有机物微粒的制造方法及有机物微粒的改性方法与流程
本发明涉及有机物微粒的制造方法及有机物微粒的改性方法。
背景技术:
通过使以往的有机物微粒化、特别是纳米微粒化,能够使其物性呈现新的功能,因此有机物的纳米微粒化技术在整个产业界中成为重要的主题。
有机物纳米微粒与数μm以上的固体相比,由于其极高的比表面积,富于反应性,在活性提高等的功能的提高的同时,作为可使原本具有的特性飞跃地提高的材料,期待着在广泛的技术领域中的应用。
为了发挥作为有机物纳米微粒所期待的特性,粒径的控制是必要的,一般地,有机物纳米微粒由于在溶剂中的溶解性提高,因此存在容易发生重结晶化引起的粒子生长、粒子的粗大化、颈缩这样的问题。
因此,如专利文献1那样,提案有通过在包含表面改性剂的不良溶剂中进行粉碎处理从而维持了粒子大小的生物体摄取物的制造方法,在专利文献9中提案有在铜酞菁等的青色有机颜料中加入有机溶剂而进行干式粉碎处理的有机颜料的制造方法。
但是,如该文献那样进行了机械的粉碎处理的情况下,多是粒子的结晶性降低,有时产生在溶剂中粉碎后的粒子凝聚、粒子粗大化的问题。另外,作为机械的粉碎手段,多采取球磨机、砂磨机、珠磨机等使用了介质的粉碎法,存在着难以避免来自装置的杂质的混入、进而为了使其成为目标粒径而需要大量的时间、动力等课题。
另外,在专利文献2中,作为用于制造在长期保存和/或高温暴露后显示粒径的稳定性和最小的结晶生长的纳米粒子组合物的方法,公开了添加表面稳定化剂、进行粉碎处理以及共沉淀法。但是,对于结晶性,停留在作为结晶相或者作为非晶质存在这样的记载,对于结晶度、晶型的控制等结晶性的控制没有示出。另外,如专利文献10中所示的、采用通过将在可溶解有机颜料的良溶剂中溶解了的有机颜料溶液和与上述良溶剂相比对于上述有机颜料的溶解度低的不良溶剂混合而使有机颜料微粒析出的方法制作的有机物纳米微粒多含有非晶质成分,存在在有机溶剂中容易粗大化的问题。
为了发挥作为有机物纳米微粒所期待的特性,不仅是粒径,而且结晶性、晶型这样的粒子的性状的控制是重要的。例如,在树脂等高分子化合物中,已知许多情况是树脂的结晶度强烈地影响二次凝聚的发生、滑动性这样的特性。因此,在专利文献3中公开了如下手法:作为经热处理的树脂微粒,通过在玻璃化转变温度以上且熔点以下的温度进行热处理,控制树脂微粒的结晶性。但是,对于该方法而言,必须进行长时间的热处理,寻求能够更为简便地控制结晶性的方法。
因此,在由本申请的申请人提出的专利文献4、8中,在对置配设的可接近·分离的至少一方相对于另一方相对地旋转的处理用面间的薄膜流体中使生物体摄取物微粒(专利文献4)或有机颜料微粒(专利文献8)析出。根据这些专利文献,能够容易地制造微细且均一的有机物纳米微粒,而且所析出的有机物纳米微粒的粒径、晶型的控制也成为可能。
但是,得到的有机物纳米微粒微细,因此与比表面积的増加相伴地溶解性提高,结果在粒子析出后由于粒子中的非晶质部分的作用,有时在溶剂中生长成粗大的粒子、有时发生粒子的颈缩。
另外,作为使微粒析出后不使析出了的微粒的粒径变化而使上述析出了的微粒的微晶径变化的技术,已知如专利文献5那样,通过使在对置配设了的、可接近·分离的、至少一方相对于另一方相对地进行旋转的至少2个处理用面之间形成的薄膜流体中析出了的粒子的核或微晶生长,得到均一且均质的粒子,具体地,使粒子析出后的流体从上述处理用面间排出后,调整使上述流体通过管状容器时的滞留时间、温度,由此控制粒子的微晶径。
但是,在专利文献5中,虽然通过滞留时间、温度来控制粒子的微晶径,但是关于进行了上述处理后的粒子,依然未能解决在溶剂中的粗大化、颈缩。
另外,在专利文献6中,为了制作测定用的试样,公开了在将分散剂溶解于有机溶剂的溶液中投入铜酞菁粉末,进行了分散处理,但这里使用的铜酞菁微粒原本结晶性极高,溶剂乃至分散剂对于有机物微粒的作用与本发明完全不同,由于出于测定目的来进行分散处理,因此不能说是对微粒进行改性、或控制微粒的性状。
在有机物中,尤其是有机颜料的情况下,由于有机颜料的色特性依赖于粒径、结晶性等粒子的性状,因此在有机颜料的用途的扩展的同时,希望精密地控制有机颜料和有机颜料微粒的性状。但是,没有用于控制结晶生长的决定性手段,对于经常使用的、与有机颜料的衍生物一起将有机颜料纳米粒子化的方法(专利文献7)而言,由于衍生物特有的色对实际使用的有机颜料的发色产生影响,因此存在难以使所要求的色发色的问题。
如以上所述,就有机物微粒而言,生物体摄取物、树脂、颜料等各种有机物已在各种领域中在各种用途中使用,但在所有的用途中粒径、结晶度等粒子的性状对其性能产生的影响大。因此,希望精密地控制有机物微粒的性状、且抑制其性状的变化直至利用实际的有机物微粒。
现有技术文献
专利文献
专利文献1:日本特表平8-501073号公报
专利文献2:日本特表2002-538199号公报
专利文献3:日本特开2007-291168号公报
专利文献4:日本再表2009-8391号公报
专利文献5:日本特开2014-23997号公报
专利文献6:日本特开2010-242104
专利文献7:国际公开wo2008/044519号公报小册子
专利文献8:日本特开2009-82902号公报
专利文献9:日本特开2004-244563号公报
专利文献10:日本特开2006-193681号公报
技术实现要素:
发明要解决的课题
因而,本发明的课题在于:提供为了有效利用有机物微粒本来的特性而可在抑制溶剂中的有机物微粒的生长且提高结晶性或者使其发生结晶转变的有机物微粒的制造方法。对于上述课题,发明人试错的结果发现:对对于至少其一部分包含非晶质的有机物微粒具有一部分溶解能力的溶剂,使将控制有机物微粒受到上述溶剂的作用的表面活性剂添加到溶剂中的粒子性状控制溶液作用于有机物微粒,由此可以基本上没有改变有机物微粒的粒径地提高有机物微粒的结晶度或者使有机物微粒发生结晶转变,完成了本发明。
用于解决课题的手段
即,本发明涉及有机物微粒的制造方法,其特征在于,在对于至少其一部分包含非晶质的有机物微粒具有一部分溶解能力的溶剂中添加表面活性剂,使上述溶剂作用于上述有机物微粒,由此基本上不改变上述有机物微粒的粒径地提高上述有机物微粒的结晶度。
另外,本发明涉及有机物微粒的制造方法,其特征在于,在对于至少其一部分包含非晶质的有机物微粒具有一部分溶解能力的溶剂中添加表面活性剂,使上述溶剂作用于上述有机物微粒,由此基本上不改变上述有机物微粒的粒径地使上述有机物微粒发生结晶转变。
另外,本发明涉及有机物微粒的制造方法,其特征在于,使添加了上述表面活性剂的上述溶剂作用于上述有机物微粒的前后的上述有机物微粒的粒径变化率(表面活性剂处理后(a)/表面活性剂处理前(b))为1~4。
另外,本发明涉及有机物微粒的制造方法,其特征在于,包含:工序1:将有机物微粒的原料溶液与用于从上述有机物微粒的原料溶液使至少1种的有机物微粒析出的析出溶剂l混合,使有机物微粒(p1)析出;和工序2:制备在对于上述有机物微粒具有一部分溶解能力的溶剂中添加了对于上述有机物微粒可进行生长抑制的表面活性剂的粒子性状控制溶液,使上述粒子性状控制溶液作用于上述有机物微粒(p1);上述工序2以控制上述有机物微粒(p1)的粒子性状的方式发挥作用。
另外,本发明涉及有机物微粒的制造方法,其特征在于,上述工序2以使上述有机物微粒(p1)的结晶度和晶型和粒径中的至少任一者变化的方式发挥作用。
另外,本发明涉及有机物微粒的制造方法,其特征在于,包含对于上述工序1中得到的上述有机物微粒(p1)进行清洗和/或溶剂置换的工序c,使上述粒子性状控制溶液作用于上述工序c中得到的有机物微粒(p2)。
另外,本发明涉及有机物微粒的制造方法,其特征在于,上述有机物微粒在至少其一部分中包含非晶质。
另外,本发明涉及有机物微粒的制造方法,其特征在于,使上述溶剂作用于上述有机物微粒时或者使上述粒子性状控制溶液作用于上述有机物微粒时进行搅拌处理,通过搅拌能量来控制上述有机物微粒、上述有机物微粒(p1)或上述有机物微粒(p2)的性状。
另外,本发明涉及有机物微粒的制造方法,其特征在于,上述有机物微粒为生物体摄取物。
另外,本发明涉及有机物微粒的制造方法,其特征在于,上述有机物微粒为树脂。
另外,本发明涉及有机物微粒的制造方法,其特征在于,上述有机物微粒为红色有机颜料、青色有机颜料等的有机颜料。
另外,本发明涉及有机物微粒的制造方法,其特征在于,用下述的微反应器进行上述工序1:将包含有机物微粒的原料溶液和析出溶剂l的至少2种的被处理流体导入到可接近和分离地相互对置配设、至少一方相对于另一方相对地进行旋转的第1处理用面与第2处理用面之间,通过在第1处理用面与第2处理用面之间所赋予的导入压力产生在使第1处理用面与第2处理用面分离的方向上发挥作用的分离力,通过上述分离力,在将第1处理用面与第2处理用面之间保持为微小的间隔的同时,使上述至少2种被处理流体在保持为上述微小的间隔的第1处理用面与第2处理用面之间合流,在上述第1处理用面与第2处理用面之间通过,由此形成薄膜流体,在上述薄膜流体中使被处理流体之间反应。能够使用具备进行旋转的搅拌叶片的搅拌机来进行上述搅拌。
通过上述制造方法得到的上述有机物微粒的粒径能够根据微粒的利用用途等各自变形来实施,例如能够使其为100nm以下、30nm以下的粒径来实施。例如,使添加了上述表面活性剂的对于上述有机物微粒具有一部分溶解能力的溶剂作用于上述有机物微粒的前后的上述有机物微粒的粒径可以为100nm以下,上述有机物微粒(p1)的粒径可以为30nm以下。
另外,本发明涉及有机物微粒的制造方法,其特征在于,在上述溶剂中基本上不含颜料衍生物。
另外,本发明涉及有机物微粒的改性方法,是基本上不改变有机物微粒的粒径的有机物微粒的改性方法,其特征在于,包含使在对于有机物微粒具有一部分溶解能力的溶剂中添加了表面活性剂的粒子性状控制溶液对于上述有机物微粒进行作用的工序,通过该工序使上述有机物微粒的结晶度提高,由此将上述有机物微粒改性为具备与目标的设定条件相符的结晶度的有机物微粒。
另外,本发明涉及有机物微粒的改性方法,是基本上不改变有机物微粒的粒径的有机物微粒的改性方法,其特征在于,包含使在对于有机物微粒具有一部分溶解能力的溶剂中添加了表面活性剂的粒子性状控制溶液对于上述有机物微粒进行作用的工序,通过该工序使上述有机物微粒的晶型变化,由此将上述有机物微粒改性为具备与目标的设定条件相符的晶型的有机物微粒。
另外,本发明涉及有机物微粒的改性方法,其特征在于,进行上述工序的处理的前后的上述有机物微粒的粒径变化率(工序的处理后(a)/工序的处理前(b))为1~4。
另外,本发明涉及有机物微粒的改性方法,其特征在于,进行上述工序的处理之前的有机物微粒在至少其一部分中包含非晶质。
发明的效果
通过使用本发明的制造方法,能够抑制有机物微粒的粒子生长,且可控制晶型、结晶性,能够制造能够完全发挥有机物微粒本来的性能的有机物微粒。另外,通过精密地控制有机物微粒的性状,能够提供可应对产业界的各种要求的有机物微粒的制造方法。进而,由于在对于上述有机物微粒具有一部分溶解能力的溶剂中能够简便地进行粒子的性状的控制,因此可由一种有机物微粒制作具备与目标相符的特性的各种各样的种类的有机物微粒,能够大幅地实现成本降低。
特别地,通过将本发明的有机物微粒的制造方法应用于可进行所析出的粒子的结晶性控制的强制薄膜型微反应器,在从微反应器使包含非晶质的有机物微粒析出后,能够进一步进行微粒的性状的控制,可以容易地制造具备非常多种多样的特性的有机物微粒。
附图说明
图1为表示本发明的实施方式涉及的处理工序的一例的图。
图2的(a)为本发明的实施方式涉及的强制薄膜型微反应器的概略剖面图,(b)为本发明的另一实施方式涉及的强制薄膜型微反应器的概略剖面图。
图3为图2(a)(b)中所示的强制薄膜型微反应器的第1处理用面的概略平面图。
图4为本发明的实施例a1、工序c中得到的吲哚美辛微粒的tem照片。
图5为本发明的实施例a1、工序c以及工序2中得到的吲哚美辛微粒的x射线衍射测定结果。
图6为本发明的实施例a1、工序2中得到的吲哚美辛微粒的tem照片。
图7为本发明的实施例a2、工序2中得到的吲哚美辛微粒的tem照片。
图8为本发明的比较例a1、工序2中得到的吲哚美辛微粒的tem照片。
图9为本发明的实施例a3、工序c以及工序2中得到的吲哚美辛微粒的x射线衍射测定结果。
图10为本发明的实施例a3、工序2中得到的吲哚美辛微粒的tem照片。
图11为本发明的比较例a2、工序2中得到的吲哚美辛微粒的tem照片。
图12为本发明的实施例a4、工序c中得到的吲哚美辛微粒的tem照片。
图13为本发明的实施例a4、工序c以及工序2中得到的吲哚美辛微粒的x射线衍射测定结果。
图14为本发明的实施例a4、工序2中得到的吲哚美辛微粒的tem照片。
图15为本发明的比较例a3、工序2中得到的吲哚美辛微粒的tem照片。
图16为本发明的实施例a5、工序c中得到的姜黄素微粒的tem照片。
图17为本发明的实施例a5以及实施例a6的工序c、以及工序2中得到的姜黄素微粒的x射线衍射测定结果。
图18为本发明的实施例a5、工序2中得到的姜黄素微粒的tem照片。
图19为本发明的比较例a4、工序2中得到的姜黄素微粒的tem照片。
图20为本发明的实施例a6、工序2中得到的姜黄素微粒的tem照片。
图21为本发明的比较例a5、工序2中得到的姜黄素微粒的tem照片。
图22为本发明的实施例a7、工序c中得到的聚丙烯微粒的sem照片。
图23为本发明的实施例a7、工序c以及工序2中得到的聚丙烯微粒的x射线衍射测定结果。
图24为本发明的实施例a7、工序2中得到的聚丙烯微粒的sem照片。
图25为本发明的比较例a6、工序2中得到的聚丙烯微粒的sem照片。
图26为本发明的实施例a8、工序1中得到的吡诺克辛微粒的tem照片。
图27为本发明的实施例a8、工序2中得到的吡诺克辛微粒的tem照片。
图28为本发明的实施例a8、工序1(工序2处理前)和工序2中得到的吡诺克辛微粒的x射线衍射测定结果。
图29为实施例b的实验编号1-1的工序1中制作的本发明的红色颜料纳米粒子的透射电子显微镜(tem)观察照片。
图30为实施例b的实验编号1-2的工序2中制作的本发明的红色颜料纳米粒子的透射电子显微镜(tem)观察照片。
图31为实施例b的实验编号1-1的工序2中制作的本发明的红色颜料纳米粒子的透射电子显微镜(tem)观察照片。
图32为在实施例c的实验编号1-7的工序1(清洗)后得到的本发明的青色有机颜料微粒的透射电子显微镜(tem)观察照片。
图33为在实施例c的实验编号1-7的工序1(清洗)后得到的本发明的青色有机颜料微粒的透射电子显微镜(tem)观察照片。
图34为在实施例c的实验编号1-7的工序2(作用)后得到的本发明的青色有机颜料微粒的透射电子显微镜(tem)观察照片。
图35为在实施例c的实验编号1-7的工序2(作用)后得到的本发明的青色有机颜料微粒的透射电子显微镜(tem)观察照片。
图36为实施例c的实验编号4-4中制作的青色有机颜料微粒的透射电子显微镜(tem)观察照片。
图37为实施例c的实验编号3-1中制作的青色有机颜料微粒的透射电子显微镜(tem)观察照片。
具体实施方式
以下对本发明的实施方式详细地说明。应予说明,本发明的方式并不只限定于以下记载的实施方式。
本发明中,有机物为含碳的化合物,主要由碳和氧形成,可以是人工合成的有机物,也可以是从天然物提取的有机物,并无特别限定。例如可列举出医药组合物、食品、食品添加物、健康食品等生物体摄取物、合成树脂、合成纤维、橡胶等高分子化合物、包含染料、颜料、涂料等的色素系化合物、香料、农药等。另外,本发明中,有机物微粒为上述的有机物的微粒。
所谓生物体摄取物,只要以生物体摄取为目的,则并无特别限定,例如可列举出如医药品中的药物那样在生物体内吸收、以呈现在生物体内的效果为目的的物质;通过体内、然后排泄的物质;药物传输系统中的药物成分的搬运用物质;或者如化妆料那样涂布于生物体皮肤的物质以及食品和上述物质的中间体等。具体地,是指医药、医药部外品、化妆品、食品、食品添加物、健康食品等中使用的有机物。作为本发明的生物体摄取物,可使用市售的产品,也可新合成。
作为上述生物体摄取物的具体例,可列举出镇痛药、抗炎症药、驱虫药、抗脉律不齐药、抗生物质、抗凝固药、抗降压药、抗糖尿病药、抗癫痫药、抗组胺药、抗恶性肿瘤药、食欲抑制药、抗肥胖药、抗毒蕈碱药、抗分枝菌药、抗新生物药、免疫抑制药、抗甲状腺药、抗菌药、抗病毒药、不安消除药、收敛剂、肾上腺素性β受体阻滞药、血液制剂、代用血浆、心肌变性力药、造影剂、皮质类固醇、咳抑制药、诊断药、诊断像形成药、利尿药、多巴胺作用药、止血药、免疫药、脂质调节药、肌肉松弛药、副交感神经刺激兴奋药、副甲状腺降血钙素、双磷酸酯类、前列腺素、放射性医药、性激素、抗过敏药、兴奋药、食欲减退物质、交感神经兴奋药、甲状腺药、血管扩张剂和黄嘌呤类、白内障治疗剂、副肾皮质荷尔蒙剂、过敏性鼻炎治疗药等医药组合物、营养药效物质、食物辅助品、维生素、矿物质、药草等食物营养辅助剂、叶酸、脂肪酸、果实和蔬菜提取物、维生素补给剂、矿物质补给剂、磷脂酰丝氨酸、硫辛酸、褪黑激素、葡糖胺/软骨素、芦荟、グッグル、谷氨酰胺、氨基酸、绿茶、番茄红素等食品或者、食品添加物、药草、植物营养素、抗氧化剂、果实的类黄酮成分、以及胶原、透明质酸、氨基酸、维生素c衍生物、氢醌类等美容辅助食品等,但并不限定于此。作为优选的性状,可列举出在水中以低溶解度可经口给药的生物体摄取物和可作为注射剂应用的生物体摄取物等。
作为医药,可列举出达那唑、他克莫司水合物、黄体酮、吲哚美辛、姜黄素、曲尼司特、苯溴马隆、萘普生、苯妥英、胡萝卜素、哌酰硫胺、哌泊舒凡、喜树碱、扑热息痛、乙酰水杨酸、胺碘酮、胆酪胺、降胆宁、色甘酸钠、沙丁胺醇、硫糖铝、柳氮磺胺吡啶、米诺地尔、替马西泮、阿普唑仑、丙氧芬、金诺芬、红霉素、环孢子菌素、阿昔洛韦、更昔洛韦、依托泊甙、美法仑、氨甲蝶呤、米托蒽醌、柔红霉素、阿霉素、甲地孕酮、他莫昔芬、甲羟孕酮、制霉菌素、特布他林、两性霉素b、阿司匹林、布洛芬、双氯芬酸、酮洛芬、氟比洛芬、二氟尼柳、薯蓣皂苷元、西洛他唑、甲糖宁、肽、色甘酸钠、吡诺克辛等。
作为医药部外品,可列举出刷牙剂、药用化妆品、生发剂、口中清凉剂、口臭预防剂等。
作为化妆品,例如可列举出化妆水、乳液、美容液等基础化妆品、防晒化妆品、上妆化妆品、头发化妆品、清洁化妆品、嘴唇化妆品、口腔化妆品、指甲化妆品、眼线化妆品、淋浴用化妆品等。
作为食品或食品添加物,可列举出维生素a·b·c·e等维生素类及其衍生物、2氨基酸类、类胡萝卜素、果实和植物提取物等。
作为健康食品,可以列举出辅酶q10、维生素a·b·c·e等维生素类及其衍生物等。这些可以单独地使用,也可以将2种以上混合而使用。
本发明中,对树脂并无特别限定,包含热塑性树脂[聚酯系树脂、聚酰胺系树脂、聚氨酯系树脂、聚(硫)醚系树脂、聚碳酸酯系树脂、聚砜系树脂、聚酰亚胺系树脂等缩合系热塑性树脂、聚烯烃系树脂、(甲基)丙烯酸系树脂、苯乙烯系树脂、乙烯基系树脂等乙烯基聚合系热塑性树脂、热塑性弹性体、纤维素衍生物等来自天然物的树脂、热塑性有机硅树脂等]、热固化性树脂[例如,环氧树脂、不饱和聚酯树脂、邻苯二甲酸二烯丙酯树脂、有机硅树脂(有机硅橡胶、有机硅清漆)]等。这些树脂可以单独地使用或者将二种以上组合使用。通常,可以优选地使用热塑性树脂、非水溶性树脂(或疏水性树脂、非水溶性热塑性树脂等)。
作为聚酯系树脂,可列举出使用了二羧酸成分、二醇成分、羟基羧酸、内酯类的各种树脂,例如聚对苯二甲酸乙二醇酯、聚对苯二甲酸1,3-丙二醇酯、聚对苯二甲酸1,2-丙二醇酯、聚对苯二甲酸丁二醇酯、聚对苯二甲酸1,4-环己烷二甲醇酯、聚萘二甲酸乙二醇酯、聚萘二甲酸丁二醇酯等聚c2-6亚烷基-芳酯系树脂、含有c2-6亚烷基-芳酯单元作为主成分(例如50重量%以上)的共聚酯(例如,共聚成分为具有氧亚烷基单元的聚氧c2-4亚烷基二醇、c6-12脂肪族二羧酸、间苯二甲酸、邻苯二甲酸等非对称性芳香族二羧酸等的共聚酯)、聚芳酯系树脂、液晶性聚酯等芳香族聚酯系树脂、聚c2-6亚烷基-草酸酯、聚c2-6亚烷基-琥珀酸酯、聚c2-6亚烷基-己二酸酯等聚c2-6亚烷基二醇-c2-10脂肪族二羧酸酯、聚羟基羧酸系树脂(例如聚乙醇酸、聚乳酸、乙醇酸-乳酸共聚物等)、聚内酯系树脂(例如,聚己内酯等聚c3-12内酯系树脂等)、它们的共聚酯(例如聚己内酯-聚琥珀酸丁二醇酯共聚树脂等)等。聚酯系树脂可含有尿烷键。进而,聚酯系树脂可具有生物降解性。
作为聚酰胺系树脂,例如可列举出脂肪族聚酰胺系树脂、脂环族聚酰胺系树脂、芳香族聚酰胺系树脂等,通常使用脂肪族聚酰胺系树脂。这些聚酰胺系树脂可以单独地使用或者将二种以上组合使用。作为脂肪族聚酰胺系树脂,可列举出脂肪族二胺成分(四亚甲基二胺、六亚甲基二胺等c4-10亚烷基二胺)与脂肪族二羧酸成分(己二酸、癸二酸、十二烷二酸等c4-20亚烷基二羧酸等)的缩合物(例如聚酰胺46、聚酰胺66、聚酰胺610、聚酰胺612、聚酰胺1010、聚酰胺1012、聚酰胺1212等)、内酰胺(ε-己内酰胺、ω-月桂内酰胺等c4-20内酰胺等)或者氨基羧酸(ω-氨基十一烷酸等碳数c4-20氨基羧酸等)的均聚物或共聚物(例如聚酰胺6、聚酰胺11、聚酰胺12、聚酰胺6/11、聚酰胺6/12等)、这些聚酰胺成分共聚而成的共聚酰胺(例如聚酰胺66/11、聚酰胺66/12等)等。聚酰胺系树脂的二羧酸成分可含有二聚酸单元。进而,聚酰胺系树脂可具有生物降解性。
作为醚系树脂、特别是聚(硫)醚系树脂,例如包含聚氧化烯系树脂(稳定化了的聚甲醛二醇或者均聚或共聚缩醛系树脂、聚氧丙烯二醇、聚氧四亚甲基二醇、聚氧乙烯-聚氧丙烯嵌段共聚物等聚氧化c2-4烯二醇)、聚苯醚系树脂、聚苯醚酮系树脂、聚硫醚系树脂(聚苯硫醚或其共聚物等聚硫醚系树脂)、聚醚酮系树脂(包含聚醚醚酮系树脂)等。
在聚烯烃系树脂中,可列举出α-c2-6烯烃的均聚物或共聚物,例如聚乙烯、聚丙烯、乙烯-丙烯共聚物、聚甲基戊烯-1等烯烃的均聚物或共聚物、烯烃与共聚性单体的共聚物[乙烯-醋酸乙烯酯共聚物、乙烯-(甲基)丙烯酸共聚物、乙烯-(甲基)丙烯酸酯共聚物等]。这些聚烯烃系树脂可以单独地使用或者将二种以上组合使用。
乙烯基系树脂中,作为含有卤素的含卤素乙烯基系树脂,例如可以例示聚氯乙烯系树脂、氯乙烯-醋酸乙烯酯共聚物、偏氯乙烯系树脂、氟树脂等。
另外,作为其他的乙烯基系树脂或其衍生物,例如可以例示羧酸乙烯酯的均聚物或共聚物(聚醋酸乙烯酯、乙烯-醋酸乙烯酯共聚物等)、它们的皂化物(聚乙烯醇、乙烯-乙烯醇共聚物等乙烯醇系树脂)、来自皂化物(乙烯醇系树脂)的衍生物(例如聚乙烯醇缩甲醛、聚乙烯醇缩丁醛等聚乙烯醇缩醛系树脂等)等。在乙烯-乙烯醇共聚物中,乙烯含量可以为5~40重量%左右。
本发明能够应用于可作为有机颜料提供并制造的各种颜料。
例如,作为本发明的红色颜料,可使用市售的产品,也可新合成。作为一例,可列举出在色指数中分类为c.i.颜料红的颜料、分类为c.i.颜料紫、c.i.颜料橙的颜料的一部分。更具体地,可列举出c.i.颜料红122、c.i.颜料紫19这样的喹吖啶酮系颜料、c.i.颜料红254、c.i.颜料橙73这样的二酮基吡咯并吡咯系颜料、c.i.颜料红150、c.i.颜料红170这样的萘酚系颜料、c.i.颜料红123这样的苝系颜料、c.i.颜料红144这样的偶氮系颜料。
另外,本发明对于青色有机颜料也能够应用。在该青色有机颜料中包含青色、藏青色、花青色等青系色的有机颜料。
作为本发明的青色有机颜料,可使用市售的产品,也可新合成。作为一例,可以列举出在色指数中分类为c.i.颜料蓝的颜料。更具体地,可以列举出c.i.颜料蓝-1、c.i.颜料蓝-2、c.i.颜料蓝-3、c.i.颜料蓝-15、c.i.颜料蓝-15:2、c.i.颜料蓝-15:3、c.i.颜料蓝-15:4、c.i.颜料蓝-16、c.i.颜料蓝-22、c.i.颜料蓝-60、c.i.颜料蓝-75等。这些可以单独地使用,也可将2种以上混合使用。
作为本发明的有机颜料,也能够使用复合酞菁微粒。就复合酞菁微粒而言,目前为止已提供各种复合酞菁微粒并已市售,也能够使用其,还可新合成。
特别地,本申请的申请人开发了能够抑制结晶生长、满足所要求的特性、最适合作为颜料等色料的、纳米级、优选地100nm级以下的铜-氧钛酞菁微粒、铜-钴酞菁微粒、和铜-氧钛-钴酞菁微粒等复合酞菁微粒及其制造方法,也能够使用采用该制造方法得到的复合酞菁微粒。
对于该新的复合酞菁微粒进行说明,该复合酞菁微粒的制造方法的特征在于,包含:使作为原料的、至少铜酞菁和氧钛酞菁和/或钴酞菁溶解于第1溶剂而制备溶解液的工序0;将上述工序0中得到的溶解液与对于上述原料成为不良溶剂的第2溶剂混合而使复合酞菁析出的工序1;和使有机溶剂作用于上述工序1中得到的复合酞菁的工序2。
上述有机溶剂优选为芳香族化合物系溶剂或杂环式化合物系溶剂,例如,上述有机溶剂优选为选自苯乙烯、二甲苯、甲苯、苯、甲酚、枯烯、四氢呋喃中的至少1种。通过使用通常诱发和促进向更为稳定的β型结晶结构等的结晶转变的芳香族化合物系溶剂或脂环族化合物系溶剂作为上述有机溶剂,令人惊奇地是,能够抑制α型铜酞菁向更为稳定的β型结晶结构等的结晶转变,另外,抑制结晶的生长成为可能。
可以作为上述工序0中的上述原料的混合重量比(铜酞菁/氧钛酞菁和/或铜酞菁/钴酞菁)为1以上且不到20的方案来实施。另外,上述工序0中,能够将氧钛酞菁和钴酞菁同时或依次溶解而实施。
上述工序1与本发明的上述的工序1同样地,能够使用使至少2种被处理流体反应的微反应器来进行。
另外,可以作为工序1中得到的复合酞菁与工序2中得到的复合酞菁为相同晶型的方案来实施。即,工序2中,即使使有机溶剂作用于工序1中得到的复合酞菁也没有发生结晶转变。另外,在上述有机溶剂中添加表面活性剂或分散剂。
上述复合酞菁微粒的长径比为1.1以上且2.5以下、粒径为5nm以上且100nm以下是适当的。上述长径比是指铜-氧钛酞菁微粒等各复合酞菁微粒中的长边与短边的比率。例如,其形状可捕捉为长方体或长方体类似体的情况下,是指3边中最长边与最短边的比率。或者,其形状可捕捉为球形或大致球形的情况的情况下,是指其最长径与最短径的比率。另外,例如,上述长径比是指由通过透射电子显微镜(tem)观察、对于100个粒子测定了长径和短径的结果的平均值求得的值。
可以作为上述复合酞菁微粒的紫外可见区域中的吸收光谱的655~700nm处的峰顶的abs(a)与550~640nm处的峰顶的abs(b)的相对值([abs(a)]/[abs(b)])为0.8以上的方案来实施。上述abs是指基于朗伯比尔定律、在紫外可见吸收光谱测定中算出的吸光度(abspbance),峰顶的abs是指特定波长的范围内的abs值中的最大值。
作为本发明的表面活性剂,能够使用以下所示的、各种市售品、新合成的产物。并无特别限定,作为一例,可以列举出阴离子性表面活性剂、阳离子性表面活性剂、非离子性表面活性剂、两性表面活性剂、各种聚合物等的分散剂等。根据用途,有时对能够使用的表面活性剂产生限制。例如,对于生物体摄取物必须考虑对生物体的毒性等。并不限定于此,可列举出ネオゲンr-k(第一工业制药)这样的十二烷基苯磺酸系、ソルスパース20000、ソルスパース24000、ソルスパース26000、ソルスパース27000、ソルスパース28000、ソルスパース41090(以上为アビシア社制造)、byk108、ディスパービックbyk160、ディスパービックbyk161、ディスパービックbyk162、ディスパービックbyk163、ディスパービックbyk166、ディスパービックbyk170、ディスパービックbyk180、ディスパービックbyk181、ディスパービックbyk182、ディスパービックbyk183、ディスパービックbyk184、ディスパービックbyk190、ディスパービックbyk191、ディスパービックbyk192、ディスパービックbyk2000、ディスパービックbyk2001、ディスパービックbyk2163、ディスパービックbyk2164(以上为毕克化学公司制造)、聚合物100、聚合物120、聚合物150、聚合物400、聚合物401、聚合物402、聚合物403、聚合物450、聚合物451、聚合物452、聚合物453、efka-46、efka-47、efka-48、efka-49、efka-1501、efka-1502、efka-4540、efka-4550(以上为efkaケミカル社制造)、カオーセラ2000、ペレックスtg、ペレックスtr(以上为花王株式会社制造)、フローレンdopa-158、フローレンdopa-22、フローレンdopa-17、フローレンg-700、フローレンtg-720w、フローレン-730w、フローレン-740w、フローレン-745w(以上为共荣社化学株式会社制造)、アジスパーpa111、アジスパーpb711、アジスパーpb811、アジスパーpb821、アジスパーpw911(以上为味之素株式会社制造)、ジョンクリル678、ジョンクリル679、ジョンクリル62(以上为ジョンソン聚合物株式会社制造)、羟甲基纤维素、羟乙基纤维素、羟丙基纤维素、羟丙基甲基纤维素、羧甲基纤维素、羧甲基纤维素钠、甲基纤维素、乙基纤维素等纤维素类、聚乙烯醇等高分子、聚乙烯基吡咯烷酮、卵磷脂、胆甾醇等磷脂质、锍化合物、阿拉伯树胶·ガディ树胶等植物性树脂、明胶、酪蛋白、磷脂、葡聚糖、甘油、黄蓍胶、硬脂酸、苯扎氯铵、硬脂酸钙、甘油单硬脂酸酯、鲸蜡硬脂醇、聚西托醇乳化蜡、span80、span60、span20等脱水山梨糖醇酯、聚氧乙烯烷基醚、聚氧乙烯蓖麻油衍生物、tween80、tween60、tween40、tween20等聚氧乙烯山梨糖醇酐脂肪酸酯、聚乙二醇、十二烷基三甲基溴化铵、聚氧乙烯硬脂酸酯、胶体二氧化硅、磷酸酯、十二烷基硫酸钠、氧化乙烯和甲醛的4-(1、1、3、3-四甲基丁基)-苯酚聚合物、lutrolf127、lutrolf108、futrolf87、futrolf68等poloxamer、poloxamine、带电磷脂质、磺基琥珀酸二辛酯、磺基琥珀酸钠的二烷基酯、十二烷基硫酸钠、烷基芳基聚醚磺酸酯、蔗糖硬脂酸酯和蔗糖二硬脂酸酯的混合物、对-异壬基苯氧基聚缩水甘油、癸酰基-n-甲基葡糖酰胺、正-癸基β-d-吡喃葡萄糖苷、正-癸基β-d-吡喃麦芽糖苷、正-十二烷基β-d-吡喃葡萄糖苷、正-十二烷基β-d-麦芽糖苷、庚酰基-n-甲基葡糖酰胺、正-庚基β-d-吡喃葡萄糖苷、正-庚基β-d-硫代葡糖苷、正-己基β-d-吡喃葡萄糖苷、壬酰基-n-甲基葡糖酰胺、正-壬基β-d-吡喃葡萄糖苷、辛酰基-n-甲基葡糖酰胺、正-辛基-β-d-吡喃葡萄糖苷、辛基β-d-硫代吡喃葡萄糖苷、溶菌酶、peg-磷脂质、peg-胆甾醇、peg-胆甾醇衍生物、peg-维生素a、以及乙酸乙烯酯和乙烯基吡咯烷酮的无规共聚物、季铵化合物、苄基-二(2-氯乙基)乙基溴化铵、椰油基三甲基氯化铵、椰油基三甲基溴化铵、椰油基甲基二羟基乙基氯化铵、椰油基甲基二羟基乙基溴化铵、癸基三乙基氯化铵、癸基二甲基羟基乙基氯化铵、癸基二甲基羟基乙基氯化溴化铵、c12-15二甲基羟基乙基氯化铵、c12-15二甲基羟基乙基氯化溴化铵、椰油基二甲基羟基乙基氯化铵、椰油基二甲基羟基乙基溴化铵、肉豆蔻基三甲基铵甲基硫酸盐、月桂基二甲基苄基氯化铵、月桂基二甲基苄基溴化铵、月桂基二甲基(乙烯氧基)4氯化铵、月桂基二甲基(乙烯氧基)4溴化铵、n-烷基(c12-18)二甲基苄基氯化铵、n-烷基(c14-18)二甲基-苄基氯化铵、n-十四烷基甲基苄基氯化铵一水合物、二甲基二癸基氯化铵、n-烷基和(c12-14)二甲基1-萘基甲基氯化铵、三甲基铵卤化物、烷基-三甲基铵盐、二烷基-二甲基铵盐、月桂基三甲基氯化铵、乙氧基化烷基酰氨基烷基二烷基铵盐、乙氧基化三烷基铵盐、二烷基苯二烷基氯化铵、n-二癸基二甲基氯化铵、n-十四烷基二甲基苄基铵、氯化物一水合物、n-烷基(c12-14)二甲基1-萘基甲基氯化铵、十二烷基二甲基苄基氯化铵、二烷基苯烷基氯化铵、月桂基三甲基氯化铵、烷基苄基甲基氯化铵、烷基苄基二甲基溴化铵、c12三甲基溴化铵、c15三甲基溴化铵、c17三甲基溴化铵、十二烷基苄基三乙基氯化铵、聚-二烯丙基二甲基氯化铵(dadmac)、二甲基氯化铵、烷基二甲基铵卤化物、三鲸蜡基甲基氯化铵、癸基三甲基溴化铵、十二烷基三乙基溴化铵、十四烷基三甲基溴化铵、甲基三辛基氯化铵、polyquat10tm、四丁基溴化铵、苄基三甲基溴化铵、胆碱酯、硬脂基二甲基苄基氯化铵化合物、溴化十六烷基吡啶鎓、氯化十六烷基吡啶鎓、季铵化聚氧乙基烷基胺的卤化物盐、mirapoltm、alkaquattm、烷基吡啶鎓盐、胺、胺盐、氧化胺、亚氨基吡咯鎓(imidoazolinium)盐、质子化季铵化丙烯酰胺、甲基化季铵化聚合物等。这些可单独地使用,也可将2种以上并用。
并不限于此,例如,在有机物微粒为实施例a中所示的吲哚美辛、姜黄素、吡诺克辛等的低分子的有机物的情况下,优选使用水溶性含氮乙烯基聚合物、非离子性纤维素衍生物等的高分子表面活性剂。更具体地,可列举出羟丙基纤维素、羟乙基纤维素、羧甲基纤维素、甲基纤维素、聚乙烯醇、聚乙烯基吡咯烷酮等。另外,从分散性的观点等出发,也有时tween80、聚氧乙烯固化蓖麻油等的非离子系的表面活性剂具有效果,也优选与高分子表面活性剂并用。在有机物微粒为实施例b、c中所示的颜料的有机物的情况下,优选使用丙烯酸系聚合物、高分子嵌段共聚物等的高分子系的表面活性剂、含有羟基的羧酸酯这样的表面活性剂、二烷基磺基琥珀酸钠、十二烷基苯磺酸钠等的阴离子表面活性剂等。
另外,在本发明的有机物微粒中,也可使用作为高分子表面活性剂的嵌段共聚物。这种情况下,作为嵌段共聚物,可列举出丙烯酸系、甲基丙烯酸系嵌段共聚物、聚苯乙烯和其他加聚系或缩聚系的嵌段共聚物、具有聚氧乙烯、聚氧化烯的嵌段的嵌段共聚物等。而且,也可以使用以往已知的嵌段共聚物,但本发明中使用的嵌段共聚物优选为两亲性。作为具体的优选的形式,可以列举出由疏水链段和具有有机酸或其离子性盐单元的亲水链段构成的二嵌段共聚物。另外,优选使用具有疏水链段和具有有机酸或其离子性盐单元的亲水链段和另外的链段的三嵌段共聚物。三嵌段的情况下,优选采用为疏水链段、非离子性的亲水链段、具有有机酸或其离子性盐单元的亲水链段的形式,在内包状态的稳定化的意义上也优选。例如如果使用上述的三嵌段共聚物,使用颜料物质等有机物和作为溶剂的水制备分散液,则可以使颜料等有机物内包于三嵌段共聚物形成的胶束中。另外,也可以使其成为其分散组合物的粒子的粒径也非常地整齐的均一的组合物。进而也可以使其分散状态极其稳定。
作为本发明的溶剂,为了使有机物微粒的原料溶解,为了从有机物微粒的原料溶液使有机物微粒析出,进而为了通过添加以下记载的表面活性剂而控制有机物微粒的性状,可以使用各种溶剂。作为这些溶剂的一例,可以列举出水(蒸馏水、纯水等)、有机溶剂(醇系溶剂、酮系溶剂、醚系溶剂、芳香族系溶剂、脂肪族烃系溶剂、腈系溶剂、亚砜系溶剂、卤素系溶剂、酯系溶剂、胺系溶剂、离子性溶液)。就这些溶剂而言,可以根据目的选择1种或2种以上的混合溶剂来实施。另外,根据需要,也能够将酸性物质、碱性物质加入各种溶剂中,调节ph。
对于上述的溶剂进一步详细地说明,作为醇系溶剂,例如可列举出甲醇、乙醇、异丙醇、正丙醇、1-甲氧基-2-丙醇(pgme)等,进而可列举出正丁醇等直链醇、2-丁醇、叔丁醇等分支状醇、乙二醇、二甘醇等多元醇等。作为酮系溶剂,例如可列举出丙酮、甲乙酮、环己酮等。作为醚系溶剂,例如可列举出二甲醚、二乙醚、四氢呋喃等。作为芳香族系溶剂,例如可列举出苯乙烯、甲苯、二甲苯、苯酚、硝基苯、氯苯、二氯苯、四氢呋喃、吡啶等。作为脂肪族系溶剂,例如可列举出戊烷、己烷、庚烷、辛烷、环己烷等。作为腈系溶剂,例如可列举出乙腈等。作为亚砜系溶剂,例如可列举出二甲基亚砜、二乙基亚砜、六亚甲基亚砜、环丁砜等。作为卤素系溶剂,例如可列举出氯仿、二氯甲烷、三氯乙烯、碘仿等。作为酯系溶剂,例如可列举出醋酸乙酯、醋酸丁酯、乳酸甲酯、乳酸乙酯、丙二醇单甲基醚乙酸酯(pgmea)等。作为离子性液体,例如可列举出苄基三甲基氢氧化铵(btma)、1-丁基-3-甲基咪唑鎓与pf6-(六氟磷酸离子)的盐等。作为胺系溶剂,例如可列举出二甲基氨基乙醇、乙二胺、甲胺、二甲胺、二甲胺、三甲胺、乙胺、二乙胺、三乙胺等。作为酰胺系溶剂,例如可列举出n,n-二甲基甲酰胺、n,n-二甲基乙酰胺等。
本发明中的“对于有机物微粒具有一部分溶解能力的溶剂”意味着在与上述有机物微粒作用后直至处理结束的期间,并非显示将上述有机物微粒的全部完全地溶解的强溶解性,而是显示出对于上述微粒的性状给予“变化”的程度的溶解性。以下也称为“具有一部分溶解能力的溶剂”。对该“变化”并无特别限定,但可以例示出使上述溶剂作用于上述有机物微粒时一部分的上述有机物微粒生长而粗大化的现象、有机物微粒之间发生颈缩等的现象等。
作为对于上述具有一部分溶解能力的溶剂的有机物微粒的溶解度的一例,在使用平均粒径1000nm的有机物微粒测定的情况下,优选为1μg/g以上且1000μg/g以下(1ppm以上且1000ppm以下),更优选为1μg/g以上且500μg/g以下(1ppm以上且500ppm以下)。即使没有完全地溶解,对于上述溶剂的有机物微粒的溶解度过高的情况下,由于粒子的生长、粒子的粗大化的程度变得过度,因此在本发明中的应用困难。
予以说明,上述溶解度的测定时,使上述溶剂作用于平均粒径1000nm的有机物微粒后,用开口径0.1~0.2μm左右的过滤器过滤,对其滤液进行uv-vis测定(可见·紫外分光测定),由此算出在溶剂中溶解的有机物的浓度。浓度的算出时,可用荧光、折射率等其他检测方法测定上述滤液。
另外,作为有机物微粒与具有一部分溶解能力的溶剂的组合的一例,在有机物微粒为吲哚美辛、姜黄素、吡诺克辛的情况下,优选使用戊烷、己烷、庚烷、辛烷、壬烷、癸烷、十一烷这样的直链的烷烃、环己烷这样的环状烷烃、水作为上述溶剂。另外,作为一例,在有机物微粒为聚丙烯的情况下,优选使用甲醇、异丙醇等的醇系溶剂、甲苯、二甲苯等的芳香族系溶剂作为上述溶剂。
作为一例,确认作为本发明的比较例a的结果的图8、图11中所看到的粒子在上述溶剂中生长、另外引起了颈缩的样子。在成为了这样的状态的情况下,即使然后进行分散处理,也不能将粒子分散,发挥通过有机物的微粒化所获得的特性(例如,溶解性的提高等物性变化、透明性的提高等光特性的变化、新型反应等化学特性变化等)非常难。
作为另一例,将本发明的实施例b的实验编号1-1中制作的红色颜料即pr254微粒投入到丙二醇单甲基醚乙酸酯(pgmea)时的tem观察结果示于图31中。另外,将投入前的tem观察结果示于图29中(为在表面活性剂水溶液中分散的情况)。与图29中看到的pr254纳米粒子相比,确认图31中看到的pr254纳米粒子在pgmea中生长、另外引起了颈缩的样子。在成为了这样的状态的情况下即使其后进行分散处理,也不能将纳米粒子分散,发挥微细的红色颜料的特性非常难。
进而,作为另一例,确认作为本发明的实施例c的实验编号4-4(图36)中制作的青色颜料的铜-氧钛-钴酞菁微粒或者作为实验编号3-1(图37)中制作的青色颜料的铜酞菁微粒在上述溶剂中生长、另外引起了颈缩的样子。在成为了这样的状态的情况下,即使其后进行分散处理,也不能将粒子分散,发挥青色有机颜料微粒的特性非常难。
本发明中,重要地是:在使粒子性状控制溶液作用之前的有机物微粒中,在至少其一部分中包含非晶质,敢于选择对于上述有机物微粒具有一部分溶解能力的溶剂,添加上述表面活性剂,具体地使混合·溶解·分子分散,使制备的粒子性状控制溶液作用于有机物微粒。由此能够控制有机物微粒的粒径、晶型、结晶性等性状。
能够控制粒子的性状的机理尚不清楚,有机物微粒的非晶质部位在固体中分子排列是无规的,与结晶质的部位相比,分子间没有紧密地接近,分子结合力也弱。因此,认为有机物微粒的非晶质部位受到对于有机物微粒具有一部分溶解能力的溶剂的作用,容易发生溶解,容易成为生长、颈缩的起点。
有机物微粒受到溶剂的影响、热的影响而在溶剂中一部分溶解了的情况下,虽然容易发生颈缩、粒子生长,但在这样的条件下保管的情况下,有时同时发生结晶度的提高、结晶转变。但是,此时与有机物微粒的一次粒径的数十倍至数百倍以上的变化相伴,结果这样粗大化了的有机物微粒失去作为有机物微粒的所期待的特性。本发明中的表面活性剂的作用之一在于抑制、控制上述有机物微粒的生长。上述表面活性剂可以抑制上述有机物微粒(特别是其非晶质的部位)受到上述具有一部分溶解能力的溶剂的作用而溶解,抑制、控制上述溶剂的对于有机物微粒的、颈缩、粒子生长这样的作用。更详细地说,如果在表面活性剂的存在下,认为没有达到由于具有一部分溶解能力的溶剂而使有机物微粒的非晶质的部位完全地溶解,非晶质的部位的无规的分子排列变化而发生压密化,由此发生结晶度的提高、结晶转变。
进而,上述表面活性剂的又一作用是上述有机物微粒在溶剂中的溶解能力的提高。更详细地说,是表面活性剂作用了的粒子全体的润湿性的提高。即,就表面活性剂而言,通过其种类、和组合的上述溶剂的种类等,可以显示出对于有机物微粒不同的性质。也可以在对上述有机物微粒具有一部分溶解能力的溶剂中添加提高上述有机物微粒在溶剂中的溶解性的表面活性剂来制备粒子性状控制溶液。另外,上述表面活性剂的进一步的作用在于可成为上述有机物微粒的晶型转变变换时的铸模、更详细地说,可成为对于有机物微粒的粒径的铸模。
认为:本发明通过使上述的作用相结合,控制有机物微粒的生长,且特别是通过将非晶质的部位压密化而使结晶性提高,由此控制了晶型的转变、结晶性。
这样,在本发明中,通过使上述粒子性状控制溶液作用于上述有机物微粒,控制有机物微粒的粒径、结晶性、晶型这样的粒子性状。所谓上述作用,具体地,为将上述有机物微粒与上述粒子性状控制溶液混合和/或搅拌、或者只是接触、喷射等的操作,通过溶剂、表面活性剂的种类、浓度、处理温度、搅拌方法等的改变,可以控制有机物微粒的性状。另外,也可通过在上述溶剂中含有下述酸性物质、碱性物质或中性物质,制备上述具有一部分溶解能力的溶剂。
对这些物质并无特别限定,作为酸性物质,可列举出王水、盐酸、硝酸、发烟硝酸、硫酸、发烟硫酸等的无机酸、甲酸、柠檬酸、苹果酸、乙酸、氯乙酸、二氯乙酸、草酸、三氟乙酸、三氯乙酸等的有机酸等。作为碱性物质,可列举出氢氧化钠、氢氧化钾等的金属氧化物、甲醇钠、异丙醇钠这样的金属醇盐、以及三乙胺、二乙基氨基乙醇、二乙胺等的胺系化合物等。进而,也可将以上述酸性物质、碱性物质的盐类为例列举出的中性物质混合。
特别是红色有机颜料、青色有机颜料等的有机颜料的情况下,即使不含一般为了抑制结晶生长所使用的颜料的衍生物也能够实施,因此也能够发挥衍生物特有的颜色不对实际使用的有机颜料的发色产生影响这样的优点。不过,并不妨碍加入这样的衍生物进行实施。
予以说明,就被对于有机物微粒具有一部分溶解能力的溶剂发挥作用的有机物微粒的浓度而言,并无特别限定,为0.001~90.00重量%,优选为0.001~50.00重量%,更优选为0.01~40.00重量%。另外,就上述表面活性剂而言,相对于上述有机物微粒,为0.01~500重量%,优选为0.1~300重量%,更优选为1.0~100重量%。
本发明中对有机物微粒的粒径并无特别限定,控制有机物微粒的粒子性状之前的有机物微粒的粒径优选为1000nm以下,更优选为500nm以下,进一步优选为200nm以下。
另外,在青色有机颜料等的有机颜料的情况下,特别优选一次粒径为500nm以下、优选为100nm以下、更优选为30nm以下的微小的粒子。对上述粒子或微粒的形状并无特别限定,例如可以是大致圆柱状、大致球状、大致圆盘状、大致三棱柱状、大致四棱柱状、大致多面体状、椭圆球状等的形态的粒体或其集合体等。
予以说明,本发明的作用的前后的粒径是指进行上述的将上述有机物微粒和上述粒子性状控制溶液混合和/或搅拌、或者只是接触、喷射等操作之前和进行之后的粒径。
在本发明中,所谓基本上不使有机物微粒的粒径改变的范围是指使添加了表面活性剂的对于有机物微粒具有一部分溶解能力的溶剂作用于有机物微粒的前后的有机物微粒的粒径变化率(表面活性剂处理后(a)/表面活性剂处理前(b))为1~4。
另外,本发明包含工序1:将用于从有机物微粒的原料溶液使有机物微粒析出的、至少1种的有机物微粒的析出溶剂l与使有机物微粒的原料溶解或分子分散的有机物微粒的原料溶液混合,使有机物微粒(p1)析出。有机物微粒的原料可使用上述的有机物,也可新合成。上述使其混合的方法例如如专利文献5中记载那样,可用强制薄膜型微反应器使其混合,能够适宜地使用有机物微粒中的公知的方法。能够通过使可使有机物微粒的原料溶解或分子分散的良溶剂与有机物微粒的原料混合而制备有机物微粒的原料溶液,使与上述良溶剂相比对于有机物的溶解度低的溶剂作为有机物微粒的析出溶剂l来实施。另外,也能够通过调节将有机物微粒的原料溶液与有机物微粒的析出溶剂l混合时的混合时的ph来实施。也可以根据需要通过将上述溶剂与酸性物质或碱性物质等组合来制备。
另外,本发明根据需要可包含对于上述工序1中得到的上述有机物微粒(p1)进行清洗和/或溶剂置换的工序c。上述进行清洗和/或溶剂置换的工序c能够适当地应用公知的手法。并无特别限定,但对于包含有机物微粒的液体,可以通过过滤·离心分离·超过滤等的操作,通过选择与目的相符的溶剂来进行上述有机物微粒的清洗和/或溶剂置换。
本发明的特征在于,通过包含使上述有机物微粒(p1)与上述粒子性状控制溶液作用的工序2,控制上述有机物微粒的性状。作为上述性状的控制,可以列举出有机物微粒的粒径和/或有机物微粒的结晶性。另外,作为上述有机物微粒的结晶性,可以列举出有机物微粒的结晶度。这里所说的粒子的性状的控制,只要能够抑制溶剂中的粒子的生长、颈缩,则并不限定于上述内容,对于有机物微粒结晶转变的情况也能够适用。另外,通过以包含非晶质的方式结晶性低地使其析出,能够精度更好地控制工序2中的晶型、结晶度等,因此本发明变得更为有效。
另外,作为本发明中的、上述工序2中的作用,例如可以列举出使其混合的工序、使其接触的工序、喷射的工序等。另外,在本发明中的上述工序2中,可将上述使其作用反复进行1次或多次(例如2次、3次、4次等)。
另外,在本发明中的上述工序2中包含搅拌处理(以下也称为采用搅拌的分散处理、分散处理)的情况下,能够通过搅拌能量来控制上述有机物微粒的性状,可以列举出使用公知的搅拌装置、搅拌手段、适当地控制搅拌能量的手法。予以说明,关于搅拌能量,在根据本申请的申请人的日本特开平04-114725号公报中已详述。
对本发明中的搅拌的方法并无特别限定。可以是使用了磁力搅拌器和搅拌子的方法,可以使用各种剪切式、摩擦式、高压喷射式、超声波式等的搅拌机、溶解机、乳化机、分散机、均化器等进行实施。作为一例,可以列举出ウルトラタラックス(ika制造)、ポリトロン(キネマティカ制造)、tkホモミキサー(プライミクス制造)、エバラマイルダー(荏原制作所制造)、tkホモミックラインフロー(プライミクス制造)、胶体磨(神钢パンテック制造)、スラッシャー(日本焦炭工业制造)、トリゴナル湿式微粉碎机(三井三池化工机制造)、キャビトロン(ユーロテック制造)、ファインフローミル(太平洋机工制造)等的连续式乳化机、クレアミックス(エム·テクニック制造)、クレアミックスディゾルバー(エム·テクニック制造)、クレアミックスダブルモーション(エム·テクニック制造)、フィルミックス(プライミクス制造)等间歇式或连续式的装置。另外,在搅拌前或搅拌后进行一部分微波处理有时是有效的。
粒子的生长有粒子之间凝聚、粒子表面溶解所引起的生长·粗大化和由在分散介质中溶解的成分、粒子的粗大化发展的情况这二种。表面活性剂的效果是保护粒子表面而抑制生长,但如果形成凝集体,有时不能有效地发挥功能。因此,利用搅拌的分散处理变得有效。本方法中,为了使表面活性剂与对于有机物微粒具有一部分溶解能力的溶剂均匀地混合反应,优选进行搅拌处理。另外,使粒子性状控制溶液作用于有机物微粒时,优选进行搅拌处理。此时,通过给予搅拌能量,可以使表面活性剂和对于有机物微粒具有一部分溶解能力的溶剂高效地作用于有机物微粒。作为上述工序2,在包含搅拌处理的情况下,能够通过搅拌能量来控制有机物微粒的性状(结晶度、晶型、粒径)。
作为一例,以下按照图1对用间歇方式的有机物微粒的制造工序(工序0~2)进行说明。
(工序0)制备有机物微粒的析出溶剂(a液:相当于有机物微粒的析出溶剂l)和有机物微粒的原料溶液(b液)。
(工序1)a液与b液的混合:使用磁力搅拌器和搅拌子对a液进行搅拌,且投入b液,使有机物微粒析出。
(工序2)将工序1中析出的有机物微粒的浆料、湿饼或干燥粉体投入到粒子性状控制溶液,进行搅拌处理。
予以说明,可将工序1中析出了的包含有机物微粒的浆料过滤,然后,使用清洗液进行清洗(工序c),制作有机物微粒的湿饼,或者通过真空干燥法等的干燥处理来制作有机物微粒的干燥粉体,将其投入到工序2的粒子性状控制溶液中,进行搅拌处理。
接着,以下按照图1对使用了后述的微反应器的情况的有机物微粒的制造工序(工序0~2)的一例进行说明。
(工序0)制备有机物微粒的析出溶剂(a液)和有机物微粒的原料溶液(b液)。
(工序1)使用图2(a)中所示的微反应器将有机物微粒的原料溶液(b液)与有机物微粒的析出溶剂(a液:相当于有机物微粒的析出溶剂l)混合,使有机物微粒析出。予以说明,作为a液、b液,除了以下的实施例中例示的实例以外,也能够应用专利文献8中记载的实例等公知的混合、析出的例子。
(工序2)将工序1中析出的有机物微粒的浆料、湿饼或干燥粉体投入到粒子性状控制溶液中,进行了搅拌处理。
予以说明,可将工序1中析出的包含有机物微粒的浆料过滤,然后,使用清洗液进行清洗(工序c),制作有机物微粒的湿饼或者通过真空干燥法等干燥处理来制作有机物微粒的干燥粉体,将其投入到工序2的粒子性状控制溶液,进行搅拌处理。
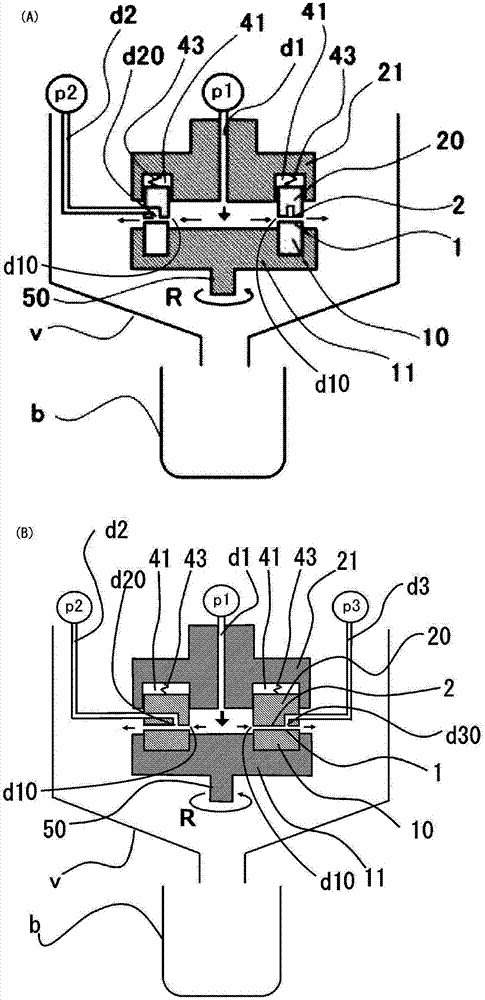
予以说明,作为微反应器,能够使用图2中所示的、与专利文献4~专利文献6中记载的装置同样的装置。以下对微反应器详细说明。图2(a)(b)以及图3中r表示旋转方向。
本实施方式中的微反应器具有对置的第1和第2的、2个处理用部10、20,第1处理用部10旋转。两处理用部10、20的对置的面分别成为处理用面。是第1处理用部10具有第1处理用面1、第2处理用部20具有第2处理用面2的强制薄膜型微反应器。
将两处理用面1、2分别连接至被处理流体的流路d1、d2,构成被处理流体的流路的一部分。将该两处理用面1、2间的间隔通常调节至1mm以下、例如0.1μm至50μm左右的微小间隔。由此,通过该两处理用面1、2间的被处理流体成为被两处理用面1、2强制了的强制薄膜流体。
而且,该装置在处理用面1、2间进行使第1、第2的被处理流体反应而进行微粒的析出的流体处理。
更具体地说明,上述装置具有:保持上述的第1处理用部10的第1支架11、保持第2处理用部20的第2支架21、接面压赋予机构43、旋转驱动机构(未图示)、第1导入部d1、第2导入部d2、和流体压赋予机构p1、p2。在流体压赋予机构p1、p2中能够采用压缩机、其他的泵。
在上述实施方式中,第1处理用部10、第2处理用部20为环状的圆盘。就第1、第2处理用部10、20的材质而言,除了金属以外,能够采用碳、陶瓷、烧結金属、耐磨损钢、蓝宝石,此外能够采用对金属实施了硬化处理的产物、对硬质材料实施了衬里、涂覆、镀敷等的产物。在上述实施方式中,对两处理用部10、20的相互对置的第1、第2的处理用面1、2进行镜面研磨,算术平均粗糙度为0.01~1.0μm。
在上述实施方式中,将第2支架21固定于装置,在同样固定于装置的旋转驱动机构的旋转轴中安装的第1支架11旋转,被该第1支架11支承的第1处理用部10相对于第2处理用部20进行旋转。当然,也可使第2处理用部20旋转,还可以使两者旋转。另外,在本发明中,能够使上述旋转速度例如为350~3600rpm。
上述实施方式中,相对于第1处理用部10,第2处理用部20在旋转轴50的方向上接近·分离,因此在设置于第2支架21的收容部41中,将与第2处理用部20的处理用面2侧相反侧的部位可出没地收容。不过,可与其相反地第1处理用部10相对于第2处理用部20接近·分离,也可两处理用部10、20相互地接近·分离。
上述收容部41为第2处理用部20的、将与处理用面2侧相反侧的部位收容的凹部,为形成为环状的沟槽。该收容部41具有可使与第2处理用部20的处理用面2侧相反侧的部位出没的足够的间隙,将第2处理用部20收容。
接面压赋予机构是用于在第1处理用部10的第1处理用面1与第2处理用部20的第2处理用面2接近的方向上产生挤压力(以下称为接面压力)的机构。通过该接面压力与由流体压力产生的使两处理用面1、2间分离的力的均衡,将两处理用面1、2间的间隔保持为规定的微小间隔,同时产生具有nm单位乃至μm单位的微小的膜厚的薄膜流体。在上述实施方式中,接面压赋予机构通过利用设置于第2支架21的弹簧43对第2处理用部20向着第1处理用部10赋能,从而赋予接面压力。另外,将被流体压赋予机构p1加压的第1被处理流体从第1导入部d1导入到两处理用部10、20的内侧的空间。
另一方面,将被流体压赋予机构p2加压的第2的被处理流体从第2导入部d2经由设置于第2处理用部20的内部的通路,从在第2处理用面形成的开口部d20导入到两处理用部10、20的内侧的空间。
在开口部d20,第1被处理流体与第2被处理流体合流、混合。此时,混合的被处理流体成为被保持上述的微小的间隙的两处理用面1、2强制的薄膜流体,要向环状的两处理用面1、2的外侧移动。由于第1处理用部10在旋转,因此混合了的被处理流体不是从环状的两处理用面1、2的内侧向外侧直线地移动,而是在环状的半径方向上的移动矢量与周向上的移动矢量的合成矢量作用于被处理流体,从内侧向外侧以大致漩涡状移动。
在此,如图3中所示那样,可在第1处理用部10的第1处理用面1形成从第1处理用部10的中心侧向外侧、即在径向上延伸的沟状的凹部13。该凹部13的平面形状可以是使第1处理用面1上弯曲或者以漩涡状延伸的形状,虽然没有图示,但也可以为笔直地在外方向上延伸的形状、以l字状等屈曲或弯曲的形状、连续的形状、断续的形状、分支的形状。另外,该凹部13也可在第2处理用面2形成而实施,也可在第1和第2处理用面1、2两者形成而实施。通过形成这样的凹部13,能够获得微泵效应,具有能够将被处理流体抽吸到第1和第2处理用面1、2间的效果。
优选上述凹部13的基端到达第1处理用部10的内周。上述凹部13的前端向着第1处理用部面1的外周面侧延伸,因此随着从基端向前端,其深度逐渐地减小。在该凹部13的前端与第1处理用面1的外周面之间设置有无凹部13的平坦面。
上述的开口部d20优选设置于与第1处理用面1的平坦面对置的位置。由此可以在层流条件下进行多个被处理流体的混合和微粒的析出。
另外,向两处理用部10、20的外侧排出的流体经由容器v,作为排出液收集在烧杯b中。在本发明的实施方式中,如后述那样,在排出液中含有有机物微粒。
予以说明,上述的被处理流体的种类及其流路的数在图2(a)的例a中设为2个,但也可以为3个以上。另外,就各处理用部中设置的导入用的开口部而言,对其形状、大小、数目并无特别限制,可适当地改变而实施。另外,可在上述第1和第2处理用面间1、2的正前面或者进一步的上游侧设置导入用的开口部。
在本发明中,只要能够在处理用面1、2间进行上述处理即可,也可以从第1导入部d1导入第2被处理流体,从第2导入部d2导入第1被处理流体。例如,各流体中的第1、第2这样的表示只不过具有为存在多个的流体的第n号这样的用于识别的含义,如上述那样也可存在第3以上的流体。
在使用了微反应器时的有机物微粒的制造工序(工序0~2)中,可使用微反应器连续地进行工序1和工序2。具体地,如图2(b)中所示那样,在第1导入部d1、第2导入部d2以外在微反应器中设置第3导入部d3,例如从第1导入部d1将作为第1流体的有机物微粒的原料溶液,从第2导入部d2将作为第2流体的有机物微粒的析出溶剂,从第3导入部d3将作为第3流体的粒子性状控制溶液各自分别地导入微反应器。这种情况下,通过将导入粒子性状控制溶液的第3导入部d3设置在第1导入部d1和第2导入部d2的下游侧,更详细地说,将第3导入部d3的开口部d30设置在第2导入部d2的开口部d20的下游侧,从而能够使在处理用面1、2间析出的有机物微粒与粒子性状控制溶液作用。具有上述的3个开口部(d10、d20和d30)的微反应器适于连续地进行工序1和工序2的情况。
不过,对本发明进行实施时,在微反应器中进行工序1、在微反应器外进行工序1以后的工序的情况下,如图1(a)中所示那样,具有至少2个开口部(d10、d20)就足以,但在对于在处理用面1、2间析出了的有机物微粒、在上述薄膜流体中实施表面处理的情况等下,并不妨碍在具有3个以上的开口部的微反应器中实施工序1。
特别是在使用了上述微反应器的情况下,对于本发明的有机物,作为微粒制造是容易的,在对于有机物微粒具有一部分溶解能力的溶剂和对于有机物微粒可进行生长抑制的表面活性剂中进行处理的前后可以设为1000nm以下,优选设为500nm以下。进而,如果是上述微反应器,所析出的微粒的结晶性的控制比较容易,因此通过在包含非晶质的结晶性低的状态下使其析出,能够精度更好地控制工序2中的晶型、结晶度等。作为这样的微反应器的具体例,可以列举出ulrea(エム·テクニック制造)。不过,对于本发明的有机物微粒的制造,并不限定于使用微反应器。
实施例
以下列举实施例对本发明进一步具体地说明。但是,本发明并不限定于下述的实施例。以下的实施例中,a液是指从图2(a)(b)中所示的装置的第1导入部d1所导入的第1被处理流体,b液是指从相同装置的第2导入部d2所导入的第2被处理流体。
本申请中,分成实施例a、实施例b、实施例c的3个组,将各实施例与比较例一起示出。
实施例a为生物体摄取物和树脂的组。
实施例b为红色有机颜料的组。
实施例c为青色有机颜料的组。
应予说明,在说明书正文中,在实施例后记载a、b、c的组记号,但在表中和附图中省略abc的组记号的记载。
(实施例a的组)
作为本发明涉及的有机物微粒的制造方法的一例,在实施例a1~4中采用吲哚美辛的微粒进行说明,在实施例a5、6中采用姜黄素的微粒进行说明,在实施例a7中采用聚丙烯的微粒进行说明,在实施例a8中采用吡诺克辛的微粒进行说明。
实施例a中,在x射线衍射测定(xrd测定)中使用粉末x射线衍射测定装置(制品名:x‘pertprompd、panalytical制造)。测定条件为测定范围:10~60°、cu对阴极、管电压45kv、管电流40ma、扫描速度16°/min。
在tem观察中使用透射型电子显微镜、jem-2100(jeol制造)。作为观察条件,使加速电压为80kv。
在sem观察中使用扫描型电子显微镜、jfm-7500f(jeol制造)。作为观察条件,设为加速电压1kv。
对于粒径评价而言,在tem观察或sem观察中使用25000倍的照片,使用粒子50个的平均值。
予以说明,这里所说的结晶度,是相对于将结晶化了的成分和非晶质合在一起的全体、结晶化了的成分的比例。
(实施例a1:吲哚美辛微粒)
<工序0:有机物微粒的析出溶剂(a液)与有机物微粒的原料溶液(b液)的制备>
<有机物微粒的析出溶剂(a液)的制备>
作为有机物微粒的析出溶剂(a液),使用磁力搅拌器制备0.1mol/l的盐酸水溶液的吲哚美辛析出溶剂。
<有机物微粒原料溶液(b液)的制备>
作为有机物微粒原料溶液(b液),制备在0.35mol/l的碳酸氢钠水溶液中混合了晶型为γ(伽马)型的吲哚美辛以致成为0.2wt%、混合了聚乙烯基吡咯烷酮(kollidon12pf(basf制造))以致成为0.2wt%的吲哚美辛原料溶液。使用作为高速旋转式分散乳化装置的クレアミックス(制品名:clm-2.2s、エム·テクニック制造),以转子转速15000rpm在约35℃搅拌30分钟,由此制备均质的吲哚美辛原料溶液。这样,在将多种成分进行混合的情况、进行溶解的情况下,优选使用作为高速旋转式分散乳化装置的クレアミックス(エム·テクニック制造)进行制备。
<工序1:混合·析出>
接着,使用图2(a)中所示的微反应器ulrea将制备的有机物微粒的析出溶剂和制备的有机物微粒的原料溶液混合。具体地,从图2(a)中所示的微反应器的第1导入部d1将作为第1被处理流体(a液)的上述制备完的有机物微粒的析出溶剂(在此,0.1mol/l的盐酸水溶液)在200ml/分钟、5℃下导入到处理用面间,一边以转速1700rpm使处理用部10运转,一边将作为第2被处理流体(b液)的制备完的吲哚美辛原料溶液在30ml/分钟、25℃下导入到处理用面1、2间,在薄膜流体中混合。然后,包含吲哚美辛微粒的排出液从流体处理用面1、2间排出。
<工序c:回收·清洗>
将上述排出液过滤,将吲哚美辛微粒回收。然后,用清洗溶剂(纯水)进行反复清洗,得到了吲哚美辛微粒的湿饼。将上述湿饼用纯水稀释,将其稀释液滴加到火棉胶膜,使用在室温下使其干燥的观察用试样进行tem观察。将tem观察结果示于图4中。另外,将上述湿饼在-0.095mpag下真空干燥16小时,得到干燥粉体。将得到的干燥粉体的、x射线衍射测定结果示于图5上段。
由tem观察结果确认了吲哚美辛微粒的平均一次粒径为76nm左右。另外,x射线衍射测定的结果确认了得到的粒子为非晶质。
<工序2:粒子性状控制>
对于工序2的粒子性状控制而言,使用纯水作为对于有机物微粒具有一部分溶解能力的溶剂,使用羟乙基纤维素(hec)作为对于有机物微粒可进行生长抑制的表面活性剂。通过在纯水中添加羟乙基纤维素、使用作为高速旋转式分散乳化装置的クレアミックスディゾルバー(制品名:clm-2.2sd、エム·テクニック制造)、以转子转速15000rpm搅拌30分钟,制备均质地被混合了的羟乙基纤维素水溶液(粒子性状控制溶液),向粒子性状控制溶液中投入工序c中得到的吲哚美辛微粒的湿饼,进行分散处理。
具体地,在0.1wt%羟乙基纤维素(粘度:200~300mpa·s、在水中2%,在20℃下、tci制造)水溶液中投入工序c中得到的吲哚美辛微粒以致成为0.2wt%后,使用超声波分散机(ヒールッシャー制造:up200s),以输出0.5%、循环0.5、在处理温度37℃±3℃下进行15分钟分散处理,得到吲哚美辛微粒分散液。用于tem观察时,将得到的吲哚美辛微粒分散液滴加到火棉胶膜,在室温下干燥,得到观察用试样。将tem观察结果示于图6中。
tem观察的结果确认了使0.1wt%羟乙基纤维素水溶液作用过的吲哚美辛微粒的平均一次粒径为98nm左右。另外,从上述分散液将吲哚美辛微粒过滤回收,使用纯水清洗后,在-0.095mpag下25℃下真空干燥16小时,得到干燥粉体。将对于得到的干燥粉体的x射线衍射测定结果示于图5下段。
x射线衍射测定的结果确认了工序2中使0.1wt%羟乙基纤维素水溶液作用过的吲哚美辛微粒转变为α型的结晶。予以说明,在图5中,为了比较,将工序c中回收·清洗后的吲哚美辛微粒(工序2处理前)的x射线衍射测定结果也一并揭示。
(实施例a2)
除了将实施例a1的工序2中的羟乙基纤维素的浓度从0.1wt%变为0.05wt%这点以外,在与实施例a1相同条件下制造了吲哚美辛微粒。将实施例a2的工序2中得到的吲哚美辛微粒的tem照片示于图7中。如图7中所看到那样,确认了吲哚美辛微粒的平均一次粒径为126nm左右。
(比较例a1)
作为实施例a1、工序2的粒子性状控制中的溶剂,没有添加表面活性剂,只使用了作为对于吲哚美辛具有一部分溶解能力的溶剂的纯水。此外,在与实施例a1相同条件下制造了吲哚美辛微粒。
将比较例a1的工序2中得到的吲哚美辛微粒的tem观察结果示于图8中。由tem观察结果确认了粒子粗大化至850nm左右。
(实施例a3:吲哚美辛微粒)
从工序0至工序c与实施例a1相同,将工序2中的对于有机物微粒具有一部分溶解能力的溶剂从实施例a1中的纯水变为己烷,作为对于有机物微粒可进行生长抑制的表面活性剂,将实施例a1中的羟乙基纤维素变为span80,制造了吲哚美辛微粒。
具体地,使用作为高速旋转式分散乳化装置的クレアミックスディゾルバー(制品名:clm-2.2sd、エム·テクニック制造),以转子转速15000rpm搅拌30分钟,由此制备均质地混合的0.01wt%的span80(wako制造)的己烷溶液(粒子性状控制溶液)。在粒子性状控制溶液中投入工序c中得到的吲哚美辛微粒以致成为0.2wt%后,使用磁力搅拌器以150rpm在温度27℃下搅拌16小时,进行分散处理,得到吲哚美辛微粒分散液。由得到的分散液将吲哚美辛微粒过滤回收,在-0.095mpag下25℃下真空干燥16小时,得到了x射线衍射测定用的干燥粉体。将x射线衍射测定结果示于图9下段。予以说明,图9上段为实施例a3的工序c中得到的吲哚美辛微粒的x射线衍射测定结果。
x射线衍射测定的结果确认了使0.01wt%的span80的己烷溶液作用过的吲哚美辛微粒转变为γ型的结晶。
然后,将上述干燥粉体用0.1%的羟乙基纤维素(0.1wt%羟乙基纤维素(粘度:200~300mpa·s、在水中2%,在20℃下、tci制造)水溶液进行分散,得到了tem观察用试样。将tem观察结果示于图10中。
tem观察的结果确认了实施例a3的工序2中使0.01wt%的span80的己烷溶液(粒子性状控制溶液)作用过的吲哚美辛微粒的平均一次粒径为138nm左右。
(比较例a2:吲哚美辛微粒)
作为实施例a3、工序2的粒子性状控制中的溶剂,没有添加表面活性剂,只使用了作为对于吲哚美辛具有一部分溶解能力的溶剂的己烷。此外在与实施例a3相同条件下制造了吲哚美辛微粒。
将比较例a2、工序2中得到的吲哚美辛微粒的tem观察结果示于图11中。由tem观察结果确认了粒子粗大化至1000nm左右。
(实施例a4:吲哚美辛微粒)
实施例a4中,改变实施例a1~3的工序0中的、吲哚美辛析出溶剂和吲哚美辛原料溶液的种类,在工序1中使α型的吲哚美辛微粒析出。另外,改变了实施例a1的工序2中的、对于有机物微粒可进行生长抑制的表面活性剂。
工序0中,对于有机物微粒的析出溶剂(a液),使用了纯水。由于为单一溶剂,因此没有进行制备。作为有机物微粒的原料溶液(b液),在乙醇中混合晶型为γ型的吲哚美辛以致成为1.0wt%、混合聚乙烯基吡咯烷酮(kollidon12pf(basf制造))以致成为1.0wt%,制备了吲哚美辛原料溶液。通过与实施例a1~3同样地使用作为高速旋转式分散乳化装置的クレアミックス(制品名:clm-2.2s、エム·テクニック制造),以转子转速15000rpm在约35℃下搅拌30分钟,制备了均质的吲哚美辛原料溶液。
工序1:混合·析出、工序c:回收·清洗的程序与实施例a1~3相同。与实施例a1~3同样地,在工序1中使吲哚美辛微粒析出,使用工序c中得到的湿饼,得到了tem观察用试样和x射线衍射测定用的干燥粉体。将tem观察结果示于图12,将x射线衍射测定结果示于图13上段。
tem观察的结果确认了工序c的清洗后的吲哚美辛微粒的平均一次粒径为146nm左右。另外,x射线衍射测定的结果确认了上述吲哚美辛微粒为α型的结晶。予以说明,结晶度为50%。
<工序2:粒子性状控制>
对于工序2的粒子性状控制而言,作为对于有机物微粒具有一部分溶解能力的溶剂,使用纯水,作为对于有机物微粒可进行生长抑制的表面活性剂,使用了lutrolf127(basf制造)。通过在纯水中添加lutrolf127,使用作为高速旋转式分散乳化装置的クレアミックスディゾルバー(制品名:clm-2.2sd、エム·テクニック制造),以转子转速15000rpm搅拌30分钟,制备了均质地混合的lutrolf127水溶液(粒子性状控制溶液)。在粒子性状控制溶液中投入工序c中得到的吲哚美辛微粒的湿饼,进行了分散处理。
具体地,在0.1wt%lutrol127水溶液中投入工序c中得到的吲哚美辛微粒以致成为0.2wt%后,使用超声波分散机(ヒールッシャー制造:up200s),以输出0.5%、循环0.5、在处理温度37℃±3℃下进行15分钟分散处理,得到了吲哚美辛分散液。将得到的吲哚美辛分散液滴加到火棉胶膜,在室温下干燥,得到了tem观察用试样。将tem观察结果示于图14中。
tem观察的结果确认了使0.1wt%lutrolf127水溶液作用过的吲哚美辛微粒的平均一次粒径为168nm左右。另外,将上述分散液过滤回收,水洗后,在-0.095mpag下25℃下真空干燥16小时,得到了x射线衍射测定用的干燥粉体。将x射线衍射测定结果示于图13下段。
x射线衍射测定的结果确认了使0.1wt%羟乙基纤维素水溶液作用过的吲哚美辛微粒是与工序c中得到的吲哚美辛微粒相同的α型。另外,确认了结晶度为62.5%,与工序c中得到的吲哚美辛微粒相比,结晶度提高了。认为是由于工序1、2中得到的吲哚美辛微粒中所含的非晶质的部位被结晶化。
(比较例a3:吲哚美辛微粒)
作为实施例a4、工序2的粒子性状控制中的溶剂,没有添加表面活性剂,只使用了作为对于吲哚美辛具有一部分溶解能力的溶剂的纯水。此外在与实施例a4相同条件下制造了吲哚美辛微粒。
将tem观察结果示于图15中。由tem观察结果确认了粒子粗大化至1160nm左右。
(实施例a5:姜黄素微粒)
<工序0:有机物微粒的析出溶剂(a液)和有机物微粒的原料溶液(b液)的制备>在工序0中,对于有机物微粒的析出溶剂(a液),使用了纯水。由于为单一溶剂,因此没有进行制备。作为有机物微粒的原料溶液(b液),在乙醇中混合晶型为1型的姜黄素以致成为0.5wt%、混合聚乙烯基吡咯烷酮(kollidon12pf(basf制造))以致成为0.5wt%,制备了姜黄素原料溶液。通过与实施例a1~4同样地使用作为高速旋转式分散乳化装置的クレアミックス(制品名:clm-2.2s、エム·テクニック制造),以转子转速15000rpm在约35℃下搅拌30分钟,制备了均质的姜黄素原料溶液。
<工序1:混合·析出>
接着,使用图2(a)中所示的微反应器将有机物微粒的析出溶剂和制备的有机物微粒的原料溶液混合。具体地,从图2(a)中所示的微反应器的第1导入部d1将作为第1被处理流体(a液)的上述制备完的有机物微粒的析出溶剂(在此为纯水)以500ml/分钟、在5℃下导入到处理用面间,一边以转速1700rpm使处理用部10运转一边将作为第2被处理流体(b液)的制备完的有机物微粒的原料溶液以30ml/分钟、在25℃下导入到处理用面1、2间,在薄膜流体中混合。然后,包含姜黄素微粒的排出液从流体处理用面1、2间排出。
<工序c:回收·清洗>
通过将上述排出液过滤、将上清液除去,将姜黄素微粒回收。然后,用清洗溶剂(纯水)进行3次的反复清洗,得到了姜黄素微粒的湿饼。将上述湿饼用纯水稀释,将其稀释液滴加到火棉胶膜,使用在室温下干燥的观察用试样,进行了tem观察。
另外,将上述湿饼在-0.095mpag下真空干燥16小时,得到了x射线衍射测定用的干燥粉体。将tem观察结果示于图16中,将x射线衍射测定结果示于图17上段。
由tem观察结果确认了姜黄素微粒的平均一次粒径为88nm左右。另外,x射线衍射测定的结果确认了得到的粒子为非晶质。
<工序2:粒子性状控制>
对于工序2的粒子性状控制而言,作为对于有机物微粒具有一部分溶解能力的溶剂,使用纯水,作为对于有机物微粒可进行生长抑制的表面活性剂,使用了聚乙烯醇。通过在纯水中添加聚乙烯醇(pva),使用作为高速旋转式分散乳化装置的クレアミックスディゾルバー(制品名:clm-2.2sd、エム·テクニック制造),以转子转速15000rpm搅拌30分钟,从而制备了均质地混合的聚乙烯醇水溶液(粒子性状控制溶液)。在粒子性状控制溶液中投入工序c中得到的姜黄素微粒的湿饼,进行了分散处理。
具体地,在作为粒子性状控制溶液的0.2wt%聚乙烯醇500(完全皂化型)水溶液中投入工序c中得到的姜黄素微粒以致成为0.2wt%后,使用超声波分散机(ヒールッシャー制造:up200s),以输出0.5%、循环0.5、在处理温度30℃±3℃下进行30分钟分散处理,得到了姜黄素微粒分散液。将得到的分散液滴加到火棉胶膜,在室温下干燥,得到了tem观察用试样。将tem观察结果示于图18中。
tem观察的结果确认了使0.2wt%聚乙烯醇500(完全皂化型)水溶液作用过的姜黄素微粒的平均一次粒径为108nm左右。另外,将上述分散处理后的液体过滤回收,水洗后,在-0.095mpag下25℃下真空干燥12小时,得到了x射线衍射测定用的干燥粉体。将x射线衍射测定结果示于图17中段。
x射线衍射测定的结果确认了使0.2wt%聚乙烯醇500(完全皂化型)水溶液作用过的姜黄素微粒从非晶质转变为3型的结晶。
(比较例a4)
作为实施例a5、工序2的粒子性状控制中的溶剂,没有添加表面活性剂,只使用了作为对于姜黄素具有一部分溶解能力的溶剂的纯水。此外在与实施例a5相同条件下制造了姜黄素微粒。
将tem观察结果示于图19中。由tem观察结果确认了虽然转变为3型的结晶,但姜黄素微粒的平均粒径为980nm左右,粒子粗大化。
(实施例a6)
从工序0至工序c与实施例a5相同,将工序2中的对于有机物微粒具有一部分溶解能力的溶剂从实施例a5中的纯水变为己烷,制造了姜黄素微粒。
作为对于有机物微粒可进行生长抑制的表面活性剂,使用span80(wako制造),通过在己烷中添加span80,使用作为高速旋转式分散乳化装置的クレアミックスディゾルバー(制品名:clm-2.2sd、エム·テクニック制造),以转子转速15000rpm搅拌30分钟,从而制备了均质地混合的溶液(粒子性状控制溶液)。在粒子性状控制溶液中投入工序c中得到的姜黄素微粒的干燥粉体,进行了分散处理。
具体地,在作为粒子性状控制溶液的0.01wt%的span80的己烷溶液中投入工序c中得到的姜黄素微粒以致成为0.2wt%后,在与实施例a5相同条件下进行分散处理,得到了姜黄素微粒。由得到的分散液将姜黄素微粒过滤回收,在-0.095mpag下25℃下真空干燥16小时,得到了x射线衍射测定用的干燥粉体。将x射线衍射测定结果示于图17下段。
x射线衍射测定的结果确认了用0.1wt%的span80的己烷溶液处理过的姜黄素微粒从非晶质转变为2型的结晶。
然后,将上述干燥粉体用0.1%的羟乙基纤维素(0.1wt%羟乙基纤维素(粘度:200~300mpa·s在水中2%在20℃下、tci制造)水溶液进行分散,得到了tem观察用试样。将tem观察结果示于图20中。tem观察的结果确认了使0.1wt%的span80的己烷溶液作用过的姜黄素微粒的一次粒径为97nm左右。
(比较例a5)
作为实施例a6、工序2的粒子性状控制中的溶剂,没有添加表面活性剂,只使用了作为对于姜黄素具有一部分溶解能力的溶剂的己烷。此外在与实施例a6相同条件下制造了姜黄素微粒。
将比较例a5、工序2中得到的姜黄素微粒的tem观察结果示于图21中。由tem观察结果确认了虽然转变为3型的结晶,但粒子粗大化至1000nm以上。
(实施例a7:聚丙烯微粒)
<工序0:有机物微粒的析出溶剂(a液)和有机物微粒的原料溶液(b液)的制备>
在工序0中,对于有机物微粒的析出溶剂(a液),使用了丙酮。由于为单一溶剂,因此没有进行制备。作为有机物微粒的原料溶液(b液),将晶型为α型的聚丙烯混合到甲苯中,制备了1wt%聚丙烯原料溶液。通过与实施例a1~6同样地使用作为高速旋转式分散乳化装置的クレアミックス(制品名:clm-2.2s、エム·テクニック制造),以转子转速15000rpm在约85℃下搅拌30分钟,制备了均质的聚丙烯原料溶液。
<工序1:混合·析出>
在实施例a7中,作为采用间歇方式的工序1的例a,使用クレアミックス将有机物微粒的析出溶剂与有机物微粒的原料溶液混合。具体地,边以15000rpm将冷却到5℃的丙酮(a液)500ml搅拌,边将30ml的制备完的有机物微粒的原料溶液(b液)在80℃下缓慢地滴入而混合。然后,从クレアミックス回收包含聚丙烯微粒的液体。
<工序c:回收·清洗>
通过将上述包含聚丙烯微粒的液体过滤,将上清液除去,从而将聚丙烯微粒回收。然后,用清洗溶剂(丙酮)进行反复清洗,得到了聚丙烯微粒的湿饼。
将上述湿饼在-0.095mpag下真空干燥16小时,得到了sem观察用和x射线衍射测定用的干燥粉体。将sem观察结果示于图22中,将x射线衍射测定结果示于图23上段中。
sem观察结果确认了聚丙烯微粒的平均一次粒径为124nm左右。另外,x射线衍射测定的结果确认了得到的粒子为α型的结晶。予以说明,结晶度为85.6%。
<工序2:粒子性状控制>
对于工序2的粒子性状控制而言,作为对于有机物微粒具有一部分溶解能力的溶剂,使用异丙醇水溶液(ipa+纯水),作为对于有机物微粒可进行生长抑制的表面活性剂,使用了tween80(wako制造)。通过在异丙醇水溶液中添加tween80,使用作为高速旋转式分散乳化装置的クレアミックスディゾルバー(制品名:clm-2.2sd、エム·テクニック制造),以转子转速15000rpm搅拌30分钟,从而制备了均质地混合的水溶液(粒子性状控制溶液)。在粒子性状控制溶液中投入工序c中得到的聚丙烯微粒的湿饼,进行了分散处理。
具体地,在作为粒子性状控制溶液的0.1wt%tween80/2.0wt%异丙醇/97.9wt%水溶液中投入工序c中得到的聚丙烯微粒以致成为0.2wt%后,使用超声波分散机(ヒールッシャー制造:up200s),以输出0.5%、循环0.5、在处理温度70℃±3℃下进行15分钟分散处理,得到了聚丙烯微粒分散液。从得到的分散液将聚丙烯微粒过滤回收,使用纯水清洗后,在-0.095mpag下25℃下真空干燥16小时,得到了sem观察用和x射线衍射测定用的干燥粉体。将sem观察结果示于图24中,将x射线衍射测定结果示于图23下段中。
sem观察的结果确认了聚丙烯微粒的平均一次粒径为197nm左右。另外,x射线衍射测定的结果确认了得到的粒子为与工序c中得到的聚丙烯微粒相同的α型的结晶。予以说明,确认了结晶度为93.5%,与工序c中得到的聚丙烯微粒相比,结晶度提高了。认为原因在于使工序1、2中得到的聚丙烯微粒中所含的非晶质的部位结晶化。
(比较例a6)
作为实施例a7、工序2的粒子性状控制中的溶剂,没有添加表面活性剂,只使用了作为对于聚丙烯具有一部分溶解能力的溶剂的异丙醇水溶液(2.0wt%异丙醇/98.0wt%水溶液)。此外在与实施例a7相同条件下制造了聚丙烯微粒。
将sem观察结果示于图25中。由sem观察结果确认了平均一次粒径为512nm左右。
(实施例a8:吡诺克辛微粒)
<工序0:有机物微粒的析出溶剂(a液)和有机物微粒的原料溶液(b液)的制备>
<有机物微粒的析出溶剂(a液)的制备>
作为有机物微粒的析出溶剂(a液),以柠檬酸成为1.9wt%的方式与纯水混合,制备了吡诺克辛析出溶剂。通过使用作为高速旋转式分散乳化装置的クレアミックス(制品名:clm-2.2s、エム·テクニック制造),以转子转速15000rpm在约35℃下搅拌30分钟,从而制备了均质的吡诺克辛析出溶剂。制备的吡诺克辛析出溶剂的ph为2.1。
<有机物微粒的原料溶液(b液)的制备>
作为有机物微粒的原料溶液(b液),在0.01mol/l的氢氧化钠水溶液中以吡诺克辛成为0.2wt%的方式混合,制备了吡诺克辛原料溶液。通过使用作为高速旋转式分散乳化装置的クレアミックス(制品名:clm-2.2s、エム·テクニック制造),以转子转速15000rpm在约35℃下搅拌30分钟,从而制备了均质的吡诺克辛原料溶液。制备的吡诺克辛原料溶液的ph为12.0。
<工序1:混合·析出>
接着,使用图2(a)中所示的微反应器将制备的有机物微粒的析出溶剂和制备的有机物微粒的原料溶液混合。具体地,从图2(a)中所示的微反应器的第1导入部d1将作为第1被处理流体(a液)的上述制备完的吡诺克辛析出溶剂(在此为1.9wt%柠檬酸水溶液)以300ml/分钟、在25℃下导入到处理用面间,一边以转速1700rpm使处理用部10运转,一边将作为第2被处理流体(b液)的制备完的吡诺克辛原料溶液以20ml/分钟、在25℃下导入到处理用面1、2间,在薄膜流体中混合。然后,包含吡诺克辛微粒的排出液从流体处理用面1、2间排出。包含吡诺克辛微粒的排出液的ph为2.48。
将包含吡诺克辛微粒的排出液滴加到火棉胶膜,在室温下干燥,准备观察用试样,进行了tem观察。由tem观察结果确认了吡诺克辛微粒的平均一次粒径为42nm左右。将tem观察结果示于图26中。
<工序2:粒子性状控制>
对于工序2的粒子性状控制而言,作为对于有机物微粒具有一部分溶解能力的溶剂,使用纯水,作为对于有机物微粒可进行生长抑制的表面活性剂,使用了tween80、苯扎氯铵。通过在纯水中添加tween80和苯扎氯铵,使用作为高速旋转式分散乳化装置的クレアミックスディゾルバー(制品名:clm-2.2sd、エム·テクニック制造),以转子转速15000rpm搅拌15分钟,从而制备均质地混合的tween80、苯扎氯铵水溶液(粒子性状控制溶液),在该粒子性状控制溶液中加入工序1中得到的吡诺克辛微粒分散液,进行了分散处理。
具体地,在0.03wt%tween80-0.01wt%苯扎氯铵-99.96wt%纯水500g中加入了工序1中得到的吡诺克辛微粒分散液500g。然后,使用クレアミックスダブルモーション(エム·テクニック制:clm-2.2/3.7w),以转子转速20000rpm、丝网转速18000rpm、在处理温度42℃±3℃下进行30分钟的分散处理,得到了吡诺克辛微粒分散液。用于tem观察时,将得到的吡诺克辛微粒分散液滴加到火棉胶膜,在室温下干燥,得到了观察用试样。将tem观察结果示于图27中。
tem观察的结果确认了用包含tween80的水溶液中处理过的吡诺克辛微粒的平均一次粒径为48nm左右。另外,从上述分散液将吡诺克辛微粒过滤回收,使用纯水清洗后,在-0.095mpag下25℃下真空干燥16小时,得到了干燥粉体。对于得到的干燥粉体,将x射线衍射测定结果示于图28下段。予以说明,为了结晶性的比较,将进行工序2的分散处理前的吡诺克辛微粒分散液(工序1排出液)与上述同样地过滤回收,使用纯水清洗后,对于在相同条件下使其干燥的粉体,进行了xrd衍射测定。将这些结果示于图28的上段中。由xrd衍射测定的结果确认了工序2的分散处理前的结晶度为58.1%,而工序2的分散处理后的结晶度上升到了63.2%。另外,x射线衍射测定的结果确认了得到的粒子的晶型在处理前后没有变化。
将以上的实施例a1~8、比较例a1~6的结果示于表1中。
[表1]
应予说明,在表1的结晶度的栏中记载的“--”表示为非晶质。另外,结晶度如上述那样使用xrd来测定,将对于工序2的采用有机物微粒的粒子性状控制溶液的处理前后、工序2处理后的平均一次粒径的值(xa)对于工序2处理前即工序c结束后(只在实施例a8中是工序1结束后)的平均一次粒径的值(xb)的比率(xa/xb)设为结晶度变化率。进而,关于吡诺克辛,由于一般不使用晶型的名称,因此表1的实施例a8的晶型的栏记为“×”,表示在工序2的处理前·处理后在晶型上没有变化。
由实施例a1、2、比较例a1的结果可知:在作为对于吲哚美辛具有一部分溶解能力的溶剂的纯水中,吲哚美辛微粒进行粗大化,但通过进行将对于有机物微粒可进行生长抑制的表面活性剂添加到上述溶剂中的工序2的粒子性状控制,吲哚美辛微粒的粒径基本上没有变化,能够抑制粗大化。进而可知,通过进行工序2的粒子性状控制,可以从非晶质向α型的结晶转变。
另外,由实施例a1、2和实施例a3的结果确认:即使改变对于吲哚美辛具有一部分溶解能力的溶剂、对于吲哚美辛可进行生长抑制的表面活性剂的种类,也获得工序2的粒子性状控制的效果。
由实施例a1~4的结果可知工序2的粒子性状控制的效果不依赖于吲哚美辛的晶型。另外可知:通过工序2的粒子性状控制,不仅结晶转变、而且结晶度提高了。
如实施例a5~8、比较例a4~6中所示那样,以上的效果表示不限于实施例a1~4的吲哚美辛,关于其他的有机物微粒也是有效的。
进而,由实施例a8的结果确认了:即使省略对于工序1中得到的有机物微粒进行清洗和/或溶剂置换的工序c,也获得工序2的粒子性状控制的效果。
(实施例b的组)
以下,对于实施例b的组(红色有机颜料)进行说明。
在实施例b的xrd测定中,使用了粉末x射线衍射测定装置(制品名:x‘pertprompd、panalytical公司制造)。测定条件为测定范围:6~60°、cu对阴极、管电压45kv、管电流40ma、扫描速度16°/min。
另外,在tem观察中使用透射型电子显微镜、jem-2100(jeol公司制造)。作为观察条件,使加速电压为80kv,使观察倍率为25000倍。
(间歇方式的情况)
与实施例a同样地,通过图1中所示的工序0~工序2而制造了颜料微粒。
(工序0)
下述的表2中所示的、颜料微粒析出溶剂(a液)和颜料微粒原料溶液(b液)的制备:a液、b液都是使液温为40℃,各自使用磁力搅拌器和搅拌子以300rpm搅拌30分钟,得到了各制备液。
(工序1)
a液与b液的混合:如下述的表2中所示那样,在使用磁力搅拌器和搅拌子以300rpm搅拌a液,且投入b液,使有机颜料微粒析出。予以说明,将工序1中得到的包含有机颜料微粒的浆料过滤,使用纯水将有机颜料微粒清洗(工序c),得到了有机颜料微粒的湿饼。或者通过真空干燥法等干燥处理得到了有机颜料微粒的干燥粉体。
(工序2)
将上述有机颜料的湿饼或干燥粉体投入到添加了表面活性剂或分散剂的对于有机物微粒具有一部分溶解能力的溶剂(粒子性状控制溶液)中,使用具有旋转的搅拌叶片的搅拌机クレアミックスclm-2.2s,进行了一定时间的搅拌处理。
[表2]
缩写的说明:dmso:二甲基亚砜、btma:苄基三甲基氢氧化铵、meoh:甲醇、soln:溶液、pr254:c.i.颜料红254、pr122:c.i.颜料红122
对于工序c中的清洗后、工序2中的搅拌后的颜料微粒分别通过tem观察算出平均一次粒径,通过xrd测定对结晶度进行测定比较(参照下述表3)。其中,结晶度是相对于将结晶化的成分和非晶质合在一起的全体、结晶化的成分所占的比例,颜料的结晶度越高,对于光、热、湿气等的耐久性就越提高。予以说明,◎、○、△、×的定义如下所述。
设为da:工序2的作用后的粒子的平均一次粒径,db:工序2之前、清洗后的平均一次粒径时,
“◎”是如下的情况:
da/db为1.0~4.0的范围,并且
在设为xa:工序2的作用后的粒子的结晶度,xb:工序2之前、清洗后的结晶度时,xa/xb为1.05以上,并且
da为80nm以下,并且
从微粒的均一性的观点出发,以25000倍进行了3视野的tem观察时,工序2(作用后)的各个颜料微粒中完全没有确认有db超过8.0倍的粒子。
“○”是如下的情况:
da/db为1.0~4.0的范围,并且
xa/xb为1.05以上,并且
da比80nm大,并且
从微粒的均一性的观点出发,以25000倍进行了3视野的tem观察时,工序2(作用后)的各个颜料微粒中完全没有确认有db超过8.0倍的粒子。
“△”是如下的情况:
da/db为1.0~4.0的范围,并且
xa/xb为1.05以上,并且
从微粒的均一性的观点出发,以25000倍进行了3视野的tem观察时,工序2(作用后)的各个颜料微粒中确认有最大1个db超过8.0倍的粒子。
“×”为不符合上述“◎”、“○”、“△”的任意者的情况。
其中,平均一次粒径的计测方法是通过tem观察中以25000倍在多个视野中所观察到的微粒合计100个的平均求出。
[表3]
(使用了微反应器的情况)
在实施例b中,a液相当于从图2(a)中所示的微反应器的第1导入部d1所导入的第1被处理流体,b液相当于同样从第2导入部d2所导入的第2被处理流体。两者的更换是任意的。予以说明,在实施例b中,作为微反应器,使用了ulrea(エム·テクニック制造)。
在以以下的表4中所示的条件进行运转的微反应器内将a液和b液混合,与实施例a同样地通过图1中所示的工序0~工序2而制造了颜料微粒。
(工序0)如表5中所示那样,颜料微粒析出溶剂(a液)和颜料微粒原料溶液(b液)的制备:a液、b液都是使液温为40℃,使用上述クレアミックスclm-2.2s,a液以10000rpm、b液以20000rpm搅拌30分钟,得到了各制备液。
(工序1)使用图1中所示的微反应器,将有机颜料溶解液(b液)与不良溶剂(a液)混合,使有机颜料微粒析出。予以说明,将(工序1)中得到的包含有机颜料微粒的浆料过滤,使用纯水将有机颜料微粒清洗(工序c),得到有机颜料微粒的湿饼或者通过真空干燥法等干燥处理得到了有机颜料微粒的干燥粉体。
(工序2)将上述有机颜料的湿饼或干燥粉体投入到添加了表面活性剂或分散剂的对于有机物微粒具有一部分溶解能力的溶剂(粒子性状控制溶液)中,使用上述クレアミックスclm-2.2s进行了一定时间的搅拌处理。
[表4]
表5
接着,与间歇方式的情况同样地,对于工序1和工序2中得到的颜料微粒分别通过tem观察算出平均一次粒径,通过xrd测定对结晶度进行测定比较(参照以下的表6)。符号、缩写的含义与间歇方式的情况相同。◎、○、△、×的定义也与间歇方式的情况相同。
[表6]
如从实施例b的实验编号1-2中得到的本发明的红色颜料纳米粒子的tem观察照片(图30)中看到那样,将本发明申请的制造方法应用于实施例b的情况下,可知由于粒子性状控制溶液的作用,得到的红色有机颜料微粒能够抑制颈缩、生长。
应予说明,对于实验编号2-1,进行了上述的tem观察时,在工序2(作用后)的各个颜料微粒中确认有2个以上的db超过8.0倍的粒子,因此将其作为了比较例。
(实施例c的组)
接着,对于实施例c的组(青色有机颜料)进行说明。
在实施例c的xrd测定中使用粉末x射线衍射测定装置(制品名:x‘pertprompd、panalytical公司制造)。测定条件为测定范围:6~60°、cu对阴极、管电压45kv、管电流40ma、扫描速度16°/min。
另外,在tem观察中使用透射型电子显微镜、jem-2100(jeol公司制造)。作为观察条件,使加速电压为80kv,使观察倍率为25000倍。
(使用了微反应器的情况)
在以下的条件下将a液与b液混合,通过下述的程序制造了青色有机颜料微粒。
予以说明,在实施例c中,作为微反应器,使用了ulrea(エム·テクニック制造)。这种情况下,a液相当于从图2(a)中所示的微反应器的第1导入部d1所导入的第1被处理流体,b液相当于同样从第2导入部d2所导入的第2被处理流体。第1导入部d1、第2导入部d2的更换是任意的。
将实施例c中的实验配方示于表7中。
[表7]
其中,cupc:铜酞菁、tiopc:氧钛酞菁、copc:钴酞菁、h2so4:浓硫酸。
在实施例c中,也与实施例a同样地通过图1中所示的工序0~工序2而制造了颜料微粒。
(工序0)
在用上述实验配方、使用ulrea实施混合·析出时,如以下那样制备了a液和b液。
有机颜料粒子析出溶剂(a液)制备条件:
如上述实验配方中记载那样在单一溶剂的情况下,不需要制备,但例如应用专利文献8中记载的实验配方的情况下,希望使用クレアミックス来进行搅拌。例如,在实施例c中使用clm-2.2s以10000rpm搅拌30分钟。
有机颜料粒子的原料溶液(b液)制备条件:
使用クレアミックスclm-2.2s以20000rpm进行了30分钟搅拌。另外,a液、b液都是使制备温度为40℃。
(工序1)
在以下的表8中所示的运转条件下使用ulrea,将有机颜料粒子析出溶剂(a液)与有机颜料粒子的原料溶液(b液)混合,使青色有机颜料微粒析出。
将工序1中得到的包含青色有机颜料微粒的浆料过滤,使用纯水将青色有机颜料微粒清洗(工序c),得到了青色有机颜料微粒的湿饼(或通过真空干燥法等干燥处理得到了青色有机颜料微粒的干燥粉体)。
(工序2)
将工序1中得到的青色有机颜料微粒的湿饼(或干燥粉体)投入到对于有机物微粒具有一部分溶解能力的溶剂的单独溶剂或者添加了表面活性剂和/或分散剂的对于有机物微粒具有一部分溶解能力的溶剂(粒子性状控制溶液)中。投入后,使用クレアミックス进行了一定时间的搅拌处理。
[表8]
对于使用了微反应器的情况下的搅拌处理前后的青色有机颜料微粒的粒径的变化和结晶度变化,示出以下的表9中。
[表9]
应予说明,关于与上述的实施例c有关的缩写、用语的定义,如以下的表10中所示那样。
[表10]
应予说明,表9中的评价结果的定义与关于红色有机颜料的实施例b相同。
对于工序c(清洗)后和工序2(作用)后的青色有机颜料微粒分别通过tem观察算出平均一次粒径,通过xrd测定对结晶度进行测定比较(表9)。另外,示出了上述实施例c的实验编号1-7中得到的tem图像(图32~图35)。另外,示出了上述实施例c的实验编号3-1中得到的比较例的tem图像(图37)。
根据上述的实验结果,在工序2中,在对于青色有机颜料微粒具有一部分溶解能力的溶剂(表9中简单地记载为溶剂)中不含表面活性剂和分散剂的比较例的实验中,发生了粒子的生长、颈缩。图37为其一例。另一方面,将本发明申请的制造方法应用于实施例c的情况下,可知得到的青色有机颜料微粒能够抑制颈缩、生长。
(间歇方式的情况)
与使用了微反应器的情况同样地,通过将用以下的表11中所示的配方制备的a液和b液混合,制造青色有机颜料微粒。
[表11]
其中,cupc:铜酞菁、tiopc:氧钛酞菁、copc:钴酞菁、h2so4:浓硫酸。
实施例c中的处理内容如以下所述。
(工序0)
在使用上述实验配方采用间歇方式实施混合·析出时,如以下那样制备了a液和b液。
有机颜料粒子析出溶剂(a液)制备条件:
如实验配方中记载那样在单一溶剂的情况下,不需要制备,但例如应用专利文献8中记载的实验配方的情况下,希望使用クレアミックス来进行搅拌。例如,在实施例c中使用clm-2.2s以10000rpm搅拌30分钟。
有机颜料粒子的原料溶液(b液)制备条件:
使用クレアミックスclm-2.2s以20000rpm进行30分钟搅拌。
另外,a液、b液都是使制备温度为40℃。
(工序1)
使用磁力搅拌器和搅拌子以300rpm对烧杯内的有机颜料粒子析出溶剂(a液)进行搅拌,且投入有机颜料粒子的原料溶液(b液),由此将a液与b液混合,使青色有机颜料微粒析出。将工序1中得到的包含青色有机颜料微粒的浆料过滤,使用纯水将青色有机颜料微粒清洗(工序c),得到青色有机颜料微粒的湿饼(或者通过真空干燥法等干燥处理得到了青色有机颜料微粒的干燥粉体)。
(工序2)
将工序1中得到的青色有机颜料微粒的湿饼(或干燥粉体)投入到对于有机物微粒具有一部分溶解能力的溶剂的单独溶剂或者添加了表面活性剂和/或分散剂的对于有机物微粒具有一部分溶解能力的溶剂(粒子性状控制溶液)中。投入后,使用クレアミックス进行一定时间的搅拌处理。
对于用间歇方式的搅拌处理前后的青色有机颜料微粒的粒径的变化和结晶度变化,示于以下的表12中。
[表12]
对于工序c(清洗)后和工序2(作用)后的青色有机颜料微粒分别通过tem观察算出平均一次粒径,通过xrd测定对结晶度进行测定比较(表12)。符号、缩写等的含义与使用了微反应器的情况相同。
根据上述的实验结果,与使用了上述微反应器的情况的实施例c同样地,即使在间歇式的情况下,也是在工序2中在对于青色有机颜料微粒具有一部分溶解能力的溶剂(表12中简单地记载为溶剂)中不含表面活性剂和分散剂的情况下,发生了粒子的生长、颈缩。作为一例,将实验编号4-4中得到的铜-氧钛-钴酞菁微粒的tem观察照片示于图36中。另一方面,将本发明申请的制造方法应用于实施例c的情况下,可知由于粒子性状控制溶液的作用,得到的青色有机颜料微粒能够抑制颈缩、生长。
以上由实施例a,实施例b、实施例c示出本发明对于全部有机物的微粒都有效。
符号的说明
1第1处理用面
2第2处理用面
10第1处理用部
11第1支架
20第2处理用部
21第2支架
d1第1导入部
d2第2导入部
d10开口部
d20开口部
- 还没有人留言评论。精彩留言会获得点赞!