d,j=2.4hz,1h),3.93(s,36h),3.92(s,3h);[m+h]
+
278.
[0324]
步骤5:5
‑
溴
‑2‑
氯
‑3‑
甲氧基苯甲酸(化合物c6)
[0325]
在0℃下向5
‑
溴
‑2‑
氯
‑3‑
甲氧基苯甲酸甲酯(25.0g,89.4mmol)在thf(100ml)、h2o(100ml)和meoh(100ml)中的溶液滴加5n naoh水溶液。将混合物在室温下搅拌1小时。向混合物添加浓hcl以酸化,并用etoac(500ml
×
2)萃取混合物。将合并的有机层用mgso4干燥,过滤并真空浓缩,得到作为橙色固体的标题化合物(22.6g,96%)。
[0326]1h nmr(400mhz,cdcl3)δ7.55(s,1h),7.13(s,1h),3.89(s,3h);[m+h]
+
265.
[0327]
步骤6:5
‑
溴
‑2‑
氯
‑3‑
甲氧基苯甲酰氯(化合物c7)
[0328]
在室温下向5
‑
溴
‑2‑
氯
‑3‑
甲氧基苯甲酸(6.0g,22.6mmol)在ch2cl2(100ml)中的悬浮液添加草酰氯(2.4ml,27.1mmol)和催化量的dmf。将混合物在室温下搅拌2小时。将混合物真空蒸发并在高真空下干燥,得到粗制标题化合物。
[0329]1h nmr(400mhz,cdcl3)δ7.49(d,j=2.4hz,1h),7.16(d,j=2.4hz,1h),3.93(s,3h),3.92(s,3h).
[0330]
步骤7:(5
‑
溴
‑2‑
氯
‑3‑
羟基苯基)(苯基)甲酮(化合物c8)
[0331]
将粗制5
‑
溴
‑2‑
氯
‑3‑
甲氧基苯甲酰氯用苯(100ml)溶解并冷却至0℃。在0℃下向反应混合物分批添加alcl3(6.9g,52.0mmol)。将混合物在90℃下搅拌15小时。将混合物冷却至室温并真空蒸发。将残余物冷却至0℃并添加1n hcl水溶液。用etoac(150ml
×
1)萃取混合物。将有机层用mgso4干燥,过滤,并真空浓缩。将残余物通过硅胶柱色谱法纯化,得到标题化合物(7.33g,定量产率)。
[0332]1h nmr(400mhz,cdcl3)δ7.85
‑
7.82(m,2h),7.70
‑
7.64(m,1h),7.55
‑
7.49(m,2h),7.37(d,j=2.2hz,1h),7.13(d,j=2.2hz,1h),5.94(s,1h).
[0333]
步骤8:3
‑
苄基
‑5‑
溴
‑2‑
氯苯酚(化合物c9)
[0334]
在0℃下向(5
‑
溴
‑2‑
氯
‑3‑
羟基苯基)(苯基)甲酮(362mg,1.16mmol)在三氟乙酸(3ml)中的混合物添加三乙基硅烷(0.37ml,2.32mmol)和催化三氟甲磺酸。将混合物在室温下搅拌12小时。将所得混合物在0℃下用饱和nahco3溶液猝灭,并用etoac萃取。将有机层用mgso4干燥,过滤,并真空浓缩。将粗制产物通过硅胶柱色谱法(biotage isolera
tm
flash纯化系统,etoac/hex)纯化,得到标题化合物(267mg,77%)。
[0335]1h nmr(400mhz,cdcl3)δ7.37
‑
7.32(m,2h),7.30
‑
7.27(m,1h),7.22
‑
7.19(m,2h),7.13(d,j=2.4hz,1h),6.92(d,j=2.0hz,1h),4.07(s,2h).[m+h]
+
297.
[0336]
步骤9:1
‑
(烯丙氧基)
‑3‑
苄基
‑5‑
溴
‑2‑
氯苯(化合物c10)
[0337]
在室温下向3
‑
苄基
‑5‑
溴
‑2‑
氯苯酚(1.72g,5.78mmol)和k2co3(1.6g,11.56mmol)在丙酮(35ml)中的混合物添加烯丙基溴(0.73ml,8.67mmol)。将反应混合物在65℃下搅拌12小时。过滤所得混合物以除去无机材料。将滤液真空浓缩。将粗制产物通过硅胶柱色谱法(biotage isolera
tm flash纯化系统,etoac/hex)纯化,得到标题化合物(1.96g,100%)。
[0338]1h nmr(400mhz,cdcl3)δ7.32
‑
7.27(m,2h),7.25
‑
7.22(m,1h),7.21
‑
7.17(m,2h),6.92(d,j=2.4hz,1h),6.92(d,j=2.0hz,1h),6.10
‑
6.00(m,1h),5.48(dq,j=17.2hz,1.6hz,1h),5.33(dq,j=12.4,1.6hz,1h),4.59(dt,j=4.4hz,1.6hz,2h),4.08(s,2h).[m+h]
+
337.
[0339]
步骤10:(3r,4s,5s,6r)
‑2‑
(3
‑
(烯丙氧基)
‑5‑
苄基
‑4‑
氯苯基)
‑6‑
(羟甲基)
‑2‑
甲氧基四氢
‑
2h
‑
吡喃
‑
3,4,5
‑
三醇(化合物c12)
[0340]
在氮气氛下于
‑
78℃下向1
‑
(烯丙氧基)
‑3‑
苄基
‑5‑
溴
‑2‑
氯苯(1.96g,5.82mmol)在四氢呋喃(5.5ml)/甲苯(11ml)中的溶液滴加正丁基锂(2.5m于己烷中,2.6ml,6.41mmol)。搅拌1小时后,在
‑
78℃下通过管经过20分钟将(3r,4s,5r,6r)
‑
3,4,5
‑
三((三甲基甲硅烷基)氧基)
‑6‑
(((三甲基甲硅烷基)氧基)甲基)四氢
‑
2h
‑
吡喃
‑2‑
酮(c11;3.54g,7.58mmol)在四氢呋喃(6.6ml)中的溶液滴加到混合物。将反应混合物在
‑
78℃下搅拌3小时。在0℃下将meoh(15ml)中的ch3so3h(0.6ml,9.25mmol)滴加到混合物。将混合物经过18小时升温至室温,然后在0℃下用饱和nahco3猝灭。将混合物减压蒸发以除去挥发物。将含水
残余物用etoac(100ml
×
2)萃取。将合并的有机层用mgso4干燥,过滤并真空浓缩,得到作为黄色固体的粗制标题产物。[m+na]
+
473.
[0341]
步骤11:(3r,4r,5s,6r)
‑2‑
(3
‑
(烯丙氧基)
‑5‑
苄基
‑4‑
氯苯基)
‑6‑
(羟甲基)四氢
‑
2h
‑
吡喃
‑
3,4,5
‑
三醇(化合物c13)
[0342]
在0℃下向(3r,4s,5s,6r)
‑2‑
(3
‑
(烯丙氧基)
‑5‑
苄基
‑4‑
氯苯基)
‑6‑
(羟甲基)
‑2‑
甲氧基四氢
‑
2h
‑
吡喃
‑
3,4,5
‑
三醇(2.55g,5.65mmol)在ch2cl2(30ml)和ch3cn(30ml)中的混合物滴加et3sih(1.82ml,11.3mmol)和bf3·
et2o(1.07ml,8.48mmol)。将反应混合物在室温下搅拌5小时。将所得混合物用饱和nahco3溶液猝灭,并用etoac萃取。将有机层用mgso4干燥,过滤,并真空浓缩。粗制产物无需进一步纯化用于下一步骤。
[0343]
步骤12:(2r,3r,4r,5s,6s)
‑2‑
(乙酰氧基甲基)
‑6‑
(3
‑
(烯丙氧基)
‑5‑
苄基
‑4‑
氯苯基)四氢
‑
2h
‑
吡喃
‑
3,4,5
‑
三基三乙酸酯(化合物c14)
[0344]
在室温下向(3r,4r,5s,6r)
‑2‑
(3
‑
(烯丙氧基)
‑5‑
苄基
‑4‑
氯苯基)
‑6‑
(羟甲基)四氢
‑
2h
‑
吡喃
‑
3,4,5
‑
三醇(c13)在ch2cl2(12ml)中的混合物添加ac2o(4.7ml,49.72mmol)、吡啶(4.0ml,49.45mmol)和dmap(35mg,0.28mmol)。将反应混合物在室温下搅拌12小时。将所得混合物用etoac稀释,并用1n hcl溶液洗涤。将有机层用mgso4干燥,过滤,并真空浓缩。将粗制产物通过硅胶柱色谱法(biotage isolera
tm flash纯化系统,etoac/hex)纯化,得到标题化合物(1.55g,47%)。
[0345]1h nmr(400mhz,dmso
‑
d6)δ7.31
‑
7.27(m,2h),7.22
‑
7.21(m,1h),7.19
‑
7.16(m,2h),7.09(d,j=1.6hz,1h),6.88(d,j=1.6hz,1h),6.12
‑
6.03(m,1h),5.47(dq,j=17.6hz,2.0hz,1h),5.35(t,j=9.6hz,1h),5.30(dq,j=10.4,1.6hz,1h),5.12(t,j=9.6hz,1h),5.06(t,j=9.6hz,1h),4.66
‑
4.62(m,3h),4.13
‑
4.03(m,5h),2.04(s,3h),2.03(s,3h),1.95(s,3h),1.70(s,3h);[m+na]
+
611.
[0346]
步骤13:(2s,3r,4r,5s,6r)
‑2‑
(3
‑
(烯丙氧基)
‑5‑
苄基
‑4‑
氯苯基)
‑6‑
(羟甲基)四氢
‑
2h
‑
吡喃
‑
3,4,5
‑
三醇(化合物c15)
[0347]
在室温下向(2r,3r,4r,5s,6s)
‑2‑
(乙酰氧基甲基)
‑6‑
(3
‑
(烯丙氧基)
‑5‑
苄基
‑4‑
氯苯基)四氢
‑
2h
‑
吡喃
‑
3,4,5
‑
三基三乙酸酯(1.55g,2.63mmol)在meoh(50ml)中的混合物添加naome(meoh中25wt%,2.34ml)。将混合物在室温下搅拌12小时。将所得混合物用冰acoh中和。将混合物用etoac稀释并用饱和nahco3溶液洗涤。将有机层用mgso4干燥,过滤,并真空浓缩。粗制产物无需进一步纯化用于下一步骤。[m+na]
+
443.
[0348]
步骤14:(2s,3s,4r,5r,6r)
‑2‑
(3
‑
(烯丙氧基)
‑5‑
苄基
‑4‑
氯苯基)
‑
3,4,5
‑
三(苄氧基)
‑6‑
((苄氧基)甲基)四氢
‑
2h
‑
吡喃(化合物c16)
[0349]
在0℃下向(2s,3r,4r,5s,6r)
‑2‑
(3
‑
(烯丙氧基)
‑5‑
苄基
‑4‑
氯苯基)
‑6‑
(羟甲基)四氢
‑
2h
‑
吡喃
‑
3,4,5
‑
三醇在dmf(26ml)中的混合物添加nah(在矿物油中的60%分散体,842mg,21.0mmol)并在室温下搅拌1小时。在0℃下将苄基溴(2.5ml,21.0mmol)滴加到反应混合物。将混合物在室温下搅拌12小时。将所得混合物用水猝灭并用etoac萃取。将有机层用mgso4干燥,过滤,并真空浓缩。将粗制产物通过硅胶柱色谱法(biotage isolera
tm
flash纯化系统,etoac/hex)纯化,得到标题化合物(1.80g,88%)。
[0350]1h nmr(400mhz,cdcl3)δ7.36
‑
7.31(m,13h),7.26
‑
7.19(m,10h),6.91(d,j=1.6hz,2h),6.91(d,j=14.8,2.0hz,2h),6.10
‑
6.00(m,1h),5.46(dq,j=17.2,1.6hz,1h),
5.31(dq,j=10.8,1.6hz,1h),4.94(abq,j
ab
=15.2hz,2h),4.90(d,j=10.8hz,1h),4.70
‑
4.64(m,2h),4.57(d,j=12.4hz,1h),4.52
‑
4.49(m,2h),4.46(d,j=10.8hz,1h),4.23
‑
4.16(m,2h),4.08(d,j=15.2hz,1h),3.89(d,j=10.8hz,1h),3.84
‑
3.75(m,4h),3.61
‑
3.57(m,1h),3.48
‑
3.44(m,1h);[m+na]
+
803.
[0351]
步骤15:3
‑
苄基
‑2‑
氯
‑5‑
((2s,3s,4r,5r,6r)
‑
3,4,5
‑
三(苄氧基)
‑6‑
((苄氧基)甲基)四氢
‑
2h
‑
吡喃
‑2‑
基)苯酚(化合物c17)
[0352]
在室温下向((2s,3s,4r,5r,6r)
‑2‑
(3
‑
(烯丙氧基)
‑5‑
苄基
‑4‑
氯苯基)
‑
3,4,5
‑
三(苄氧基)
‑6‑
((苄氧基)甲基)四氢
‑
2h
‑
吡喃(1.80g,2.30mmol)在thf(25ml)中的混合物添加nabh4(700mg,18.4mmol)和pd(pph3)4(266mg,0.23mmol)。将反应混合物在室温下搅拌12小时。所得混合物用饱和nahco3溶液猝灭,并用etoac萃取。将有机层用mgso4干燥,过滤,并真空浓缩。将粗制产物通过硅胶柱色谱法(biotage isolera
tm flash纯化系统,etoac/hex)纯化,得到标题化合物(1.62g,95%)。
[0353]1h nmr(400mhz,cdcl3)δ7.37
‑
7.31(m,13h),7.27
‑
7.21(m,8h),7.18
‑
7.16(m,2h),7.08(d,j=2.0hz,1h),6.98(dd,j=7.6,2.0hz,2h),6.89(d,j=2.0hz,1h),4.93(abq,j
ab
=16.0hz,2h),4.89(d,j=10.8hz,1h),4.67(d,j=4.8hz,1h),4.64(d,j=6.0hz,1h),4.57(d,j=12.4hz,1h),4.46(d,j=10.4hz,1h),4.19
‑
4.12(m,2h),4.03(d,j=15.2hz,1h),3.95(d,j=10.4hz,1h),3.82
‑
3.75(m,4h),3.61
‑
3.57(m,1h),3.49
‑
3.45(m,1h);[m+na]
+
763.
[0354]
步骤16:3
‑
苄基
‑6‑
溴
‑2‑
氯
‑5‑
((2s,3s,4r,5r,6r)
‑
3,4,5
‑
三(苄氧基)
‑6‑
((苄氧基)甲基)四氢
‑
2h
‑
吡喃
‑2‑
基)苯酚(化合物c18)
[0355]
在0℃下向3
‑
苄基
‑2‑
氯
‑5‑
((2s,3s,4r,5r,6r)
‑
3,4,5
‑
三(苄氧基)
‑6‑
((苄氧基)甲基)四氢
‑
2h
‑
吡喃
‑2‑
基)苯酚(1.62g,2.18mmol)在acoh(11ml)中的混合物添加三乙胺(0.46ml,3.27mmol)和溴(0.11ml,2.18mmol)。将反应混合物在室温下搅拌12小时。将所得混合物用饱和nahco3溶液猝灭并用etoac萃取。将有机层用mgso4干燥,过滤,并真空浓缩。将粗制产物通过硅胶柱色谱法(biotage isolera
tm flash纯化系统,etoac/hex)纯化,得到标题化合物(1.04g,58%)。[m+na]
+
841.
[0356]
步骤17:3
‑
(3
‑
苄基
‑6‑
溴
‑2‑
氯
‑5‑
((2s,3s,4r,5r,6r)
‑
3,4,5
‑
三(苄氧基)
‑6‑
((苄氧基)甲基)四氢
‑
2h
‑
吡喃
‑2‑
基)苯氧基)丙
‑1‑
醇(化合物c19)
[0357]
在室温下向3
‑
苄基
‑6‑
溴
‑2‑
氯
‑5‑
((2s,3s,4r,5r,6r)
‑
3,4,5
‑
三(苄氧基)
‑6‑
((苄氧基)甲基)四氢
‑
2h
‑
吡喃
‑2‑
基)苯酚(1.04g,1.27mmol)和k2co3(0.35g,2.54mmol)在丙酮(13ml)中的混合物添加2
‑
溴乙醇(0.14ml,1.90mmol)。将反应混合物在50℃下搅拌12小时。过滤所得混合物以除去无机材料。将滤液真空浓缩。将粗制产物通过硅胶柱色谱法(biotageisolera
tm flash纯化系统,etoac/hex)纯化,得到化合物c19(1.10g,100%)。[m+na]
+
899.
[0358]
步骤18:(2s,3s,4r,5r,6r)
‑2‑
(5
‑
苄基
‑2‑
溴
‑4‑
氯
‑3‑
(2
‑
氯乙氧基)苯基)
‑
3,4,5
‑
三(苄氧基)
‑6‑
((苄氧基)甲基)四氢
‑
2h
‑
吡喃(化合物c20)
[0359]
在室温下向3
‑
(3
‑
苄基
‑6‑
溴
‑2‑
氯
‑5‑
((2s,3s,4r,5r,6r)
‑
3,4,5
‑
三(苄氧基)
‑6‑
((苄氧基)甲基)四氢
‑
2h
‑
吡喃
‑2‑
基)苯氧基)丙
‑1‑
醇(1.09g,1.25mmol)和三苯基膦(1.64g,6.28mmol)在ch3cn(12ml)中的混合物添加四氯化碳(12ml,134mmol)。将反应混合
物在55℃下搅拌12小时。蒸发所得混合物以除去溶剂。将粗制产物通过硅胶柱色谱法(biotage isolera
tm flash纯化系统,etoac/hex)纯化,得到标题化合物(0.61g,55%)。[m+na]
+
903.
[0360]
步骤19:6
‑
苄基
‑7‑
氯
‑4‑
((2s,3s,4r,5r,6r)
‑
3,4,5
‑
三(苄氧基)
‑6‑
((苄氧基)甲基)四氢
‑
2h
‑
吡喃
‑2‑
基)
‑
2,3
‑
二氢苯并呋喃(化合物c21)
[0361]
在
‑
78℃下向(2s,3s,4r,5r,6r)
‑2‑
(5
‑
苄基
‑2‑
溴
‑4‑
氯
‑3‑
(2
‑
氯乙氧基)苯基)
‑
3,4,5
‑
三(苄氧基)
‑6‑
((苄氧基)甲基)四氢
‑
2h
‑
吡喃(10.02g,11.4mmol)在thf(114ml)中的混合物滴加正丁基锂(己烷中2.5m,6.8ml,17.0mmol)。将反应混合物在
‑
78℃下搅拌3小时。将所得混合物用1n hcl溶液(100ml)猝灭并用etoac萃取。将有机层用mgso4干燥,过滤,并真空浓缩。将粗制产物通过硅胶柱色谱法(biotage isolera
tm flash纯化系统,etoac/hex)纯化,得到标题化合物(6.9g,69%)。[m+na]
+
789.
[0362]
步骤20:(2s,3r,4r,5s,6r)
‑2‑
(6
‑
苄基
‑7‑
氯
‑
2,3
‑
二氢苯并呋喃
‑4‑
基)
‑6‑
(羟甲基)四氢
‑
2h
‑
吡喃
‑
3,4,5
‑
三醇(化合物c22)
[0363]
在h2下室温下,将6
‑
苄基
‑7‑
氯
‑4‑
((2s,3s,4r,5r,6r)
‑
3,4,5
‑
三(苄氧基)
‑6‑
((苄氧基)甲基)四氢
‑
2h
‑
吡喃
‑2‑
基)
‑
2,3
‑
二氢苯并呋喃(6.0g,7.82mmol)和pd/c(0.35g,2.54mmol)在meoh(220ml)/thf(220ml)中的混合物搅拌5小时。通过硅藻土过滤所得混合物以除去无机材料。将滤液真空浓缩,得到标题化合物(定量量)。粗制产物无需进一步纯化用于下一步骤。
[0364]1h nmr(400mhz,cd3od)δ7.28
‑
7.14(m,5h),6.89(s,1h),4.65(t,j=8.6hz,2h),4.17(d,j=8.8hz,1h),4.07(abq,δv
ab
=18.0hz,j
ab
=15.0hz,2h),3.90(dd,j=11.8,1.4hz,1h),3.72
‑
3.67(m,1h),3.53
‑
3.42(m,3h),3.40
‑
3.37(m,2h);[m+na]
+
507.
[0365]
步骤21:(2r,3r,4r,5s,6s)
‑2‑
(乙酰氧基甲基)
‑6‑
(6
‑
苄基
‑7‑
氯
‑
2,3
‑
二氢苯并呋喃
‑4‑
基)四氢
‑
2h
‑
吡喃
‑
3,4,5
‑
三基三乙酸酯(化合物c23)
[0366]
在室温下向(2s,3r,4r,5s,6r)
‑2‑
(6
‑
苄基
‑7‑
氯
‑
2,3
‑
二氢苯并呋喃
‑4‑
基)
‑6‑
(羟甲基)四氢
‑
2h
‑
吡喃
‑
3,4,5
‑
三醇在ch2cl2(78ml)中的混合物添加ac2o(5.9ml,62.6mmol)、吡啶(5.0ml,62.6mmol)和dmap(48mg,0.39mmol)。将反应混合物在室温下搅拌12小时。将所得混合物用etoac稀释,并用1n hcl溶液洗涤。将有机层用mgso4干燥,过滤,并真空浓缩。将粗制产物通过硅胶柱色谱法(biotage isolerat
m flash纯化系统,etoac/hex)纯化,得到标题化合物(4.54g,100%)。
[0367]1h nmr(400mhz,cdcl3)δ7.32
‑
7.28(m,2h),7.25
‑
7.18(m,3h),6.59(s,1h),5.30(t,j=9.2hz,2h),5.19(t,j=9.6hz,1h),4.77
‑
4.68(m,2h),4.35
‑
4.32(m,1h),4.31
‑
4.26(m,1h),4.21
‑
4.14(m,1h),4.11(m,1h),4.02(d,j=15.6hz,1h),3.83
‑
3.79(m,1h),3.42(td,j=8.8,1,6hz,2h),2.10(s,3h),2.09(s,3h),2.03(s,3h),1.70(s,3h);[m+na]
+
597.
[0368]
步骤22:(2r,3r,4r,5s,6s)
‑2‑
(乙酰氧基甲基)
‑6‑
(6
‑
(4
‑
乙酰基苄基)
‑7‑
氯
‑
2,3
‑
二氢苯并呋喃
‑4‑
基)四氢
‑
2h
‑
吡喃
‑
3,4,5
‑
三基三乙酸酯(化合物c24)
[0369]
在0℃下向(2r,3r,4r,5s,6s)
‑2‑
(乙酰氧基甲基)
‑6‑
(6
‑
苄基
‑7‑
氯
‑
2,3
‑
二氢苯并呋喃
‑4‑
基)四氢
‑
2h
‑
吡喃
‑
3,4,5
‑
三基三乙酸酯(3.75g,6.52mmol)在ch2cl2(78ml)中的混合物滴加乙酰氯(3.71ml,52.16mmol)和氯化铝(6.95mg,52.16mmol)。将反应混合物在室温下搅拌12小时。将所得混合物用冰水猝灭并用ch2cl2萃取。将有机层用mgso4干燥,过滤,
干燥,过滤,并真空浓缩。将粗制产物通过硅胶柱色谱法(biotage isolera
tm flash纯化系统,etoac/hex)纯化,得到标题化合物(285mg,60%)。
[0379]1h nmr(400mhz,cdcl3)δ7.04
‑
7.02(m,2h),6.98
‑
6.95(m,2h),6.53(s,1h),5.29
‑
5.24(m,1h),5.18
‑
5.12(m,2h),4.71
‑
4.65(m,2h),4.31
‑
4.26(m,1h),4.25
‑
4.22(m,1h),4.15
‑
4.11(m,1h),4.05
‑
3.91(m,2h),3.79
‑
3.74(m,1h),3.40
‑
3.35(m,2h),2.60(s,3h),2.05(s,3h),1.99(s,3h),1.88
‑
1.81(m,1h),1.66(s,3h),0.94
‑
0.89(m,2h),0.66
‑
0.61(m,2h);[m+na]
+
637.
[0380]
步骤26:(2s,3r,4r,5s,6r)
‑2‑
(7
‑
氯
‑6‑
(4
‑
环丙基苄基)
‑
2,3
‑
二氢苯并呋喃
‑4‑
基)
‑6‑
(羟甲基)四氢
‑
2h
‑
吡喃
‑
3,4,5
‑
三醇(化合物c28)
[0381]
将(2r,3r,4r,5s,6s)
‑2‑
(乙酰氧基甲基)
‑6‑
(7
‑
氯
‑6‑
(4
‑
环丙基苄基)
‑
2,3
‑
二氢苯并呋喃
‑4‑
基)四氢
‑
2h
‑
吡喃
‑
3,4,5
‑
三基三乙酸酯(298mg,0.48mmol)和k2co3(536mg,3.88mmol)在meoh(20ml)中的混合物搅拌12小时。过滤所得混合物以除去无机材料。将滤液真空浓缩。将粗制产物通过制备型hplc(gilson系统,ch3cn/h2o)纯化,得到标题化合物(101mg,47%)。
[0382]
根据上述步骤1至26的合成途径,比较实施例1的最终化合物的总产率计算为1%或更低。
[0383]1h nmr(400mhz,cd3od)δ7.02(d,j=8.0hz,2h),6.92(d,j=8.0hz,2h),6.81(s,1h),4.59(t,j=8.8hz,2h),4.11(d,j=9.2hz,1h),3.96(abq,δv
ab
=19.0hz,j
ab
=15.2hz,2h),3.87
‑
3.84(m,1h),3.67
‑
3.63(m,1h),3.47
‑
3.37(m,3h),3.35
‑
3.33(m,3h),1.85
‑
1.79(m,1h),0.91
‑
0.86(m,2h),0.61
‑
0.57(m,2h);[m+na]
+
469.
[0384]
实施例1:(2s,3r,4r,5s,6r)
‑2‑
(7
‑
氯
‑6‑
(4
‑
环丙基苄基)
‑
2,3
‑
二氢苯并呋喃
‑4‑
基)
‑6‑
(羟甲基)四氢
‑
2h
‑
吡喃
‑
3,4,5
‑
三醇的合成
[0385][0386]
步骤1:5
‑
溴
‑2‑
氯
‑3‑
羟基苯甲酸甲酯(化合物c29)
[0387]
在氮气氛下于0℃下向5
‑
溴
‑2‑
氯
‑3‑
甲氧基苯甲酸甲酯(化合物c5;30.0g,107.3mmol)在ch2cl2(300ml)中的溶液缓慢添加bbr3(25.9ml,268.3mmol)。将混合物缓慢升
温至室温并在室温下搅拌15小时。将反应混合物在0℃下用meoh(100ml)猝灭。减压蒸发混合物以除去ch2cl2,然后向其提供meoh(150ml)。将所得混合物在室温下搅拌16小时。将反应混合物真空浓缩,得到标题化合物(29.4g,110.8mmol,103%)。
[0388]1h nmr(400mhz,cdcl3)δ7.60(d,j=2.4hz,1h),7.36(d,j=2.4hz,1h),6.00(s,1h),3.94(s,1h);[m+h]
+
265.
[0389]
步骤2:3
‑
(烯丙氧基)
‑5‑
溴
‑2‑
氯苯甲酸甲酯(化合物c30)
[0390]
在室温下向5
‑
溴
‑2‑
氯
‑3‑
羟基苯甲酸甲酯(38.2g,143.9mmol)在丙酮(700ml)中的溶液添加烯丙基溴(14.9ml,172.7mmol)和k2co3(29.8g,215.9mmol)。将混合物在60℃下搅拌12小时,然后冷却至室温。通过硅藻土滤除不溶性盐后,减压蒸发滤液,将残余物溶于etoac(500ml)中。将有机溶液用盐水洗涤,用mgso4干燥,过滤,并真空浓缩(44.1g,144.3mmol,100%)。粗制残余物不经进一步纯化用于下一步骤。
[0391]1h nmr(400mhz,cdcl3)δ7.49(d,j=2.4hz,1h),7.16(d,j=2.4hz,1h),6.09
‑
6.00(m,1h),5.48(dd,j=17.2hz,1.2hz,1h),5.35(dd,j=10.6hz,1.4hz,1h),4.63
‑
4.61(m 2h),3.93(s,3h);[m+h]
+
305.
[0392]
步骤3:4
‑
烯丙基
‑5‑
溴
‑2‑
氯
‑3‑
羟基苯甲酸甲酯(化合物c31)
[0393]
在氮气氛下于0℃下向3
‑
(烯丙氧基)
‑5‑
溴
‑2‑
氯苯甲酸甲酯(10.0g,32.7mmol)在ch2cl2(150ml)中的溶液滴加(0.5至1小时)二异丁基氯化铝(己烷中25%,64.0ml)。将混合物缓慢升温至室温,并在室温下另外搅拌12小时。将反应混合物冷却至0℃并用1m hcl(50ml)猝灭,然后用etoac(150ml
×
2)萃取。将合并的有机层用mgso4干燥,过滤,并真空浓缩(9.9g,32.3mmol,99%)。将粗制残余物不经进一步纯化用于下一步骤,得到标题化合物。
[0394]1h nmr(400mhz,cdcl3)δ7.23(s,1h),6.20(s,1h),5.96
‑
5.86(m,1h),5.11
‑
5.07(m,2h),3.92(s,3h),3.65(dt,j=5.4hz,1.4hz,2h);[m+h]
+
305.
[0395]
步骤4:5
‑
溴
‑2‑
氯
‑3‑
羟基
‑4‑
(2
‑
羟乙基)苯甲酸甲酯(化合物c33)
[0396]
方法a)通过醛还原的合成
[0397]
在0℃下向4
‑
烯丙基
‑5‑
溴
‑2‑
氯
‑3‑
羟基苯甲酸甲酯(9.9g,32.3mmol)在thf/h2o(100ml/100ml)中的混合物添加naio4(20.8g,97.0mmol)和oso4(82mg,0.32mmol)。在0℃下搅拌1小时后,将反应混合物升温至室温并在室温下搅拌2小时。过滤混合物以除去不溶物质。将滤液倒入饱和na2s2o3溶液(100ml)中,并用etoac(200ml
×
2)萃取混合物。将有机层用mgso4干燥,过滤,并真空浓缩(9.0g,29.4mmol,91%)。粗制残余物不经进一步纯化用于下一步骤,得到5
‑
溴
‑2‑
氯
‑3‑
羟基
‑4‑
(2
‑
氧代乙基)苯甲酸甲酯(化合物c32)。[m+h]
+
307.
[0398]
在氮气氛下于0℃下向5
‑
溴
‑2‑
氯
‑3‑
羟基
‑4‑
(2
‑
氧代乙基)苯甲酸甲酯(20.1g,65.3mmol)在thf(200ml)中的溶液添加nabh4(2.72g,71.8mmol)。将反应混合物在0℃下搅拌2小时。将混合物用饱和nh4cl(100ml)猝灭并用etoac(100ml
×
2)萃取[一次萃取可能不够]。将合并的有机层用mgso4干燥,过滤,并真空浓缩(19.9g)。向残余物在etoac(20ml)中的悬浮液添加己烷(10至20ml)。通过过滤收集所得沉淀物并用己烷(50ml)洗涤。将沉淀物在高真空下干燥,得到标题化合物(14.3g,72%)。
[0399]1h nmr(400mhz,cdcl3)δ7.68(s,1h),7.35(s,1h),3.97
‑
3.93(m,2h),3.92(s,3h),3.21(t,j=6.2hz,2h);[m+h]
+
309.
[0400]
方法b)通过臭氧化然后还原的合成
[0401]
在
‑
78℃下,将臭氧气体鼓泡通过ch2cl2/meoh(150ml/35ml)中的4
‑
烯丙基
‑5‑
溴
‑2‑
氯
‑3‑
羟基苯甲酸甲酯(10.2g,33.4mmol,80%纯度),持续4小时(溶液颜色从黄色变为至浅绿色)。停止添加臭氧后,用氮气吹扫反应溶液直至放出绿色(溶液颜色恢复为黄色)。在
‑
78℃下分批添加硼氢化钠(2.5g,66.8mmol)。将所得混合物缓慢升温至室温持续2小时,浓缩,悬浮于etoac中,并浓缩。向残余物添加1n hcl水溶液(200ml),并搅拌30分钟。通过过滤收集沉淀物。将(定量,80%纯度)沉淀物悬浮在etoac中并搅拌。向所得混合物缓慢添加己烷。通过过滤得到沉淀物,从而得到标题化合物(7.9g,76.4%,92%纯度)。
[0402]1h nmr(400mhz,meod)δ7.57(s,1h),3.93(s,3h),3.76(t,j=7.24hz,2h),3.20(t,j=7.28hz,2h);[m+h]
+
309.
[0403]
步骤5:4
‑
溴
‑7‑
氯
‑
2,3
‑
二氢苯并呋喃
‑6‑
甲酸甲酯(化合物c34)
[0404]
vilsmeier试剂的制备;在室温下向n,n
‑
二甲基甲酰胺(7.9ml,102.2mmol)的溶液添加socl2(7.5ml,102.2mmol)。将反应混合物在40℃下搅拌2小时。将所得混合物真空浓缩,得到吸湿性白色固体。
[0405]
在0℃下向vilsmeier试剂(13.08g,102.2mmol)在dmf(100ml)中的混合物缓慢添加dmf(130ml)中的5
‑
溴
‑2‑
氯
‑3‑
羟基
‑4‑
(2
‑
羟乙基)苯甲酸甲酯(21.08g,68.10mmol)。将混合物在0℃至15℃(梯度加热)下搅拌1小时。在0℃下,将反应混合物用dmf(38ml)中的三乙胺(38ml,272.4mmol)猝灭。搅拌10分钟后,将混合物倒入0℃的水(1400ml)中并在室温下搅拌2小时。通过过滤收集所得沉淀物,用水洗涤并真空干燥浓缩,得到作为浅黄色固体的标题化合物(13.0g,44.6mmol,65%)。
[0406]1h nmr(400mhz,cdcl3)δ7.53(s,1h),4.75(t,j=8.8hz,2h),3.91(s,3h),3.33(t,j=8.8hz,2h);[m+h]
+
291.
[0407]
步骤6:(4
‑
溴
‑7‑
氯
‑
2,3
‑
二氢苯并呋喃
‑6‑
基)甲醇(化合物c35)
[0408]
在室温下向4
‑
溴
‑7‑
氯
‑
2,3
‑
二氢苯并呋喃
‑6‑
甲酸甲酯(13.0g,44.7mmol)在thf/etoh(150ml/75ml)中的混合物缓慢添加硼氢化钠(5.07g,133.98mmol)。将混合物在室温下搅拌12小时。将所得混合物在0℃下用饱和nh4cl猝灭并用etoac(水性,ph约7.0)萃取。将有机层用mgso4干燥,过滤,并真空浓缩,得到作为白色固体的标题化合物(11.7g,44.4mmol,99%)。粗制产物无需进一步纯化用于下一步骤。
[0409]1h nmr(400mhz,cdcl3)δ7.15(s,1h),4.73(m,4h),3.29(t,j=8.8hz,2h),1.91(t,j=6.4hz,1h);[m
‑
h2o]
+
245.
[0410]
步骤7:4
‑
溴
‑7‑
氯
‑
2,3
‑
二氢苯并呋喃
‑6‑
甲醛(化合物c36)
[0411]
在室温下向(4
‑
溴
‑7‑
氯
‑
2,3
‑
二氢苯并呋喃
‑6‑
基)甲醇(11.7g,44.4mmol)在ch2cl2(450ml)中的溶液缓慢添加pcc(14.4g,66.6mmol,氯铬酸吡啶)。搅拌8小时后,使用硅胶垫滤出沉淀物并用ch2cl2洗涤。将滤液真空浓缩,得到作为白色固体的标题化合物(10.4g,39.8mmol,90%)。粗制产物无需进一步纯化用于下一步骤。
[0412]1h nmr(400mhz,cdcl3)δ10.33(s,1h),7.59(s,1h),4.79(t,j=8.8hz,2h),3.35(t,j=8.8hz,2h);[m+h]
+
261.
[0413]
步骤8:(4
‑
溴
‑7‑
氯
‑
2,3
‑
二氢苯并呋喃
‑6‑
基)(4
‑
环丙基苯基)甲醇(化合物c39)
[0414]
(4
‑
环丙基苯基)溴化镁(化合物c38)的制备:将含有镁(屑,1.1g,46.6mmol)的250ml三颈烧瓶火焰干燥。在氮气氛下向烧瓶配备冷凝器和加料漏斗。将无水thf(32.4ml)
中的4
‑
环丙基苯基溴(peptech,usa)(6.0ml,42.4mmol)转移到加料漏斗。用约5ml的4
‑
环丙基苯基溴溶液引发格氏反应。在室温下经过4小时添加剩余的溴化物溶液。所得溶液直接用于下一步骤。
[0415]
在氮气氛下于0℃下向4
‑
溴
‑7‑
氯
‑
2,3
‑
二氢苯并呋喃
‑6‑
甲醛(4.6g,17.6mmol)在无水thf(170ml)中的溶液添加新制备的(4
‑
环丙基苯基)溴化镁溶液(化合物c38)(30.0ml,thf中0.85m,26.4mmol)。将反应混合物在0℃下搅拌30分钟。将反应混合物用水(100ml)猝灭并用etoac(100ml)萃取。将有机层用mgso4干燥,过滤,并真空浓缩,得到粗制标题产物(7.6g,20.0mmol,114%)。将粗制残余物不经进一步纯化用于下一步骤,得到粗制标题化合物。[m
‑
h2o]
+
361.
[0416]
步骤9:4
‑
溴
‑7‑
氯
‑6‑
(4
‑
环丙基苄基)
‑
2,3
‑
二氢苯并呋喃(化合物c40)
[0417]
在氮气氛下于
‑
20℃下向(4
‑
溴
‑7‑
氯
‑
2,3
‑
二氢苯并呋喃
‑6‑
基)(4
‑
环丙基苯基)甲醇(7.6g,20.0mmol)在ch2cl2/ch3cn(100ml/100ml)中的溶液添加三乙基硅烷(4.6ml,40mmol)和三氟化硼二乙基醚合物(3.8ml,30mmol)。将混合物逐渐升温至室温,并在室温下再搅拌50分钟。通过缓慢添加饱和nahco3溶液(200ml)猝灭反应混合物,并用etoac(100ml)萃取。将有机层用mgso4干燥,过滤,并真空浓缩。将残余物通过硅胶柱色谱法纯化,得到标题产物(4.4g,12.1mmol,2步85%)。
[0418]1h nmr(400mhz,cdcl3)δ7.07(d,j=8.0hz,2h),6.99(d,j=8.0hz,2h),6.80(s,1h),4.70(t,j=8.8hz,2h),3.97(s,2h),3.26(t,j=8.8hz,2h),1.88
‑
1.84(m,1h),0.95
‑
0.90(m,2h),0.68
‑
0.64(m,2h).
[0419]
步骤10:(3r,4s,5r,6r)
‑2‑
(7
‑
氯
‑6‑
(4
‑
环丙基苄基)
‑
2,3
‑
二氢苯并呋喃
‑4‑
基)
‑
3,4,5
‑
三(三甲基甲硅烷氧基)
‑6‑
((三甲基甲硅烷氧基)甲基)四氢
‑
2h
‑
吡喃
‑2‑
醇(化合物c41)
[0420]
在氮气氛下于
‑
78℃下向4
‑
溴
‑7‑
氯
‑6‑
(4
‑
环丙基苄基)
‑
2,3
‑
二氢苯并呋喃(5.16g,14.2mmol)在四氢呋喃(80ml)中的溶液滴加正丁基锂(己烷中2.5m,7.38ml,18.4mmol)。在相同温度下搅拌40至60分钟后(黄色溶液),通过管经过20分钟将预冷(在
‑
78℃)的(3r,4s,5r,6r)
‑
3,4,5
‑
三((三甲基甲硅烷基)氧基)
‑6‑
(((三甲基甲硅烷基)氧基)甲基)四氢
‑
2h
‑
吡喃
‑2‑
酮(化合物c11;8.6g,18.4mmol)在四氢呋喃(20ml)中的溶液滴加到该混合物。将反应混合物在相同温度下搅拌2至3小时(淡黄色溶液)。
[0421]
将反应混合物在
‑
78℃下用1%乙酸(20ml)猝灭,然后在减压下蒸发以除去挥发物。用etoac(150ml
×
2)萃取含水残余物。将合并的有机层用mgso4干燥,过滤,并真空浓缩,得到作为浅黄色油状物的粗制标题化合物(11.8g,定量)。粗制残余物不经进一步纯化用于下一步骤。
[0422]
步骤11:(3r,4s,5s,6r)
‑2‑
(7
‑
氯
‑6‑
(4
‑
环丙基苄基)
‑
2,3
‑
二氢苯并呋喃
‑4‑
基)
‑6‑
(羟甲基)
‑2‑
甲氧基四氢
‑
2h
‑
吡喃
‑
3,4,5
‑
三醇(化合物c42)
[0423]
在0℃下向(3r,4s,5r,6r)
‑2‑
(7
‑
氯
‑6‑
(4
‑
环丙基苄基)
‑
2,3
‑
二氢苯并呋喃
‑4‑
基)
‑
3,4,5
‑
三(三甲基甲硅烷氧基)
‑6‑
((三甲基甲硅烷氧基)甲基)四氢
‑
2h
‑
吡喃
‑2‑
醇;11.8g)在meoh(150ml)中的溶液滴加ch3so3h(1.5ml,23.5mmol)。将混合物经过18小时升温至室温,然后在0℃下用饱和nahco3猝灭。减压蒸发混合物以除去挥发物。用etoac(100ml
×
2)萃取含水残余物。将合并的有机层用mgso4干燥,过滤并真空浓缩,得到作为黄色固体的
粗制标题化合物(6.0g,2步88%)。[m+na]
+
499和[m
‑
ome]
+
445。
[0424]
步骤12:(3r,4r,5s,6r)
‑2‑
(7
‑
氯
‑6‑
(4
‑
环丙基苄基)
‑
2,3
‑
二氢苯并呋喃
‑4‑
基)
‑6‑
(羟甲基)四氢
‑
2h
‑
吡喃
‑
3,4,5
‑
三醇(化合物c43)
[0425]
在
‑
50至
‑
45℃下向(3r,4s,5s,6r)
‑2‑
(7
‑
氯
‑6‑
(4
‑
环丙基苄基)
‑
2,3
‑
二氢苯并呋喃
‑4‑
基)
‑6‑
(羟甲基)
‑2‑
甲氧基四氢
‑
2h
‑
吡喃
‑
3,4,5
‑
三醇(6.0g,12.6mmol)在ch2cl2/ch3cn(v∶v=1∶1,120ml)中的搅拌溶液滴加et3sih(6.0ml,37.8mmol),然后bf3·
oet2(3.2ml,25.2mmol)。将反应混合物经过3至3.5小时升温至
‑
10至0℃,然后用饱和nahco3(130ml)猝灭。减压蒸发混合物以除去挥发物,并将所得残余物用etoac(150ml
×
2)萃取。将合并的有机层用mgso4干燥,过滤并真空浓缩,得到作为黄色固体的粗制标题化合物(5.8g,12.9mmol,102%)。[m+na]
+
469.
[0426]
步骤13:(2r,3r,4r,5s,6s)
‑2‑
(乙酰氧基甲基)
‑6‑
(7
‑
氯
‑6‑
(4
‑
环丙基苄基)
‑
2,3
‑
二氢苯并呋喃
‑4‑
基)四氢
‑
2h
‑
吡喃
‑
3,4,5
‑
三基三乙酸酯(化合物c27)
[0427]
在室温下向(3r,4r,5s,6r)
‑2‑
(7
‑
氯
‑6‑
(4
‑
环丙基苄基)
‑
2,3
‑
二氢苯并呋喃
‑4‑
基)
‑6‑
(羟甲基)四氢
‑
2h
‑
吡喃
‑
3,4,5
‑
三醇(5.8g,12.9mmol)在ch2cl2(120ml)中的溶液添加dmap(1.9g,15.5mmol)和ac2o(9.7ml,103.76mmol)。在室温下搅拌18小时后,用水(120ml)猝灭反应。用ch2cl2(100ml
×
2)萃取所得混合物。用1m hcl和盐水洗涤后,将合并的有机层用mgso4干燥,过滤并减压蒸发(7.0g,粗制品)。将etoh(45ml)中的浆化残余物在80℃下加热以回流1小时。将混合物冷却至室温并搅拌18小时。过滤所得沉淀物,用etoh洗涤并真空干燥,得到作为白色固体的标题化合物(4.7g,7.6mmol,59%)。
[0428]1h nmr(400mhz,cdcl3)δ7.04
‑
7.02(m,2h),6.98
‑
6.95(m,2h),6.53(s,2h),5.29
‑
5.24(m,1h),5.18
‑
5.12(m,2h),4.71
‑
4.65(m,2h),4.31
‑
4.26(m,1h),4.25
‑
4.22(m,1h),4.15
‑
4.11(m,1h),4.15
‑
4.11(m,1h),4.05
‑
3.91(m,2h),3.79
‑
3.74(m,1h),3.40
‑
3.35(m,2h),2.60(s,3h),2.05(s,3h),1.99(s,3h),1.88
‑
1.81(m,1h),1.66(s,3h),0.94
‑
0.89(m,2h),0.66
‑
0.61(m,2h);[m+na]
+
637.
[0429]
步骤14:(2s,3r,4r,5s,6r)
‑2‑
(7
‑
氯
‑6‑
(4
‑
环丙基苄基)
‑
2,3
‑
二氢苯并呋喃
‑4‑
基)
‑6‑
(羟甲基)四氢
‑
2h
‑
吡喃
‑
3,4,5
‑
三醇(化合物c28)
[0430]
向(2r,3r,4r,5s,6s)
‑2‑
(乙酰氧基甲基)
‑6‑
(7
‑
氯
‑6‑
(4
‑
环丙基苄基)
‑
2,3
‑
二氢苯并呋喃
‑4‑
基)四氢
‑
2h
‑
吡喃
‑
3,4,5
‑
三基三乙酸酯(1.5g,2.44mmol)在thf/meoh(5.4ml/10.8ml;0.15m)中的溶液添加4m naoh水溶液(2.8ml)。将反应混合物在室温下搅拌1.5小时。将溶液冷却至0℃,然后用1n hcl中和。将反应溶液用etoac和水稀释。分离有机层,水层用etoac萃取两次。将合并的有机层用mgso4干燥,过滤并真空浓缩,得到粗制标题化合物。
[0431]
将粗制标题化合物在甲苯(8ml)中的悬浮液在40℃下加热30分钟(粘稠溶液
‑
>澄清溶液
‑
>产生白色固体)并冷却至室温。将浆液在过滤漏斗中过滤,并用2个滤饼体积的甲苯洗涤滤饼。将湿滤饼真空干燥,得到1.0g(2.24mmol;定量)标题化合物。
[0432]
根据上述步骤1至14的合成途径,实施例1的最终化合物的总产率计算为约12%。
[0433]1h nmr(400mhz,cd3od)δ7.02(d,j=8.0hz,2h),6.92(d,j=8.0hz,2h),6.81(s,1h),4.59(t,j=8.8hz,2h),4.11(d,j=9.2hz,1h),3.96(abq,av
ab
=19.0hz,j
ab
=15.2hz,2h),3.87
‑
3.84(m,1h),3.67
‑
3.63(m,1h),3.47
‑
3.37(m,3h),3.35
‑
3.33(m,3h),1.85
‑
1.79(m,1h),0.91
‑
0.86(m,2h),0.61
‑
0.57(m,2h);[m+na]
+
469.
[0434]
实施例2:(2s,3r,4r,5s,6r)
‑2‑
(7
‑
氯
‑6‑
(4
‑
环丙基苄基)
‑
2,3
‑
二氢苯并呋喃
‑4‑
基)
‑6‑
(羟甲基)四氢
‑
2h
‑
吡喃
‑
3,4,5
‑
三醇的合成
[0435][0436]
步骤1:(3r,4s,5r,6r)
‑2‑
(7
‑
氯
‑6‑
(4
‑
环丙基苄基)
‑
2,3
‑
二氢苯并呋喃
‑4‑
基)
‑
3,4,5
‑
三(三甲基甲硅烷氧基)
‑6‑
((三甲基甲硅烷氧基)甲基)四氢
‑
2h
‑
吡喃
‑2‑
醇(化合物c41)
[0437]
在
‑
78℃,在氮气氛下,向4
‑
溴
‑7‑
氯
‑6‑
(4
‑
环丙基苄基)
‑
2,3
‑
二氢苯并呋喃(化合物c40,5.00g,13.8mmol)在四氢呋喃(140ml)中的溶液逐滴添加正丁基锂(2.5m在己烷中,8.28ml,20.7mmol)。在相同温度下进行搅拌5分钟后,向混合物逐滴添加四氢呋喃中的tms
‑
保护的内酯(化合物c11;7.70g,16.6mmol)的溶液30分钟。在相同温度下将反应混合物搅拌1小时。在0℃下将反应混合物用饱和nh4cl水溶液(300ml)猝灭,并用etoac萃取。将有机层用na2so4干燥,过滤,并真空浓缩,获得作为黄色油状物的粗标题化合物(10.3g,定量)。粗残余物不经进一步纯化用于下一步骤。
[0438]
步骤2:(3r,4r,5s,6r)
‑2‑
(7
‑
氯
‑6‑
(4
‑
环丙基苄基)
‑
2,3
‑
二氢苯并呋喃
‑4‑
基)
‑6‑
(羟甲基)四氢
‑
2h
‑
吡喃
‑
3,4,5
‑
三醇(化合物c43)
[0439]
在
‑
78℃下,向在ch2cl2(70ml)和ch3cn(70ml)中的粗制(3r,4s,5r,6r)
‑2‑
(7
‑
氯
‑6‑
(4
‑
环丙基苄基)
‑
2,3
‑
二氢苯并呋喃
‑4‑
基)
‑
3,4,5
‑
三(三甲基甲硅烷氧基)
‑6‑
((三甲基甲硅烷氧基)甲基)四氢
‑
2h
‑
吡喃
‑2‑
醇(10.3g)添加三乙基硅烷(8.8ml,55.2mmol)和tmsotf(10ml,55.2mmol)。在
‑
78℃下进行搅拌1小时后,在0℃下将反应混合物用水(200ml)猝灭,并用ch2cl2(300ml)萃取。将有机层用na2so4干燥,过滤,并真空浓缩,获得作为黄色油状物的粗制标题化合物(6.3g,定量)。粗制残余物不经进一步纯化用于下一步骤。
[0440]
步骤3:(2r,3r,4r,5s,6s)
‑2‑
(乙酰氧基甲基)
‑6‑
(7
‑
氯
‑6‑
(4
‑
环丙基苄基)
‑
2,3
‑
二氢苯并呋喃
‑4‑
基)四氢
‑
2h
‑
吡喃
‑
3,4,5
‑
三基三乙酸酯(化合物c27)
[0441]
在室温下,向(3r,4r,5s,6r)
‑2‑
(7
‑
氯
‑6‑
(4
‑
环丙基苄基)
‑
2,3
‑
二氢苯并呋喃
‑4‑
基)
‑6‑
(羟甲基)四氢
‑
2h
‑
吡喃
‑
3,4,5
‑
三醇(6.3g,13.8mmol)在ch2cl2(140ml)中的溶液添加dmap(0.84g,6.9mmol)和ac2o(13.0ml,13.8mmol)。在室温下进行搅拌18小时后,将反应混合物用水(120ml)猝灭,并用dcm(200ml)进行萃取。用nahco3水溶液(100ml)洗涤有机层,用na2so4干燥,过滤,并真空浓缩。将异丙醇(20ml)中的浆化残余物在80℃下加热10分钟并冷却至室温。然后,过滤所得沉淀物并真空浓缩,获得作为白色固体的β
‑
形式的标题化合物(4.52g,7.35mmol,53%)。
[0442]1h nmr(400mhz,cdcl3)δ7.04
‑
7.02(m,2h),6.98
‑
6.95(m,2h),6.53(s,1h),5.29
‑
5.24(m,1h),5.18
‑
5.12(m,2h),4.71
‑
4.65(m,2h),4.31
‑
4.26(m,1h),4.25
‑
4.22(m,1h),4.15
‑
4.11(m,1h),4.05
‑
3.91(m,2h),3.79
‑
3.74(m,ih),3.40
‑
3.35(m,2h),2.06(s,3h),2.05(s,3h),1.99(s,3h),1.88
‑
1.81(m,1h),1.66(s,3h),0.94
‑
0.89(m,2h),0.66
‑
0.61(m,2h);[m+na]+637.
[0443]
步骤4:(2s,3r,4r,5s,6r)
‑2‑
(7
‑
氯
‑6‑
(4
‑
环丙基苄基)
‑
2,3
‑
二氢苯并呋喃
‑4‑
基)
‑6‑
(羟甲基)四氢
‑
2h
‑
吡喃
‑
3,4,5
‑
三醇(化合物c28)
[0444]
向(2r,3r,4r,5s,6s)
‑2‑
(乙酰氧基甲基)
‑6‑
(7
‑
氯
‑6‑
(4
‑
环丙基苄基)
‑
2,3
‑
二氢苯并呋喃
‑4‑
基)四氢
‑
2h
‑
吡喃
‑
3,4,5
‑
三基三乙酸酯(4.52g,7.35mmol)在meoh(70ml)中的溶液添加naome(25wt%,0.35ml)。在室温下进行搅拌18小时后,将反应混合物真空浓缩,用水(200ml)稀释,并用etoac(300ml)萃取。将有机层用na2so4干燥,过滤,并真空浓缩。通过在甲苯中重结晶使残余物纯化,获得作为黄色固体的β
‑
形式的标题化合物(3.16g,96%)。
[0445]1h nmr(400mhz,cd3od)δ7.02(d,j=8.0hz,2h),6.92(d,j=8.0hz,2h),6.81(s,1h),4.59(t,j=8.8hz,2h),4.11(d,j=9.2hz,1h),3.96(abq,δνab=19.0hz,jab=15.2hz,2h),3.87
‑
3.84(m,1h),3.67
‑
3.63(m,1h),3.47
‑
3.37(m,3h),3.35
‑
3.33(m,3h),1.85
‑
1.79(m,1h),0.91
‑
0.86(m,2h),0.61
‑
0.57(m,2h);[m+na]+469.
[0446]
实施例3:(2s,3r,4r,5s,6r)
‑2‑
(7
‑
氯
‑6‑
(4
‑
甲氧基苄基)
‑
2,3
‑
二氢苯并呋喃
‑4‑
基)
‑6‑
(羟甲基)四氢
‑
2h
‑
噻喃
‑
3,4,5
‑
三醇的合成
[0447][0448]
步骤1:(3r,4s,5s,6r)
‑
3,4,5
‑
三(苄氧基)
‑6‑
((苄氧基)甲基)
‑2‑
(7
‑
氯
‑6‑
(4
‑
甲氧基苄基)
‑
2,3
‑
二氢苯并呋喃
‑4‑
基)四氢
‑
2h
‑
噻喃
‑2‑
醇(化合物c52)
[0449]
重复实施例4中步骤8的合成过程,只是使用4
‑
溴
‑7‑
氯
‑
2,3
‑
二氢苯并呋喃
‑6‑
甲醛作为原料,并使用(4
‑
甲氧基苯基)溴化镁(化合物c48)作为格氏试剂,以提供(4
‑
溴
‑7‑
氯
‑
2,3
‑
二氢苯并呋喃
‑6‑
基)(4
‑
甲氧基苯基)甲醇(化合物c49)。然后重复实施例4中步骤9的合成过程,只是使用(4
‑
溴
‑7‑
氯
‑
2,3
‑
二氢苯并呋喃
‑6‑
基)(4
‑
甲氧基苯基)甲醇(化合物c49)作为原料,以提供4
‑
溴
‑7‑
氯
‑6‑
(4
‑
甲氧基苄基)
‑
2,3
‑
二氢苯并呋喃(化合物c50)。
[0450]
在
‑
78℃氮气氛下,向4
‑
溴
‑7‑
氯
‑6‑
(4
‑
甲氧基苄基)
‑
2,3
‑
二氢苯并呋喃(化合物c50,859mg,2.43mmol)在四氢呋喃(8ml)中的溶液逐滴添加正丁基锂(2.5m在己烷中,1.3ml,3.24mmol),并在相同温度下将混合物搅拌1.5小时。然后逐滴添加四氢呋喃(4ml)中的(3r,4s,5s,6r)
‑
3,4,5
‑
三(苄氧基)
‑6‑
((苄氧基)甲基)四氢
‑
2h
‑
噻喃
‑2‑
酮(化合物c51,898mg,1.62mmol,该化合物是通过参考h.driguez和b.henrissat,tetrahedron lett.1981,22,5061
‑
5062;kakinuma,h.,等,j.med.chem.2010,53,3247
‑
3261而合成的)的溶液,并在相同温度下将混合物搅拌1.5小时。通过添加饱和氯化铵溶液猝灭反应混合物。添加完成后,将溶液逐渐升至室温。分离有机层,并将水层用乙酸乙酯萃取。将合并的有机层用盐水洗涤,用硫酸镁干燥,过滤并真空浓缩,提供粗制化合物(3r,4s,5s,6r)
‑
3,4,5
‑
三(苄氧基)
‑6‑
((苄氧基))甲基)
‑2‑
(7
‑
氯
‑6‑
(4
‑
甲氧基苄基)
‑
2,3
‑
二氢苯并呋喃
‑4‑
基)四氢
‑
2h
‑
噻喃
‑2‑
醇(定量产率)。
[0451]
步骤2:7
‑
氯
‑6‑
(4
‑
甲氧基苄基)
‑4‑
((2s,3r,4r,5s,6r)
‑
3,4,5
‑
三(苄氧基)
‑6‑
((苄氧基)甲基)四氢
‑
2h
‑
噻喃
‑2‑
基)
‑
2,3
‑
二氢苯并呋喃(化合物c53)
[0452]
在
‑
20℃下,向经搅拌的内酯(c52)在二氯甲烷(16ml)中的溶液添加三乙基硅烷(1.6ml,9.72mmol),随后添加三氟化硼二乙醚合物(0.8ml,6.48mmol),添加速率使得反应温度维持在
‑
20℃至0℃。经过1.5小时将溶液升温至0℃,然后用饱和碳酸氢钠溶液猝灭。在减压下除去有机挥发物后,将残余物在乙酸乙酯和水之间分配。将水层用乙酸乙酯萃取后,
将合并的有机层用盐水洗涤,用硫酸镁干燥,过滤并真空浓缩。将粗残余物通过硅胶色谱法(硅胶,3%至25%乙酸乙酯在己烷中)纯化,以提供作为白色固体的粗制化合物7
‑
氯
‑6‑
(4
‑
甲氧基苄基)
‑4‑
((2s,3r,4r,5s,6r)
‑
3,4,5
‑
三(苄氧基)
‑6‑
((苄氧基)甲基)四氢
‑
2h
‑
噻喃
‑2‑
基)
‑
2,3
‑
二氢苯并呋喃(603mg,46%,2个步骤)。[m+na]+835。
[0453]
步骤3:(2s,3r,4r,5s,6r)
‑2‑
(7
‑
氯
‑6‑
(4
‑
甲氧基苄基)
‑
2,3
‑
二氢苯并呋喃
‑4‑
基)
‑6‑
(羟甲基)四氢
‑
2h
‑
噻喃
‑
3,4,5
‑
三醇(化合物c47)
[0454]
在0℃下,向7
‑
氯
‑6‑
(4
‑
甲氧基苄基)
‑4‑
((2s,3r,4r,5s,6r)
‑
3,4,5
‑
三(苄氧基)
‑6‑
((苄氧基)甲基)四氢
‑
2h
‑
噻喃
‑2‑
基)
‑
2,3
‑
二氢苯并呋喃(c53,570mg,0.70mmol)在二氯甲烷(8ml)中的溶液添加bcl3(1.0m,在二氯甲烷中,2.8ml)。在0℃下将反应混合物搅拌1小时。用甲醇猝灭反应后,在减压下蒸发溶剂。通过反相制备型hplc(gilson,sunfire
tm prep,5%至50%乙腈在水中梯度)的纯化提供作为白色固体的标题化合物(18mg,6%)。
[0455]1h nmr(400mhz,cdcl3)δ7.07(d,j=8.4hz,2h),6.79(d,j=8.8hz,2h),6.76(s,1h),4.63(td,j=8.0,1.6hz,2h),3.95(s,2h),3.92(d,j=3.6hz,1h),3.79
‑
3.75(m,3h),3.74(s,3h),3.71(d,j=6.4hz,1h),3.56(dd,j=10.0,8.8hz,1h),3.42
‑
3.35(m,2h),3.24
‑
3.20(m,1h),3.01
‑
2.96(m,1h),0.90(t,j=7.2hz,3h);[m+na]
+
475.
[0456]
实施例4:(2s,3r,4r,5s,6r)
‑2‑
(7
‑
氯
‑6‑
(4
‑
乙氧基苄基)
‑
2,3
‑
二氢苯并呋喃
‑4‑
基)
‑6‑
(甲硫基)四氢
‑
2h
‑
吡喃
‑
3,4,5
‑
三醇的制备
[0457][0458]
步骤1:((3as,5s,6r,6as)
‑5‑
(羟甲基)
‑
2,2
‑
二甲基四氢呋喃并[3,2
‑
d][1,3]二氧杂环戊烯
‑6‑
醇
[0459]
在室温下,向l
‑
(
‑
)
‑
木糖(19.15g,127.5mmol)和mgso4(30.72g,255.0mmol)在丙酮(190ml)中的悬浮液添加浓h2so4(1.9ml)。12小时后,过滤反应混合物(其中所有l
‑
(
‑
)
‑
木糖已被消耗),并将合并的固体用丙酮(每次洗涤20ml)洗涤两次。在进行搅拌的同时用nh4oh溶液将黄色滤液中和至约ph 9。通过过滤除去悬浮的固体。浓缩滤液,以获得作为黄色油状物的双缩丙酮化合物中间体。将黄色油状物悬浮于水(5ml)中,然后用1n hcl水溶液将ph从9调节至2。在室温下将反应混合物搅拌12小时。通过添加25%(w/w)水中的k3po4来中和所得混合物直至ph变为约7。将混合物用etoac萃取。将有机层用mgso4干燥,过滤,并真空
浓缩。将粗制产物通过硅胶柱色谱法纯化,获得作为黄色油状物的标题化合物(12.63g,52%)。
[0460]1h nmr(400mhz,cd3od)δ5.88(d,j=4.0hz,1h),4.47(d,j=4.0hz,1h),4.18
‑
4.14(m,1h),4.11(d,j=2.8hz,1h),3.83
‑
3.71(m,2h),1.45(s,3h),1.29(s,3h).
[0461]
步骤2:(3as,5r,6s,6as)
‑6‑
羟基
‑
2,2
‑
二甲基四氢呋喃并[3,2
‑
d][1,3]二氧杂环戊烯,5
‑
甲酸
[0462]
在室温下,向((3as,5s,6r,6as)
‑5‑
(羟甲基)
‑
2,2
‑
二甲基四氢呋喃并[3,2
‑
d][1,3]二氧杂环戊烯
‑6‑
醇(14.6g,76.7mmol)、nahco3(19.3g,230.3mmol)和nabr(1.6g,15.4mmol)在丙酮/水(120ml/40ml)中的溶液添加tempo(0.24g,1.5mmol)。将混合物冷却至0℃,然后以小份添加三氯异氰尿酸(17.8g,76.7mmol)。在室温下搅拌悬浮液12小时。添加甲醇(2.0ml),并在室温下将混合物搅拌2小时。将混合物过滤并用丙酮(两次,每次洗涤20ml)洗涤。真空除去有机溶剂,将水层用etoac萃取,并将有机层真空浓缩。向其添加丙酮,并将混合物过滤。将滤液浓缩,获得作为浅黄色固体的期望的酸(9.0g,58%)。
[0463]1h nmr(400mhz,cd3od)δ5.98(d,j=3.6hz,1h),4.71(d,j=3.2hz,1h),4.51(d,j=3.6hz,1h),4.36(d,j=3.6hz,1h),1.45(s,3h),1.31(s,3h).
[0464]
步骤3:((3as,5r,6s,6as)
‑6‑
羟基
‑
2,2
‑
二甲基四氢呋喃并[3,2
‑
d][1,3]二氧杂环戊烯
‑2‑
基)(吗啉代)甲酮(化合物c56)
[0465]
在室温下,向(3as,5r,6s,6as)
‑6‑
羟基
‑
2,2
‑
二甲基四氢呋喃并[3,2
‑
d][1,3]二氧杂环戊烯
‑5‑
甲酸(9.0g,44.2mmol)和hbtu(25.1g,66.3mmol,n,n,n
′
,n
′‑
四甲基
‑
o
‑
(1h
‑
苯并三唑
‑1‑
基)脲六氟磷酸盐)在四氢呋喃中的悬浮液添加4
‑
甲基吗啉(7.3ml,66.3mmol)。1小时后,在室温下向混合物添加吗啉(5.8ml,66.3mmol)。12小时后,过滤所得混合物,并将滤饼用四氢呋喃洗涤。将滤液真空浓缩,并将粗制材料通过硅胶柱色谱法纯化,获得作为黄色固体的标题化合物(5.8g,48%)。
[0466]1h nmr(400mhz,cd3od)δ6.01(d,j=3.6hz,1h),5.10(s,1h),4.59(d,j=2.4hz,1h),4.57(d,j=3.6hz,1h),4.47(d,j=2.4hz,1h),3.85
‑
3.62(m,6h),3.53
‑
3.49(m,2h),1.49(s,3h),1.33(s,3h).[m+h]
+
274.
[0467]
步骤4:(7
‑
氯
‑6‑
(4
‑
乙氧基苄基)
‑
2,3
‑
二氢苯并呋喃
‑4‑
基)((3as,5r,6s,6as)
‑6‑
羟基
‑
2,2
‑
二甲基四氢呋喃并[2,3
‑
d][1,3]二氧杂环戊烯
‑5‑
基)甲酮(化合物c58)
[0468]
在
‑
78℃下,向4
‑
溴
‑7‑
氯
‑6‑
(4
‑
乙氧基苄基)
‑
2,3
‑
二氢苯并呋喃(化合物c57,0.7g,1.90mmol)在thf(17.5ml)中的溶液添加正丁基锂(2.5m在己烷中的溶液,0.9ml,2.28mmol)。1小时后,在
‑
78℃下,向混合物逐滴添加在thf(8.0ml)中的((3as,5r,6s,6as)
‑6‑
羟基
‑
2,2
‑
二甲基四氢呋喃并[3,2
‑
d][1,3]二氧杂环戊烯
‑5‑
基)(吗啉代)甲酮(化合物c56,0.17g,0.63mmol)。4小时后,将所得混合物用饱和nh4cl溶液猝灭,并用etoac进行萃取。将有机层用mgso4干燥,过滤,并真空浓缩。将粗制产物通过硅胶柱色谱法(biotage isolera
tm flash纯化系统)纯化,获得标题化合物(0.13g,43%);[m+h]
+
475。
[0469]
步骤5:(3as,5s,6r,6as)
‑5‑
((s)
‑
(7
‑
氯
‑6‑
(4
‑
乙氧基苄基)
‑
2,3
‑
二氢苯并呋喃
‑4‑
基)(羟基)甲基)
‑
2,2
‑
二甲基四氢呋喃并[2,3
‑
d][1,3]二氧杂环戊烯
‑6‑
醇(化合物c59)
[0470]
向(7
‑
氯
‑6‑
(4
‑
乙氧基苄基)
‑
2,3
‑
二氢苯并呋喃
‑4‑
基)((3as,5r,6s,6as)
‑6‑
羟基
‑
2,2
‑
二甲基四氢呋喃[2,3
‑
d][1,3]二氧杂环戊烯
‑5‑
基)甲酮(0.13g,0.27mmol)在甲醇
(18ml)中的溶液添加cecl3.7h2o(0.12g,0.32mmol),并在室温下将混合物搅拌直至所有固体溶解。然后,将混合物冷却至
‑
78℃,并以小份添加nabh4(0.012g,0.32mmol)。在
‑
78℃下将混合物搅拌2小时,缓慢升温至0℃,并用饱和nh4cl溶液猝灭。在减压下浓缩混合物以除去ch3oh,用etoac萃取,并用饱和nacl溶液洗涤。将有机层用mgso4干燥,过滤,并真空浓缩。将粗制标题化合物在高真空下干燥,并不需纯化作为白色固体(0.13g)用于下一步骤。
[0471]
[m+na]+499。
[0472]
步骤6:(3s,4r,5s,6s)
‑6‑
(7
‑
氯
‑6‑
(4
‑
乙氧基苄基)
‑
2,3
‑
二氢苯并呋喃
‑4‑
基)四氢
‑
2h
‑
吡喃
‑
2,3,4,5
‑
四基四乙酸酯(化合物c60)
[0473]
在100℃下,将(3as,5s,6r,6as)
‑5‑
((s)
‑
(7
‑
氯
‑6‑
(4
‑
乙氧基苄基)
‑
2,3
‑
二氢苯并呋喃
‑4‑
基)(羟基)甲基]
‑
2,2
‑
二甲基四氢呋喃并[2,3
‑
d][1,3]二氧杂环戊烯
‑6‑
醇(0.13g,0.27mmol)在acoh/水(4.0/2.5ml)中的溶液搅拌12小时。将所得混合物冷却至室温并在减压下浓缩。在0℃下用在吡啶(0.7ml)中的乙酸酐(0.2ml,2.16mmol)处理粗制油状物。在室温下将混合物搅拌8小时。将所得混合物用水猝灭,用etoac进行萃取,并用盐水进行洗涤。将有机层用mgso4干燥,过滤,并真空浓缩。将残余物通过硅胶柱色谱法纯化,获得作为白色固体的标题化合物(0.16g,96%)。[m+na]
+
627。
[0474]
步骤7:(2s,3s,4r,5s,6r)
‑2‑
(7
‑
氯
‑6‑
(4
‑
乙氧基苄基)
‑
2,3
‑
二氢苯并呋喃
‑4‑
基)
‑6‑
(甲硫基)四氢
‑
2h
‑
吡喃
‑
3,4,5
‑
三基三乙酸酯(化合物c61)
[0475]
向(3s,4r,5s,6s)
‑6‑
(7
‑
氯
‑6‑
(4
‑
乙氧基苄基)
‑
2,3
‑
二氢苯并呋喃
‑4‑
基)四氢
‑
2h
‑
吡喃
‑
2,3,4,5
‑
四基四乙酸酯(160mg,0.26mmol)和硫脲(39mg,0.52mmol)在1,4
‑
二氧六环(3.1ml)中的溶液添加tmsotf(70μl,0.39mmol),并将反应混合物在80℃下加热4小时。将混合物冷却至室温,向其添加mei(40μl,0.65mmol)和dipea(452μl,2.60mmol),并进行搅拌3小时。将所得混合物用etoac稀释并用水洗涤。将有机层用mgso4干燥,过滤,并真空浓缩。将粗制标题化合物在高真空下干燥,并不需纯化作为白色固体(150mg,48%)用于下一步骤。[m+na]
+
615。
[0476]
步骤8:(2s,3r,4r,5s,6r)
‑2‑
(7
‑
氯
‑6‑
(4
‑
乙氧基苄基)
‑
2,3
‑
二氢苯并呋喃
‑4‑
基)
‑6‑
(甲硫基)四氢
‑
2h
‑
吡喃
‑
3,4,5
‑
三醇(化合物c54)
[0477]
在室温下,向(2s,3s,4r,5s,6r)
‑2‑
(7
‑
氯
‑6‑
(4
‑
乙氧基苄基)
‑
2,3
‑
二氢苯并呋喃
‑4‑
基)
‑6‑
(甲硫基)四氢
‑
2h吡喃
‑
3,4,5
‑
三基三乙酸酯(150mg,0.25mmol)在ch3oh(0.7ml)中的悬浮液添加naome(催化量,25%溶液,在ch3oh中)。20小时后,将所得混合物真空浓缩。将粗制产物用etoac稀释并通过膜过滤。将粗制产物通过制备型hplc(gilson system,ch3cn/h2o)纯化,获得标题化合物(41mg,35%)。
[0478]1h nmr(400mhz,cdcl3)δ7.09(d,j=8.4hz,2h),6.80(d,j=8.8hz,2h),6.68(s,1h),4.69
‑
4.64(m,2h),4.35(d,j=10.0hz,1h),4.20(d,j=9.2hz,1h),4.02
‑
3.88(m,4h),3.67
‑
3.65(m,2h),3.61
‑
3.58(m,1h),3.56
‑
3.52(m,1h),3.42
‑
3.40(m,2h),3.29
‑
3.27(m,2h),2.17(s,3h),1.40(t,j=7.0hz,3h);[m+na]
+
489.
[0479]
实施例5:(2s,3r,4r,5s,6r)
‑2‑
(7
‑
氯
‑6‑
(4
‑
乙基苄基)
‑
2,3
‑
二氢苯并呋喃
‑4‑
基)
‑6‑
(甲硫基)四氢
‑
2h
‑
吡喃
‑
3,4,5
‑
三醇的制备
[0480]
通过重复实施例4的合成过程获得标题化合物,不同之处在于使用4
‑
溴
‑7‑
氯
‑6‑
(4
‑
乙基苄基)
‑
2,3
‑
二氢苯并呋喃代替步骤4中的4
‑
溴
‑7‑
氯
‑6‑
(4
‑
乙氧基苄基)
‑
2,3
‑
二氢
苯并呋喃。
[0481]1h nmr(400mhz,cdcl3)δ7.10
‑
7.90(m,4h),6.71(s,1h),4.68
‑
4.62(m,2h),4.33(d,j=9.6hz,1h),4.18(d,j=9.2hz,1h),4.07
‑
4.00(m,2h),3.63
‑
3.57(m,3h),3.52
‑
3.49(m,1h),3.39
‑
3.37(m,2h),3.27
‑
3.25(m,2h),2.60(q,j=7.4hz,2h),2.15(s,3h),1.20(t,j=7.6hz,3h);[m+na]+473.
[0482]
实施例6:(2s,3r,4r,5s,6r)
‑2‑
(7
‑
氯
‑6‑
(4
‑
环丙基苄基)
‑
2,3
‑
二氢苯并呋喃
‑4‑
基)
‑6‑
(羟甲基)四氢
‑
2h
‑
吡喃
‑
3,4,5
‑
三醇的制备
[0483][0484]
步骤1:(3r,4s,5s,6r)
‑2‑
[7
‑
氯
‑6‑
(4
‑
环丙基苄基)
‑
2,3
‑
二氢苯并呋喃
‑4‑
基])
‑6‑
(羟甲基)
‑2‑
甲氧基四氢
‑
2h
‑
吡喃
‑
3,4,5
‑
三醇(化合物c42)
[0485]
通过以下1a或1b的途径合成标题化合物。
[0486]
(1a)在室温、氮气下,向反应容器逐滴添加4
‑
溴
‑7‑
氯
‑6‑
(4
‑
环丙基苄基)
‑
2,3
‑
二氢苯并呋喃(250g,0.687mol)、(3r,4s,5r,6r)
‑
3,4,5
‑
三((三甲基甲硅烷基)氧基)
‑6‑
(((三甲基甲硅烷基)氧基)甲基)四氢
‑
2h
‑
吡喃
‑2‑
酮(642g,1.38mol)和无水四氢呋喃(2.00l),并使混合物完全溶解。将反应容器冷却至
‑
78℃后,向其逐滴添加正丁基锂(550ml,2.0m,在己烷中,1.38mol)1小时,同时保持内部温度在
‑
60℃或更低。一旦完成正丁基锂的逐滴添加,在
‑
78℃下将混合物进一步搅拌40分钟。向反应混合物逐滴添加浓盐酸/甲醇(152ml/1750ml)溶液20分钟,同时保持内部温度在
‑
30℃或更低。一旦完成逐滴添加,将反应容器移至室温并搅拌18小时。确定反应完成后,将反应容器冷却至0℃,向其添加饱和nahco3水溶液(2.5l),用ph计将ph调节至9至10,然后使用真空浓缩器除去反应溶剂。将浓缩物用etoac(2.5l)、蒸馏水(1.25l)和盐水(1.25l)稀释,并分层。然后,将有机层合并,并将水层用etoac(2
×
1.25l)萃取。将有机层合并,并用蒸馏水(2.5l)和盐水(2.5l)清洗。将有机层用mgso4(50g)干燥并过滤。然后,将滤液在减压下浓缩以除去溶剂。重复两次将残余物用甲苯(500ml)稀释并通过在减压下蒸馏进行除去的操作,获得作为黄色液体的标题化合物(328g)。粗制残余物不经进一步纯化用于下一步骤。
[0487]
(1b)在室温、氮气下,向反应容器逐滴添加4
‑
溴
‑7‑
氯
‑6‑
(4
‑
环丙基苄基)
‑
2,3
‑
二
氢苯并呋喃(10.0g,27.5mol)、(3r,4s,5r,6r)
‑
3,4,5
‑
三((三甲基甲硅烷基)氧基)
‑6‑
(((三甲基甲硅烷基)氧基)甲基)四氢
‑
2h
‑
吡喃
‑2‑
酮(25.7g,54.9mol)和无水四氢呋喃(80ml),并使混合物完全溶解。将反应容器冷却至
‑
78℃后,向其逐滴添加正丁基锂(22.1ml,2.5m,在己烷中,54.9mmol)15分钟,同时保持内部温度在
‑
60℃或更低。一旦完成正丁基锂的逐滴添加,在
‑
78℃下将混合物进一步搅拌30分钟。向反应混合物逐滴添加浓盐酸/甲醇(7.01ml/70ml)溶液10分钟,同时保持内部温度在
‑
30℃或更低。一旦完成逐滴添加,将反应容器移至室温并搅拌18小时。确定反应完成后,将反应容器冷却至0℃,向其添加饱和nahco3水溶液(60ml),用ph计将ph调节至9至10,然后使用真空浓缩器除去反应溶剂。将浓缩物用etoac(60ml)、蒸馏水(60ml)和盐水(60ml)稀释,并分层。然后,将有机层合并,并将水层用etoac(2
×
30ml)萃取。将有机层合并,并用蒸馏水(60ml)和盐水(60ml)清洗。将有机层用mgso4(5g)干燥,过滤,并将滤液在减压下浓缩以除去溶剂。将残余物用甲苯(50ml)稀释,然后,在室温下搅拌己烷的同时将甲苯溶液缓慢逐滴添加到己烷(200ml)。在相同温度下将所得悬浮液搅拌1小时,然后真空过滤。将所得滤液用己烷(10ml)洗涤,然后在真空烘箱(40℃)中干燥直至其水分含量通过卡尔
‑
费休(karl
‑
fischer)分析变为1%或更低,获得作为黄色固体的标题化合物(12.6g,96%)。
[0488]1h nmr(500mhz,cdcl3):δ7.02(d,j=8.0hz,2h),6.92(d,j=8.0hz,2h),6.81(s,1h),4.64(m,1h),4.57(m,1h),4.05(d,j=15.0hz,1h),3.96(d,j=15.0hz,1h),3.93(dd,j=11.8,3.0hz,1h),3.87(m,2h),3.65(m,2h),3.51(m,1h),3.30(d,j=9.5hz,1h),3.14(s,3h),1.83(m,1h),0.91(m,2h),0.63(m,2h);lc
‑
ms:[m
‑
ome]
+
445.
[0489]
步骤2:(3r,4r,5s,6r)
‑2‑
[7
‑
氯
‑6‑
(4
‑
环丙基苄基)
‑
2,3
‑
二氢苯并呋喃
‑4‑
基]
‑6‑
(羟甲基)四氢
‑
2h
‑
吡喃
‑
3,4,5
‑
三醇(化合物c43)
[0490]
在反应容器中,在室温、氮气下进行搅拌的同时,使根据步骤1中1a的反应途径获得的粗制残余物(328g,0.687mol)的ch2cl2/ch3cn(=v/v,1∶1,5.00l)溶液完全溶解。将反应容器冷却至
‑
50℃,然后逐滴添加et3sih(329ml,2.08mol)和bf3‑
oet2(170ml,1.37mol)10分钟,同时保持内部温度在
‑
45℃或更低。将反应混合物缓慢升温至
‑
10℃,保持1小时,并将所得混合物升温至0℃。在0℃下进行搅拌3小时后,向反应混合物添加饱和nahco3水溶液(5.5l),并用ph计将ph调节至7.0至7.5。使用真空浓缩器从混合物中除去有机溶剂,并将浓缩物用etoac(2.5l)稀释。然后,分离出有机层。将水层用etoac(2
×
125l)稀释并进行萃取。将所有有机层合并,用无水mgso4(50g)干燥,过滤,然后将滤液在减压下浓缩以除去溶剂。将残余物真空干燥,获得作为黄色液体的标题化合物(307g)。由此获得的粗制残余物不经进一步纯化用于下一步骤。
[0491]1h nmr(500mhz,cd3od):δ7.04(d,j=8.0hz,2h),6.93(d,j=8.0hz,2h),6.83(s,1h),4.61(t,j=9.0hz,2h),4.13(d,j=9.0hz,1h),3.99(d,j=15.0hz,1h),3.94(d,j=15.0hz,1h),3.87(d,j=12.0hz,1h),3.66(m,1h),3.44(m,1h),3.41(t,j=9.0hz,2h),3.36(m,2h),3.31(m,ih),1.83(m,1h),0.91(m,2h),0.63(m,2h);lc
‑
ms:[m+na]
+
469.
[0492]
步骤3:(2r,3r,4r,5s,6s)
‑2‑
(乙酰氧基甲基)
‑6‑
[7
‑
氯
‑6‑
(4
‑
环丙基苄基)
‑
2,3
‑
二氢苯并呋喃
‑4‑
基]四氢
‑
2h
‑
吡喃
‑
3,4,5
‑
三基三乙酸酯(化合物c27)
[0493]
在室温下,在搅拌步骤2中的粗制残余物(307g,0.687mol)的ch2cl2(5.00l)溶液的同时,向反应容器依次逐滴添加dmap(101g,0.825mol)和ac2o(520ml,5.50mol)。在室温下
将所得的黄色反应混合物搅拌2小时。将反应混合物用蒸馏水(500ml)猝灭。将混合物分层。储存有机层,并将水层用二氯甲烷(2
×
1.25l)萃取。将所有有机层合并并用1nhcl水溶液(2.5l)和盐水(2.5l)清洗。将有机层用mgso4(50g)干燥,过滤,然后将滤液在减压下浓缩。将残余物用meoh(2.5l)稀释,并在室温下搅拌30分钟。在减压下过滤所得固体,并将滤液用meoh(500ml)清洗。将经过滤的固体干燥,获得作为白色固体的标题化合物(357g,产率:84%,纯度:>97.6%)。
[0494]1h nmr(500mhz,cdcl3):δ7.04(d,j=8.0hz,2h),6.95(d,j=8.0hz,2h),6.53(s,1h),5.24(dd,j=9.5,9.5hz,1h),5.12(m,2h),4.67(m,2h),4.29(d,j=10.0hz,1h),4.24(dd,j=12.5,4.5hz,1h),4.13(dd,j=12.5,1.5hz,1h),4.02(d,j=15.0hz,1h),3.92(d,j=15.0hz,1h),3.77(m,1h),3.38(m,2h),2.07(s,3h),2.06(s,3h),1.99(s,3h),1.84(m,1h),1.66(s,3h),0.92(m,2h),0.63(m,2h);lc
‑
ms:[m+na]
+
637.
[0495]
步骤4:(2s,3r,4r,5s,6r)
‑2‑
[7
‑
氯
‑6‑
(4
‑
环丙基苄基)
‑
2,3
‑
二氢苯并呋喃
‑4‑
基]
‑6‑
(羟甲基)四氢
‑
2h
‑
吡喃
‑
3,4,5
‑
三醇(化合物c28)
[0496]
在室温下,在搅拌悬浮液的同时,向(2r,3r,4r,5s,6s)
‑2‑
(乙酰氧基甲基)
‑6‑
(7
‑
氯
‑6‑
(4
‑
环丙基苄基)
‑
2,3
‑
二氢苯并呋喃
‑4‑
基)四氢
‑
2h
‑
吡喃
‑
3,4,5
‑
三基三乙酸酯(357g,0.580mol)的thf/meoh(=v/v,1∶2,4.28l)悬浮液逐滴添加4m naoh水溶液(668ml,2.67mol)20分钟。在室温下将所得悬浮液进一步搅拌2小时。将反应容器冷却至0℃后,向反应混合物缓慢地逐滴添加1n hcl水溶液(1.18l),并用ph计将ph调节至6.5至7.0。使用真空浓缩器除去反应溶剂,并将浓缩物用etoac(5.36l)和蒸馏水(5.36l)稀释。将混合物分层。储存有机层,并将水层用etoac(2
×
1.79l)萃取。将所有有机层合并并用蒸馏水(1.79l)清洗。将有机层用mgso4(710g)干燥,过滤,然后将滤液在减压下浓缩,获得粗制标题化合物。
[0497]
将粗制标题化合物用etoac(3.89l)稀释,然后在回流下搅拌30分钟以完全溶解固体。然后,将生成物冷却至室温。向所得悬浮液逐滴添加异丙醚(1.29l)10分钟,并进行搅拌30分钟(包括逐滴添加时间)。重复添加异丙醚的过程两次。然后,将反应容器冷却至0℃并搅拌30分钟。在减压下过滤所得固体,并将滤液用etoac/异丙醚(=v/v,1∶1,357ml)的混合液清洗。将经过滤的固体在真空烘箱(40℃,18小时)中干燥,获得作为白色固体的标题化合物(236g,产率:92%,纯度:>99.7%)。此外,计算出根据上述步骤1至4合成途径的实施例6的最终化合物的总产率为约77%。
[0498]1h nmr(500mhz,cd3od):δ7.04(d,j=8.0hz,2h),6.93(d,j=8.0hz,2h),6.83(s,1h),4.61(t,j=9.0hz,2h),4.13(d,j=9.0hz,1h),3.99(d,j=15.0hz,1h),3.94(d,j=15.0hz,1h),3.87(d,j=12.0hz,1h),3.66(m,1h),3.44(m,1h),3.41(t,j=9.0hz,2h),3.36(m,2h),3.31(m,1h),1.83(m,1h),0.91(m,2h),0.63(m,2h);lc
‑
ms:[m+na]
+
469.
[0499]
实施例7:(2s,3r,4r,5s,6r)
‑2‑
(7
‑
氯
‑6‑
(4
‑
环丙基苄基)
‑
2,3
‑
二氢苯并呋喃
‑4‑
基)
‑6‑
(羟甲基)四氢
‑
2h
‑
吡喃
‑
3,4,5
‑
三醇的制备
[0500][0501]
步骤1:(3r,4s,5s,6r)
‑2‑
[7
‑
氯
‑6‑
(4
‑
环丙基苄基)
‑
2,3
‑
二氢苯并呋喃
‑4‑
基]
‑6‑
(羟甲基)
‑2‑
甲氧基四氢
‑
2h
‑
吡喃
‑
3,4,5
‑
三醇(化合物c42)
[0502]
根据实施例6的步骤1中1b的反应途径,使用4
‑
溴
‑7‑
氯
‑6‑
(4
‑
环丙基苄基)
‑
2,3
‑
二氢苯并呋喃(21.6g)获得作为浅黄色固体的标题化合物(26.2g,93%)。
[0503]
步骤2:(3r,4s,5r,6r)
‑6‑
(乙酰氧基甲基)
‑2‑
[7
‑
氯
‑6‑
(4
‑
环丙基苄基)
‑
2,3
‑
二氢苯并呋喃
‑4‑
基]
‑2‑
甲氧基四氢
‑
2h
‑
吡喃
‑
3,4,5
‑
三基三乙酸酯(化合物c44)
[0504]
在室温下,在搅拌(3r,4s,5s,6r)
‑2‑
[7
‑
氯
‑6‑
(4
‑
环丙基苄基)
‑
2,3
‑
二氢苯并呋喃
‑4‑
基]
‑6‑
(羟甲基)
‑2‑
甲氧基四氢
‑
2h
‑
吡喃
‑
3,4,5
‑
三醇(26.2g,55.0mmol)的ch2cl2(65.5l)溶液的同时,向反应容器依次逐滴添加dmap(8.06g,66.0mmol)和ac2o(51.8ml,550mol)。在室温下将所得黄色反应混合物搅拌6小时。将反应混合物用蒸馏水(50ml)猝灭。将混合物分层。储存有机层,并将水层用ch2cl2(2
×
30ml)萃取。将所有有机层合并并用1n hcl水溶液(50ml)和盐水(30ml)清洗。将有机层用mgso4(6g)干燥,过滤,然后将滤液在减压下浓缩。将残余物用meoh(100ml)稀释,并在室温下搅拌12小时。将所得固体在减压下过滤,并将滤液用meoh(30ml)清洗。将经过滤的固体干燥,获得作为白色固体的标题化合物(29.2g,71%,总共经过两个步骤)。
[0505]1h nmr(500mhz,cdcl3):δ7.01(d,j=8.5hz,2h),6.95(d,j=8.5hz,2h),6.65(s,1h),5.53(dd,j=10.0,9.5hz,1h),5.19(dd,j=10.0,9.5hz,1h),4.99(d,j=10.0hz,1h),4.63(m,2h),4.34(dd,j=12.0,4.5hz,1h),4.14(dd,j=12.0,2.0hz,1h),4.05(d,j=15.5hz,1h),4.01(m,1h),3.99(d,j=15.5hz,1h),3.49(m,1h),3.33(m,1h),3.17(s,3h),2.08(s,3h),2.05(s,3h),1.94(s,3h),1.83(m,1h),1.61(s,3h),0.90(m,2h),0.62(m,2h).
[0506]
步骤3:(2r,3r,4r,5s,6s)
‑2‑
(乙酰氧基甲基)
‑6‑
[7
‑
氯
‑6‑
(4
‑
环丙基苄基)
‑
2,3
‑
二氢苯并呋喃
‑4‑
基]四氢
‑
2h
‑
吡喃
‑
3,4,5
‑
三基三乙酸酯(化合物c27)
[0507]
在反应容器中,通过在室温下搅拌使(3r,4s,5r,6r)
‑6‑
(乙酰氧基甲基)
‑2‑
[7
‑
氯
‑6‑
(4
‑
环丙基苄基)
‑
2,3
‑
二氢苯并呋喃
‑4‑
基]
‑2‑
甲氧基四氢
‑
2h
‑
吡喃
‑
3,4,5
‑
三基三乙酸酯(5.00g,7.75mmol)的ch2cl2/ch3cn(=v/v,1∶10,550ml)溶液完全溶解。将反应容器
冷却至0℃后,向其依次逐滴添加et3sih(9.89ml,62.0mmol)和bf3‑
oet2(4.96ml,40.3mmol)10分钟,同时保持内部温度在5℃或更低。在5℃或更低的温度下将反应混合物搅拌1小时。然后,将反应混合物升温至10℃并进一步搅拌2小时。向反应混合物添加饱和nahco3水溶液,并使用ph计确定ph是否为7.0至7.5。然后,使用真空浓缩器除去有机溶剂。将浓缩物用etoac(50ml)稀释,并分离出有机层。将水层用etoac(2
×
30ml)萃取。将所有有机层合并,用mgso4(5g)干燥,过滤,然后将滤液在减压下浓缩。将残余物溶解在ch2cl2(5ml)中,向其添加己烷(10ml),并在室温下进行搅拌5分钟。向混合物添加异丙醚(20ml)并进行搅拌30分钟。向所得悬浮液进一步添加异丙醚(20ml)。将反应容器冷却至0℃并搅拌2小时。将所得固体在减压下过滤,将滤液用异丙醚(10ml)清洗。将经过滤的固体真空干燥,获得作为白色固体的标题化合物(4.38g,92%)。
[0508]
步骤4:(2s,3r,4r,5s,6r)
‑2‑
[7
‑
氯
‑6‑
(4
‑
环丙基苄基)
‑
2,3
‑
二氢苯并呋喃
‑4‑
基]
‑6‑
(羟甲基)四氢
‑
2h
‑
吡喃
‑
3,4,5
‑
三醇(化合物c28)
[0509]
根据如上所述的实施例1的步骤4中描述的合成方法,使用(2r,3r,4r,5s,6s)
‑2‑
(乙酰氧基甲基)
‑6‑
[7
‑
氯
‑6‑
(4
‑
环丙基苄基)
‑
2,3
‑
二氢苯并呋喃
‑4‑
基]四氢
‑
2h
‑
吡喃
‑
3,4,5
‑
三基三乙酸酯(4.38g,7.12mmol),以获得粗制标题化合物。将残余物用etoac(45.3ml)稀释,然后在回流下搅拌10分钟以完全溶解固体。将生成物冷却至室温。向所得悬浮液逐滴添加异丙醚(15.1ml)10分钟,并进行搅拌30分钟(包括逐滴添加时间)。重复添加异丙醚的过程两次。然后,将反应容器冷却至0℃并搅拌30分钟。将所得固体在减压下过滤,并将滤液用异丙醚(10ml)清洗。将经过滤的固体在真空烘箱(40℃,18小时)中干燥,获得作为白色固体的标题化合物(2.93g,产率:92%,纯度:>99.5%)。计算出根据上述步骤1至4合成途径的实施例2的最终化合物的总产率为约60%。
[0510]
实验实施例1:根据重排反应条件评价产率
[0511][0512]
(1)使用基于胺的溶剂或不使用溶剂的反应
[0513]
将作为原料的3
‑
(烯丙氧基)
‑5‑
溴
‑2‑
氯苯甲酸甲酯(化合物c30)在160℃下进行重排反应,以获得4
‑
烯丙基
‑5‑
溴
‑2‑
氯
‑3‑
羟基苯甲酸甲酯(化合物c31)并计算其产率。
[0514]
此时,如下表1中所示调节原料的量和反应条件。使用5m二乙胺(dea)作为反应溶剂,或在没有任何溶剂的情况下进行反应。
[0515]
结果,产率最大为67%,并且没有任何进一步的提高。
[0516]
[表1]
[0517][0518]
(2)添加路易斯酸的反应
[0519]
将作为原料的3
‑
(烯丙氧基)
‑5‑
溴
‑2‑
氯苯甲酸甲酯(化合物c30)溶解在溶剂中,并进行重排反应。然后,将残余物通过硅胶柱色谱法纯化,以获得4
‑
烯丙基
‑5‑
溴
‑2‑
氯
‑3‑
羟基苯甲酸甲酯(化合物c31),并计算其产率。
[0520]
此时,如下表2中所示调节反应溶剂、反应温度和路易斯酸,并在添加或不添加路易斯酸的情况下进行反应。
[0521]
结果,在添加二异丁基氯化铝((i
‑
bu)2alcl)或二乙基氯化铝(et2alcl)(其为路易斯酸)的情况下,产率提高至80%。
[0522]
[表2]
[0523]
测试编号路易斯酸反应溶剂反应温度产率1bcl3ch2cl2‑
25℃至室温17%2(i
‑
bu)2alcl己烷/ch2cl20℃至室温71%3ticl4甲基咪唑室温未反应4ticl4ch2cl2室温未反应5
‑
水室温未反应
6bf3.oet2ch2cl20℃至室温未反应7alcl3ch2cl20℃至室温35%8(i
‑
bu)2alcl己烷/ch2cl20℃至室温80%9et2alcl己烷/ch2cl20℃至室温72%
[0524]
实验实施例2:根据环化反应条件评价产率
[0525]
(1)使用vilsmeier试剂进行环化
[0526][0527]
制备vilsmeier试剂(制备试剂a);在室温下,向n,n
‑
二甲基甲酰胺的溶液(5.6ml,74.24mmol)添加socl2(5.3ml,72.24mmol)。在40℃下将混合物搅拌2小时,然后真空浓缩,获得vilsmeier试剂,其为吸湿性白色固体。
[0528]
制备vilsmeier试剂(制备试剂b);在室温下,向n,n
‑
二甲基甲酰胺的溶液(2.9ml,37.07mmol)添加socl2(2.7ml,37.07mmol)。在40℃下将混合物搅拌2小时,然后真空浓缩,获得vilsmeier试剂,其为吸湿性白色固体。
[0529]
然后,向vilsmeier试剂在dmf中的混合物缓慢添加在dmf中的5
‑
溴
‑2‑
氯
‑3‑
羟基
‑4‑
(2
‑
羟乙基)苯甲酸甲酯(化合物c33)。在进行搅拌1小时的同时将该混合物进行环化反应,获得4
‑
溴
‑7‑
氯
‑
2,3
‑
二氢苯并呋喃
‑6‑
甲酸甲酯(化合物c34),并计算其产率。
[0530]
此时,如下表3中所示调节原料、反应温度等,并进行反应。
[0531]
结果,以总体上地高产率获得环化产物。但是,在原料不纯的情况下,产物以低产率回收。
[0532]
[表3]
[0533][0534]
(2)使用离去基团进行环化
[0535][0536]
向作为原料的5
‑
溴
‑2‑
氯
‑3‑
羟基
‑4‑
(2
‑
羟乙基)苯甲酸甲酯(化合物c33)添加甲磺酰氯(mscl)和溶剂。将混合物进行环化反应,获得4
‑
溴
‑7‑
氯
‑
2,3
‑
二氢苯并呋喃
‑6‑
甲酸甲酯(化合物c34),并计算其产率。
[0537]
此时,如下表4中所示调节反应物的使用量、添加方法和反应条件,并进行反应。
[0538]
结果,有时以高产率获得期望的化合物。但是,存在由mscl的当量控制和逐滴添加引起的不便。
[0539]
[表4]
[0540][0541]
(3)使用卤化物或mitsunobu反应进行环化
[0542]
将作为原料的5
‑
溴
‑2‑
氯
‑3‑
羟基
‑4‑
(2
‑
羟乙基)苯甲酸甲酯(化合物c33)进行mitsunobu反应或通过卤化物进行环化反应,以获得4
‑
溴
‑7‑
氯
‑
2,3
‑
二氢苯并呋喃
‑6‑
甲酸甲酯(化合物c34),并计算其产率。
[0543]
此时,其中将三苯基膦(pph3)、咪唑、碘(i2)和甲苯组合的试剂用于进行使用卤化物的环化反应,并且将其中将三苯基膦(pph3)、偶氮二甲酸二异丙酯(diad)和四氢呋喃(thf)组合的试剂用于进行mitsunobu反应。
[0544]
结果,获得达约60%的产率。但是,不便之处在于必须除去三苯基氧化膦,其是反应后所产生的副产物。
[0545]
实验实施例3:根据还原反应条件评价产率
[0546][0547]
向作为原料的4
‑
溴
‑7‑
氯
‑
2,3
‑
二氢苯并呋喃
‑6‑
甲酸甲酯(化合物c34)添加nabh4(3当量),并且还添加或不添加路易斯酸。然后,在室温下使混合物在溶剂中进行还原反应,
以获得(4
‑
溴
‑7‑
氯
‑
2,3
‑
二氢苯并呋喃
‑6‑
基)甲醇(化合物c35),并计算其产率。另外,还测量了副产物1(化合物c35
‑
1)的量。
[0548]
(1)如下表5中所示调节反应溶剂和路易斯酸的添加量,并进行反应。结果是,在使用thf/etoh(1∶1)作为反应溶剂的情况下,可以以最高产率获得产物,并且即使在仅使用nabh4而不使用路易斯酸的情况下也获得了95%的良好产率。
[0549]
[表5]
[0550][0551]
(2)不添加路易斯酸,如下表6中所示调节反应溶剂和nabh4的添加量,并进行反应。结果,在thf/etoh(1∶1)或thf/etoh(2∶1)作为反应溶剂的条件下,以3当量的量添加nabh4的情况下,获得95%的良好产率,没有副产物。
[0552]
[表6]
[0553][0554][0555]
(3)如下表7中所示调节反应溶剂、nabh4的添加量和路易斯酸的添加量,并进行反应。结果,在仅使用thf或使用thf/i
‑
proh作为反应溶剂的情况下,反应不进行。另一方面,在使用thf/乙醇作为反应溶剂的情况下,即使仅使用nabh4而不使用路易斯酸,也获得95%
或更高的良好产率。
[0556]
[表7]
[0557][0558]
实验实施例4:晶形的制备和分析
[0559]
通过在不同溶剂中的结晶过程,从根据本发明的方法制备的化合物(具体地,根据实施例1中的步骤1至14获得的粗制(2s,3r,4r,5s,6r)
‑2‑
(7
‑
氯
‑6‑
(4
‑
环丙基苄基)
‑
2,3
‑
二氢苯并呋喃
‑4‑
基)
‑6‑
(羟甲基)四氢
‑
2h
‑
吡喃
‑
3,4,5
‑
三醇(化合物c28))中制备晶体并对其进行分析。
[0560]
对于xrd光谱,根据普通方法,通过用cu
‑
k
α
辐射(波长,辐射(波长,)照射,使用x射线衍射仪测量粉末x射线衍射。
[0561]
对于差示扫描量热法(dsc),使用差示扫描量热计以+1℃/分钟的速率进行测量。
[0562]
(1)使用甲苯溶剂制备晶体
[0563]
使用甲苯的结晶类似于实施例1中步骤14过程的后面部分中所述。具体地,通过在40℃下加热30分钟来溶解粗制化合物c28的甲苯(8倍于c28的重量)溶液,然后使其冷却至室温。在室温下形成悬浮液后,进一步进行搅拌30分钟。将所得沉淀物过滤,用甲苯(滤液体积的2倍)洗涤,然后在真空烘箱(50℃,12小时)中干燥,获得白色晶体(产率:91.8%)。
[0564]
所制备晶体的xrd光谱显示出如图1中所示的晶形(晶形a),并且衍射角(2θ)、晶面间距(d)和特征峰的相对强度(i/i
o
×
100)总结于表8中。
[0565]
[表8]
[0566][0567]
如图2中所示,dsc光谱使得确定晶体的熔化吸热峰成为可能。
[0568]
(2)使用乙酸乙酯溶剂制备晶体
[0569]
使用乙酸乙酯的结晶类似于实施例6中步骤4的后面部分中所述。具体地,通过在回流下进行搅拌来溶解粗制化合物c28的乙酸乙酯(15倍于c28的重量)溶液,然后使其冷却至室温。在室温下形成悬浮液后,进一步进行搅拌30分钟。经过30分钟向所得混合物逐滴添加异丙醚(15倍于c28的重量),并在室温下进一步进行搅拌30分钟。将所得沉淀物过滤,用0℃乙酸乙酯(2倍于c28的重量)洗涤,然后在真空烘箱(50℃,12小时)中干燥,获得白色晶体(产率:88.3%)。所制备晶体的xrd光谱分析显示出与实验实施例4的(1)中相同的晶形(晶形a)。
[0570]
(3)使用二氯甲烷溶剂制备晶体
[0571]
通过在40℃下加热来溶解粗制化合物c28的二氯甲烷(170倍于c28的重量)溶液,然后使其冷却至室温。在室温下将混合物搅拌30分钟。向反应混合物添加实施例6的步骤4中获得的化合物c28(10mg,晶种),然后进行搅拌12小时。将所得沉淀物过滤,用二氯甲烷(两倍于c28的重量)洗涤,然后在真空烘箱(50℃,12小时)中干燥,获得白色晶体(产率:50.1%)。所制备晶体的xrd光谱分析显示出与实验实施例4的(1)中相同的晶形(晶形a)。
[0572]
(4)使用丙酮溶剂制备晶体
[0573]
通过在回流下进行加热来溶解粗制化合物c28的丙酮(35倍于c28的重量)溶液,然后使其冷却至室温。在室温下形成悬浮液后,进一步进行搅拌3小时。将所得沉淀物过滤,用丙酮(滤液体积的2倍)洗涤,然后在真空烘箱(50℃,12小时)中干燥,获得白色晶体(产率:38.2%)。所制备晶体的xrd光谱分析显示出与实验实施例4的(1)中相同的晶形(晶形a)。
[0574]
(5)使用乙腈溶剂制备晶体
[0575]
通过在60℃下加热来溶解粗制化合物c28的乙腈(10倍于c28的重量)溶液,然后使其冷却至室温。在室温下形成悬浮液后,进一步进行搅拌1小时。将所得沉淀物过滤,用乙腈(滤液体积的2倍)洗涤,然后在真空烘箱(50℃,12小时)中干燥,获得白色晶体(产率:39.4%)。所制备晶体的xrd光谱分析显示出与实验实施例4的(1)中相同的晶形(晶形a)。
[0576]
(6)使用2
‑
丙醇溶剂制备晶体
[0577]
通过在60℃下加热来溶解粗制化合物c28的2
‑
丙醇(10倍于c28的重量)溶液,然后使其冷却至室温。在室温下形成悬浮液后,进一步进行搅拌30分钟。向反应混合物进一步逐滴添加2
‑
丙醇(5倍于c28的重量),然后进行搅拌30分钟。将所得沉淀物过滤,用2
‑
丙醇(滤液体积的2倍)洗涤,然后在真空烘箱(50℃,12小时)中干燥,获得白色晶体(产率:9.5%)。
所制备晶体的xrd光谱分析显示出与实验实施例4的(1)中相同的晶形(晶形a)。
[0578]
(7)使用四氢呋喃/二氯甲烷溶剂制备晶体
[0579]
通过在室温下进行搅拌30分钟来溶解粗制化合物c28的四氢呋喃(5倍于c28的重量)溶液。在减压下浓缩反应混合物以除去有机溶剂。将浓缩的残余物用二氯甲烷(30倍于c28的重量)稀释并在室温下搅拌。形成悬浮液后,进一步进行搅拌30分钟。将所得沉淀物过滤,用二氯甲烷(滤液体积的2倍)洗涤,然后在真空烘箱(50℃,12小时)中干燥,获得白色晶体(产率:65.3%)。所制备晶体的xrd光谱分析显示出与实验实施例4的(1)中相同的晶形(晶形a)。
[0580]
(8)使用四氢呋喃/正己烷溶剂制备晶体
[0581]
通过在室温下进行搅拌30分钟来溶解粗制化合物c28的四氢呋喃(5倍于c28的重量)溶液。向反应混合物逐滴添加正己烷(10倍于c28的重量),并进行搅拌1小时。向所得悬浮液进一步逐滴添加正己烷(10倍于c28的重量),然后进行搅拌30分钟。向反应悬浮液重复逐滴添加正己烷(5倍于c28的重量),并进行搅拌30分钟。将所得沉淀物过滤,用正己烷(滤液体积的5倍)洗涤,然后在真空烘箱(50℃,12小时)中干燥,获得白色晶体(产率:99.6%)。所制备晶体的xrd光谱分析显示出与实验实施例4的(1)中相同的晶形(晶形a)。
[0582]
(9)使用甲醇/蒸馏水溶剂制备晶体
[0583]
通过在室温下进行搅拌30分钟来溶解粗制化合物c28的甲醇(5倍于c28的重量)溶液。向反应混合物逐滴添加蒸馏水(10倍于c28的重量),然后进行搅拌30分钟。向所得悬浮液中进一步逐滴添加蒸馏水(10倍于c28的重量),然后进行搅拌1小时。将所得沉淀物过滤,用蒸馏水(滤液体积的2倍)洗涤,然后在真空烘箱(50℃,12小时)中干燥,获得白色晶体(产率:100%)。
[0584]
所制备晶体的xrd光谱显示出与图3中所示相同的晶形(晶形b),并且衍射角(2θ)、晶面间距(d)和特征峰的相对强度(i/i
o
×
100)总结于表9中。
[0585]
[表9]
[0586][0587]
如图4中所示,dsc光谱使得确定晶体的熔化吸热峰成为可能。
[0588]
(10)使用甲醇/正己烷溶剂制备晶体
[0589]
通过在室温下进行搅拌30分钟来溶解粗制化合物c28的甲醇(5倍于c28的重量)溶液。向反应混合物逐滴添加正己烷(15倍于c28的重量),并进行搅拌30分钟。向所得悬浮液进一步逐滴添加正己烷(10倍于c28的重量),并进行搅拌1小时。将所得沉淀物过滤,用正己烷(滤液体积的5倍)洗涤,然后在真空烘箱(50℃,12小时)中干燥,获得白色晶体(产率:97.1%)。所制备晶体的xrd光谱分析显示出与实验实施例4的(9)中相同的晶形(晶形b)。
[0590]
(11)使用甲醇/二氯甲烷/正己烷溶剂制备晶体
[0591]
通过在室温下进行搅拌30分钟溶解粗制化合物c28的二氯甲烷/甲醇(20∶1倍于c28的重量)溶液。向反应混合物逐滴添加正己烷(10倍于c28的重量),并进行搅拌1小时。向所得悬浮液进一步逐滴添加正己烷(10倍于c28的重量),并进行搅拌30分钟。向反应悬浮液重复逐滴添加正己烷(5倍于c28的重量),并进行搅拌30分钟。将所得沉淀物过滤,用正己烷(滤液体积的5倍)洗涤,然后在真空烘箱(50℃,12小时)中干燥,获得白色晶体(产率:99.2%)。所制备晶体的xrd光谱分析显示出与实验实施例4的(9)中相同的晶形(晶形b)。
[0592]
(12)使用四氢呋喃/甲苯溶剂制备晶体
[0593]
通过在室温下进行搅拌30分钟来溶解粗制化合物c28的四氢呋喃(5倍于c28的重量)溶液。在减压下浓缩反应混合物以除去有机溶剂。将经浓缩的残余物用甲苯(30倍于c28的重量)稀释,然后在室温下搅拌1小时。将所得沉淀物过滤,用甲苯(滤液体积的2倍)洗涤,然后在真空烘箱(50℃,12小时)中干燥,获得白色晶体(产率:76.1%)。
[0594]
所制备晶体的xrd光谱显示出与图5中所示相同的晶形(晶形c),并且衍射角(2θ)、晶面间距(d)和特征峰的相对强度(i/i
o
×
100)总结于表10中。
[0595]
[表10]
[0596][0597]
如图6中所示,dsc光谱使得确定晶体的熔化吸热峰成为可能。
[0598]
(13)使用乙醇/蒸馏水/正己烷溶剂制备晶体
[0599]
通过在50℃下加热来溶解粗制化合物c28的乙醇(5倍于c28的重量)溶液,然后使其冷却至室温。向反应混合物逐滴添加蒸馏水(10倍于c28的重量)并进行搅拌1小时。向所得悬浮液进一步逐滴添加蒸馏水(10倍于c28的重量),并进行搅拌30分钟。向反应悬浮液逐滴添加正己烷(1倍于c28的重量),并进行搅拌1小时。将所得沉淀物过滤,用蒸馏水(滤液体积的2倍)洗涤,然后在真空烘箱(50℃,12小时)中干燥,获得白色晶体(产率:95.8%)。所制备晶体的xrd光谱分析显示出与实验实施例4的(12)中相同的晶形(晶形c)。
[0600]
(14)使用乙醇/正己烷溶剂制备晶体
[0601]
通过在50℃下加热来溶解粗制化合物c28的乙醇(5倍于c28的重量)溶液,然后使其冷却至室温。向反应混合物逐滴添加正己烷(5倍于c28的重量),并进行搅拌30分钟。向所得悬浮液进一步逐滴添加正己烷(10倍于c28的重量),并进行搅拌30分钟。向反应悬浮液重复逐滴添加正己烷(5倍于c28的重量),并进行搅拌30分钟。将所得沉淀物过滤,用正己烷(滤液体积的5倍)洗涤,然后在真空烘箱(50℃,12小时)中干燥,获得白色晶体(产率:96.5%)。
[0602]
所制备晶体的xrd光谱显示出与图7中所示相同的晶形(晶形d),并且衍射角(2θ)、
晶面间距(d)和特征峰的相对强度(i/i
o
×
100)总结于表11中。
[0603]
[表11]
[0604][0605]
如图8中所示,dsc光谱使得确定晶体的熔化吸热峰成为可能。
[0606]
实验实施例5:确定晶形的稳定性
[0607]
使用实验实施例4的(2)中制备的晶形,在加速(温度:40
±
2℃,相对湿度:75
±
5%)和长期(温度:25
±
2℃,相对湿度:60
±
5%)的测试条件下,进行与其特性、鉴定测试、水分含量、比旋光度、有关物质、和含量相关的稳定性确定测试,持续3个月。稳定性确定测试的结果示于表12中。
[0608]
[表12]
[0609][0610]
(1)特性测试确定,晶形的特性在加速和长期条件下没有变化。
[0611]
(2)使用韩国药典中规定的一般测试方法中的红外分光光度法和液相色谱法进行鉴定测试,并确定该晶形在这两种测试方法中均显示出与标准产物相同的光谱。
[0612]
<分析条件>
[0613]
‑
柱:capcdell
‑
pak c18 mg(uspl1),250
×
4.6mm,5μm
[0614]
‑
温度:35℃
[0615]
‑
检测器:光二极管阵列(pda)检测器(测量波长:225nm)
[0616]
‑
流量:1.0ml/min
[0617]
‑
流动相:缓冲剂/甲醇(25∶75)
[0618]
‑
缓冲剂:通过将1.36g磷酸二氢钾溶解在1,000ml蒸馏水中,然后用磷酸将ph调节至3.0而获得的缓冲剂。
[0619]
(3)在xrd光谱的情况下,可以确定,晶形在加速和长期条件下没有变化。
[0620]
(4)在根据韩国药典规定的一般测试方法中的湿度测量方法进行测量的情况下,水分含量测试可以确定的结果是,在加速和长期条件下,晶形几乎没有表现出含水分特性。
[0621]
(5)比旋光度确定测试可以确定晶形在加速和长期条件下的结构稳定性。
[0622]
(6)通过韩国药典中规定的一般测试方法中的液相色谱法进行与有关物质相关的稳定性确定测试。样品溶液的分析时间为使得测量进行达主峰保持时间的三倍的时间。除了在空白测试溶液中出现的所有峰之外,根据计算表达式计算标准溶液(0.05mg/ml)和样品溶液(1mg/ml)的峰面积。结果,可以确定与有关物质相关的晶形在加速和长期条件下的稳定性。
[0623]
‑
分析条件:实验实施例5的(2)中的确定测试的液相色谱法分析条件
[0624]
‑
计算表达式:其他个别有关物质(%)=(测试样品中每个有关物质的峰面积
×
标准产物的使用量
×
标准产物的纯度)/(标准溶液的主峰峰面积
×
样品的使用量
×
稀释因子)。
[0625]
(7)对于含量测试,使用标准产物的甲醇溶液作为标准溶液(0.2mg/ml),并使用晶形的甲醇溶液作为样品溶液(1mg/ml)。通过韩国药典中规定的一般测试方法中的液相色谱法测试样品溶液和标准溶液,并且使用标准溶液和样品溶液的峰面积通过以下计算表达式进行计算。结果,可以确定在加速和长期条件下几乎没有表现出含量变化的晶形的稳定性。
[0626]
‑
分析条件:实验实施例5的(2)中的确定测试的液相色谱法分析条件
[0627]
‑
计算表达式:其他个别有关物质(%)=(测试样品主峰的峰面积
×
标准产物的使用量
×
标准产物的纯度)/[标准溶液主峰的峰面积
×
样品的使用量
×
(100
‑
样品水分含量)]。
[0628]
以下内容对应于母案申请中的原始权利要求书,现作为说明书的一部分并入此处:
[0629]
1.用于制备下式1a的化合物的方法,所述方法包括以下步骤:
[0630]
(1)使下式2的化合物与下式3的化合物反应,并使生成物进行环化反应,得到下式4的化合物;
[0631]
(2)使式4化合物进行醛基化或酰胺化,然后使生成物与下式5的化合物反应并进行还原,得到下式6的化合物;以及
[0632]
(3)使式6化合物与下式7的化合物反应,并进行脱保护和还原,
[0633][0634]
在所述式中,
[0635]
a是氧(o)或硫(s);
[0636]
n是1或2;
[0637]
pg是保护基;
[0638]
x
′
是卤素或c1‑7烷基;
[0639]
x、y和hal各自独立地为卤素;
[0640]
b是(b
‑
1)或(b
‑
2)
[0641]
其中ra、rb、rc和rd各自独立地为氢、卤素、羟基、巯基、氰基、硝基、氨基、羧基、氧代、c1‑7烷基、c1‑7烷硫基、c2‑7烯基、c2‑7炔基、c1‑7烷氧基、c1‑7烷氧基
‑
c1‑7烷基、c2‑7烯基
‑
c1‑7烷氧基、c2‑7炔基
‑
c1‑7烷氧基、c3‑
10
环烷基、c3‑7环烷硫基、c5‑
10
环烯基、c3‑
10
环烷氧基、c3‑
10
环烷氧基
‑
c1‑7烷氧基、苯基
‑
c1‑7烷基、c1‑7烷硫基
‑
苯基、苯基
‑
c1‑7烷氧基、单c1‑7烷基氨基或二c1‑7烷基氨基、单c1‑7烷基氨基
‑
c1‑7烷基或二c1‑7烷基氨基
‑
c1‑7烷基、c1‑7烷酰基、c1‑7烷酰基氨基、c1‑7烷基羰基、c1‑7烷氧基羰基、氨甲酰基、单c1‑7烷基氨甲酰基或二c1‑7烷基氨甲酰基、c1‑7烷基磺酰基氨基、苯基磺酰基氨基、c1‑7烷基亚磺酰基、c6‑
14
芳基硫烷基、c6‑
14
芳基磺酰基、c6‑
14
芳基、5至13元杂芳基、5至10元杂环烷基、5至10元杂环烷基
‑
c1‑7烷基、或5至10元杂环烷基
‑
c1‑7烷氧基;
[0642]
环c为c3‑
10
环烷基、c5‑
10
环烯基、c6‑
14
芳基、5至13元杂芳基、或5至10元杂环烷基;
[0643]
烷基、烯基、炔基和烷氧基各自独立地未经取代,或具有一个或更多个选自卤素、羟基、氰基、硝基、氨基、巯基、c1‑7烷基和c2‑7炔基的取代基;
[0644]
环烷基、环烯基、芳基、杂芳基和杂环烷基各自独立地未经取代,或具有一个或更多个选自卤素、羟基、氰基、硝基、氨基、巯基、c1‑4烷基和c1‑4烷氧基的取代基;并且
[0645]
杂芳基和杂环烷基各自独立地含有一个或更多个选自n、s和o的杂原子。
[0646]
2.根据项1所述的用于制备式1a化合物的方法,
[0647]
其中步骤(1)包括以下步骤:
[0648]
(i)使式2化合物与式3化合物反应,得到下式3a的化合物;
[0649]
(ii)使式3a化合物的烯丙基进行重排反应,并使生成物进行氧化或臭氧化反应,然后进行还原,得到下式3d的化合物;以及
[0650]
(iii)使式3d化合物进行环化反应,得到式4化合物,
[0651][0652]
在所述式中,n、x和y如项1中所限定。
[0653]
3.根据项2所述的用于制备式1a化合物的方法,
[0654]
其中所述环化反应是使用vilsmeier试剂的环化反应、使用离去基团的环化反应、使用卤化物的环化反应、或使用mitsunobu反应的环化反应。
[0655]
4.根据项1所述的用于制备式1a化合物的方法,
[0656]
其中步骤(2)包括使式4化合物进行醛基化,得到下式4a的化合物,然后使式4a化合物与式5化合物反应,
[0657]
[式4a]
[0658][0659]
在该式中,n、x和y如项1中所限定。
[0660]
5.根据项1所述的用于制备式1a化合物的方法,
[0661]
其中步骤(2)包括使式4化合物进行酰胺化,得到下式4b的化合物,然后使式4b化合物与式5化合物反应,
[0662]
[式4b]
[0663][0664]
其中,n、x和y如项1中所限定。
[0665]
6.根据项1所述的用于制备式1a化合物的方法,
[0666]
其中所述方法还包括步骤(3)之后的烷基化反应,并且x
′
是c1‑7烷基。
[0667]
7.根据项1所述的用于制备式1a化合物的方法,
[0668]
其中步骤(3)还包括在所述脱保护和还原过程之后或期间仅分离其中葡萄糖为β形式的化合物。
[0669]
8.根据项1所述的用于制备式1a化合物的方法,
[0670]
其中步骤(3)通过包括以下步骤的方法进行:
[0671]
(3a
‑
1)使式6化合物与式7化合物在正丁基锂、仲丁基锂、叔丁基锂或异丙基氯化
镁存在下进行反应,得到下式7a的化合物;
[0672]
(3a
‑
2)使式7a化合物在酸性条件下在甲醇存在下进行脱保护和甲基化反应,得到下式7d的化合物;
[0673]
(3b)还原式7b化合物,得到下式7e的化合物;以及
[0674]
(3c)向式7e化合物中引入保护基,在醇、乙酸乙酯或二氯甲烷中加热生成物,分离所得沉淀物并进行脱保护,得到仅β形式,
[0675][0676]
在所述式中,pg是保护基;并且a、b、n和x如项1所限定。
[0677]
9.根据项1所述的用于制备式1a化合物的方法,
[0678]
其中步骤(3)通过包括以下步骤的方法进行:
[0679]
(3a
′
)使式6化合物与式7化合物在正丁基锂、仲丁基锂、叔丁基锂或异丙基氯化镁存在下进行反应,并使生成物在酸性条件下在甲醇存在下进行脱保护和甲基化反应,无需单独进行纯化,得到下式7d的化合物;
[0680]
(3b
′
)还原式7d化合物,得到下式7e的化合物;以及
[0681]
(3c
′
)向式7e化合物中引入保护基以仅分离β形式,并进行脱保护,
[0682][0683]
在所述式中,a、b、n和x如项1中所限定。
[0684]
10.根据项1所述的用于制备式1a化合物的方法,
[0685]
其中步骤(3)通过包括以下步骤的方法进行:
[0686]
(3a
″
)使式6化合物与式7化合物在正丁基锂、仲丁基锂、叔丁基锂或异丙基氯化镁存在下进行反应,并使生成物在酸性条件下在甲醇存在下进行脱保护和甲基化反应,无需单独进行纯化,得到下式7d的化合物;
[0687]
(3b
″
)向式7d化合物中引入保护基,得到下式7f的化合物;以及
[0688]
(3c“)仅分离β形式的式7f化合物的并进行还原,然后进行脱保护,
[0689][0690]
在所述式中,pg是保护基;并且a、b、n和x如项1所限定。
[0691]
11.根据项9或10所述的用于制备式1a化合物的方法,其中相对于1当量的式6化合物,式7化合物和正丁基锂各自以1.5至2.5当量的量使用。
[0692]
12.根据项1所述的用于制备式1a化合物的方法,
[0693]
其中所述方法还包括在步骤(3)之后使用选自以下的溶剂进行结晶的步骤:甲苯、乙酸乙酯、二氯甲烷,四氢呋喃与二氯甲烷的混合物,以及四氢呋喃与正己烷的混合物。
[0694]
13.根据项1所述的用于制备式1a化合物的方法,
[0695]
其中n是1。
[0696]
14.根据项1所述的用于制备式1a化合物的方法,
[0697]
其中a是氧;
[0698]
n是1;
[0699]
x
′
是卤素;并且
[0700]
b是未经取代的或被一个或两个选自以下的取代基取代的苯基:卤素、羟基、氰基、硝基、氨基、巯基、c1‑7烷基、c3‑
10
环烷基和c1‑7烷氧基。
[0701]
15.用于制备下式1b的化合物的方法,所述方法包括以下步骤:
[0702]
(1)使下式2的化合物与下式3的化合物反应,并使生成物进行环化反应,得到下式4的化合物;
[0703]
(2)使式4化合物进行醛基化或酰胺化,然后使生成物与式5化合物反应并进行还原,得到下式6的化合物;
[0704]
(3)使式6化合物与下式8的化合物反应然后进行还原,得到下式9的化合物;
[0705]
(4)在酸性条件下使式9化合物的呋喃糖环形成吡喃糖环然后向其中引入保护基,得到下式10的化合物;以及
[0706]
(5)用硫脲处理式10化合物,使生成物与c1‑7烷基卤化物反应,然后进行还原,
[0707][0708]
在所述式中,
[0709]
r是c1‑7烷硫基;
[0710]
n是1或2;
[0711]
pg是保护基;
[0712]
x
′
是卤素或c1‑7烷基;
[0713]
x、y和hal各自独立地为卤素;
[0714]
b是(b
‑
1)或(b
‑
2)
[0715]
其中ra、rb、rc和rd各自独立地为氢、卤素、羟基、巯基、氰基、硝基、氨基、羧基、氧代、c1‑7烷基、c1‑7烷硫基、c2‑7烯基、c2‑7炔基、c1‑7烷氧基、c1‑7烷氧基
‑
c1‑7烷基、c2‑7烯基
‑
c1‑7烷氧基、c2‑7炔基
‑
c1‑7烷氧基、c3‑
10
环烷基、c3‑7环烷硫基、c5‑
10
环烯基、c3‑
10
环烷氧基、c3‑
10
环烷氧基
‑
c1‑7烷氧基、苯基
‑
c1‑7烷基、c1‑7烷硫基
‑
苯基、苯基
‑
c1‑7烷氧基、单c1‑7烷基氨基或二c1‑7烷基氨基、单c1‑7烷基氨基
‑
c1‑7烷基或二c1‑7烷基氨基
‑
c1‑7烷基、c1‑7烷酰基、c1‑7烷酰基氨基、c1‑7烷基羰基、c1‑7烷氧基羰基、氨甲酰基、单c1‑7烷基氨甲酰基或二c1‑7烷基氨甲酰基、c1‑7烷基磺酰基氨基、苯基磺酰基氨基、c1‑7烷基亚磺酰基、c6‑
14
芳基硫烷基、c6‑
14
芳基磺酰基、c6‑
14
芳基、5至13元杂芳基、5至10元杂环烷基、5至10元杂环烷基
‑
c1‑7烷基、或5至10元杂环烷基
‑
c1‑7烷氧基;
[0716]
环c为c3‑
10
环烷基、c5‑
10
环烯基、c6‑
14
芳基、5至13元杂芳基、或5至10元杂环烷基;
[0717]
烷基、烯基、炔基和烷氧基各自独立地未经取代,或具有一个或更多个选自卤素、羟基、氰基、硝基、氨基、巯基、c1‑7烷基和c2‑7炔基的取代基;
[0718]
环烷基、环烯基、芳基、杂芳基和杂环烷基各自独立地未经取代,或具有一个或更
多个选自卤素、羟基、氰基、硝基、氨基、巯基、c1‑4烷基和c1‑4烷氧基的取代基;并且
[0719]
杂芳基和杂环烷基各自独立地含有一个或更多个选自n、s和o的杂原子。
[0720]
16.下式c28的化合物的晶形,
[0721]
[式c28]
[0722][0723]
17.根据项16所述的晶形,
[0724]
其中在使用cu
‑
k
α
光源照射的情况下,所述晶形具有x射线衍射(xrd)光谱,所述光谱包含在6.2
°±
0.2
°
、7.2
°±
0.2
°
、8.8
°±
0.2
°
、17.6
°±
0.2
°
、19.0
°±
0.2
°
、22.5
°±
0.2
°
和25.1
°±
0.2
°
的衍射角(2θ)处的峰。
[0725]
18.根据项16所述的晶形,
[0726]
其中在使用cu
‑
k
α
光源照射的情况下,所述晶形具有x射线衍射(xrd)光谱,所述光谱包含在7.0
°±
0.2
°
、14.9
°±
0.2
°
、17.7
°±
0.2
°
、18.8
°±
0.2
°
、20.6
°±
0.2
°
、21.8
°±
0.2
°
和23.5
°±
0.2
°
的衍射角(2θ)处的峰。
[0727]
19.根据项16所述的晶形,
[0728]
其中在使用cu
‑
k
α
光源照射的情况下,所述晶形具有x射线衍射(xrd)光谱,所述光谱包含在5.6
°±
0.2
°
、7.3
°±
0.2
°
、15.7
°±
0.2
°
、17.2
°±
0.2
°
、18.9
°±
0.2
°
、21.2
°±
0.2
°
和21.9
°±
0.2
°
的衍射角(2θ)处的峰。
[0729]
20.根据项16所述的晶形,
[0730]
其中在使用cu
‑
k
α
光源照射的情况下,所述晶形具有x射线衍射(xrd)光谱,所述光谱包含在5.5
°±
0.2
°
、7.2
°±
0.2
°
、15.3
°±
0.2
°
、17.2
°±
0.2
°
、17.6
°±
0.2
°
、18.9
°±
0.2
°
和21.1
°±
0.2
°
的衍射角(2θ)处的峰。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1