一种适用于近紫外/蓝光激发的白光LED用绿/黄色荧光材料及其制备方法与流程
本发明属于发光材料技术领域,具体涉及一种适用于近紫外/蓝光激发的白光LED用绿/黄色荧光材料及其制备方法。
背景技术:
传统照明光源,如白炽灯、荧光灯,因其能效低,污染环境,寿命短等缺点逐渐被新一代照明光源所取代。白光发光二极管(LED)作为一种新型的固态照明技术,发光效率高、使用寿命长、环境友好,其应用从显示领域已逐渐发展到背光照明、装饰照明、交通信号照明,并逐渐取代传统照明光源,成为现代社会主要的普通照明技术。目前市场上常用的获得白光LED的方法是在蓝光LED芯片上涂覆黄色荧光粉,黄光与部分蓝光组合实现白光。但是目前广泛使用的YAG黄色荧光粉缺乏红色组分,致使获得的白光LED光源色温偏高,显色指数偏低。难以满足普通照明的需求。
(氮)氧化物荧光粉在热稳定性、化学稳定性及发光效率方面具有优异的性能,且激发范围宽,适用于蓝光、近紫外或紫外光激发,打破了白光LED用红色荧光粉长期以来的沉寂和瓶颈,受到科学界和产业界极大关注。然而(氮)氧化物荧光粉的合成一般需要气氛条件下高温/高压热处理的苛刻条件,目前市面销售不多,价格昂贵,这也极大地制约了该系列荧光粉的应用。
技术实现要素:
有鉴于此,本发明要解决的技术问题在于提供一种适用于近紫外/蓝光激发的白光LED用绿/黄色荧光材料及其制备方法,本发明提供的荧光材料制备方法操作步骤简单,反应烧结温度较低,烧结时间短,所使用的原料成本低廉,可重复性强,可以实现批量制备。
本发明提供了一种适用于近紫外/蓝光激发的白光LED用绿/黄色荧光粉,其特征在于,通式如式(I)所示:
(Ba0.97-ySry)3Si6-xAlxO15-zNδ:0.09Eu2+,式(I);
式(I)中,0≤x≤0.3,0≤y≤0.5,z=3δ/2,0≤δ≤2。
优选的,化学式如式(II)~式(V)中的一种:
Ba2.91Si6O15:0.09Eu2+,式(II);
Ba2.91Si6O15-zNδ:0.09Eu2+,式(III);式(III)中,z=3δ/2,0<δ≤2;
Ba2.91Si6-xAlxO15-zNδ:0.09Eu2+,式(IV);式(IV)中,0.03≤x≤0.3,0<δ≤2,z=3δ/2;
(Ba0.97-y-Sry)3Si6O15-zNδ:0.09Eu2+,式(V);式(V)中,0.1≤y≤0.5,z=3δ/2,0<δ≤2。
本发明还提供了一种具有式(II)所示化学式的适用于近紫外/蓝光激发的白光LED用绿/黄色荧光粉的制备方法,包括以下步骤:
A)按照化学组成称取钡源化合物、铕源化合物、硅源化合物、柠檬酸、乙醇、聚乙二醇和水混合搅拌,得到溶胶;
B)将所述溶胶加热,得到凝胶;
C)将所述凝胶在空气条件下预烧后煅烧,再在氨气条件下1100~1300℃烧结10~24h,得到式(II)所示的适用于近紫外/蓝光激发的白光LED用绿/黄色荧光粉。
本发明还提供了一种具有式(III)、式(IV)或式(V)所示化学式的适用于近紫外/蓝光激发的白光LED用绿/黄色荧光粉的制备方法,包括以下步骤:
a)按照化学组成称取钡源化合物、铕源化合物、硅源化合物、铝源化合物、锶源化合物、柠檬酸、乙醇、聚乙二醇和水混合搅拌,得到溶胶;
b)将所述溶胶加热,得到凝胶;
c)将所述凝胶在空气条件下预烧后,再在氨气条件下1100~1300℃烧结10~24h,得到具有式(III)、式(IV)或式(V)所示化学式的适用于近紫外/蓝光激发的白光LED用绿/黄色荧光粉。
优选的,所述钡源化合物选自钡的硝酸盐、钡的醋酸盐或钡的氯化物;
所述铕源化合物选自铕的硝酸盐、铕的醋酸盐或铕的氯化物;
所述硅源化合物选自正硅酸四乙酯。
优选的,所述铝源化合物选自铝的硝酸盐、铝的醋酸盐或铝的氯化物;
所述锶源化合物选自锶的硝酸盐、锶的醋酸盐或锶的氯化物。
优选的,所述预烧的温度为400~600℃,所述预烧的时间为4~8小时。
优选的,所述煅烧的温度为700~900℃,所述煅烧的时间为4~8小时。
优选的,所述氨气的流量为0.1~1.5L/min。
优选的,所述加热的温度为70~85℃,所述加热的时间为12~24h。
与现有技术相比,本发明提供了一种具有式(II)所示化学式的适用于近紫外/蓝光激发的白光LED用绿/黄色荧光粉的制备方法,包括以下步骤:A)按照化学组成称取钡源化合物、铕源化合物、硅源化合物、柠檬酸、乙醇、聚乙二醇和水混合搅拌,得到溶胶;B)将所述溶胶加热,得到凝胶;C)将所述凝胶在空气条件下预烧后煅烧,再在氨气条件下1100~1300℃烧结10~24h,得到式(II)所示的适用于近紫外/蓝光激发的白光LED用绿/黄色荧光粉。本发明提供的制备方法操作步骤简单,反应烧结温度较低,烧结时间短,所使用的原料成本低廉,可重复性强,可以实现批量制备,减少了工作量和能耗。避免了传统制备方法所需的高温、高压、长时间烧结等条件。另外,本发明提供的荧光材料激发范围更广,不仅与蓝光LED芯片相匹配,更可以与近紫外光LED芯片匹配,并且具有高的热稳定性和化学稳定性改善材料在近紫外/蓝光区的吸收强度,更好地与商业LED芯片匹配,并导致光谱红移,有效地改善WLEDs的显色指数与色温,实现“暖白光”照明的要求。
附图说明
图1为本发明实施例1、2制备的荧光材料的XRD衍射图谱;
图2是本发明实施例1制备的荧光材料的激发、发射光谱图;
图3是本发明实施例2制备的荧光材料的激发、发射光谱图;
图4是本发明实施例3、4、5制备的荧光材料的XRD衍射图谱;
图5是本发明实施例3、4、5制备的荧光材料的激发、发射光谱图;
图6是本发明实施例6、7、8制备的荧光材料的XRD衍射图谱;
图7是本发明实施例6、7、8制备的荧光材料的激发、发射光谱图;
图8是本发明实施例1~8制备的荧光材料的热猝灭曲线及与商业Ba2SiO4:Eu2+绿粉、YAG:Ce3+黄粉的热衰减对比图。
具体实施方式
本发明提供了一种适用于近紫外/蓝光激发的白光LED用绿/黄色荧光粉,其化学式如式(I)所示:
(Ba0.97-ySry)3Si6-xAlxO15-zNδ:0.09Eu2+,式(I);
式(I)中,0≤x≤0.3,0≤y≤0.5,z=3δ/2,0≤δ≤2。
优选的,其化学式如式(II)~式(V)中的一种:
Ba2.91Si6O15:0.09Eu2+,式(II);
Ba2.91Si6O15-zNδ:0.09Eu2+,式(III);式(III)中,z=3δ/2,0<δ≤2;
Ba2.91Si6-xAlxO15-zNδ:0.09Eu2+,式(IV);式(IV)中,0.03≤x≤0.3,0<δ≤2,z=3δ/2;
(Ba0.97-y-Sry)3Si6O15-zNδ:0.09Eu2+,式(V);式(V)中,0.1≤y≤0.5,z=3δ/2,0<δ≤2。
具体的,当式(I)中的x/y/z/δ=0时,所述适用于近紫外/蓝光激发的白光LED用绿/黄色荧光粉的化学组成为Ba2.91Si6O15:0.09Eu2+。
当式(I)中的x/y=0时,所述适用于近紫外/蓝光激发的白光LED用绿/黄色荧光粉的化学组成为Ba2.91Si6O15-zNδ:0.09Eu2+,并且z=3δ/2,0<δ≤2。
当式(I)中的y=0时,所述适用于近紫外/蓝光激发的白光LED用绿/黄色荧光粉的化学组成为Ba2.91Si6-xAlxO15-zNδ:0.09Eu2+,并且0.03≤x≤0.3,0<δ≤2,z=3δ/2。
当式(I)中的x=0时,所述适用于近紫外/蓝光激发的白光LED用绿/黄色荧光粉的化学组成为(Ba0.97-y-Sry)3Si6O15-zNδ:0.09Eu2+,并且0.1≤y≤0.5,z=3δ/2,0<δ≤2。
本发明还提供了一种具有式(II)所示化学式的适用于近紫外/蓝光激发的白光LED用绿/黄色荧光粉的制备方法,包括以下步骤:
A)按照化学组成称取钡源化合物、铕源化合物、硅源化合物、柠檬酸、乙醇、聚乙二醇和水混合搅拌,得到溶胶;
B)将所述溶胶加热,得到凝胶;
C)将所述凝胶在空气条件下预烧后煅烧,再在氨气条件下1100~1300℃烧结10~24h,得到式(II)所示的适用于近紫外/蓝光激发的白光LED用绿/黄色荧光粉。
本发明首先按照化学组成称取钡源化合物、铕源化合物、硅源化合物、柠檬酸、乙醇、聚乙二醇和水混合搅拌,得到溶胶;
所述溶胶具体按照如下方法进行制备:
将钡源化合物和铕源化合物溶解于水中,得到混合溶液;
向所述混合溶液中加入柠檬酸进行搅拌,接着加入无水乙醇、硅源化合物以及聚乙二醇混合搅拌,得到溶胶。
其中,所述钡源化合物选自钡的硝酸盐、钡的醋酸盐或钡的氯化物,优选为钡的硝酸盐;所述铕源化合物选自铕的硝酸盐、铕的醋酸盐或铕的氯化物,优选为铕的硝酸盐;所述硅源化合物选自正硅酸四乙酯。
所述柠檬酸与所述混合溶液中金属离子的摩尔比优选为2:1。
所述聚乙二醇的数均分子量为两万,所述聚乙二醇在所述溶胶中的浓度为0.01mol/L。
将所述溶胶进行加热,得到凝胶。在本发明中,优选将所述溶胶置于烘箱中进行静置加热,所述加热的温度优选为70~85℃,更优选为80~85℃所述加热的时间优选为12~24h,更优选为18~20h。
接着,将得到的凝胶置于空气条件下预烧后煅烧,再在氨气条件下进行烧结,得到式(II)所示的适用于近紫外/蓝光激发的白光LED用绿/黄色荧光粉。
在本发明中,所述预烧的温度为400~600℃,优选为500℃;所述预烧的时间为4~8小时,优选为5~7小时。
所述煅烧的温度为700~900℃,优选为750~850℃,更优选为800℃;所述煅烧的时间为4~8小时,优选为5~7小时。
所述烧结的温度为1100~1300℃,优选为1150~1250℃,更优选为1200℃;所述烧结的时间为10~24小时,优选为12~15小时。
烧结时,所述氨气的流量为0.1~1.5L/min,优选为0.5~1.0L/min。
在本发明中,所述预烧、烧结和煅烧都在常压下进行。
本发明还提供了一种具有式(III)、式(IV)或式(V)所示化学式的适用于近紫外/蓝光激发的白光LED用绿/黄色荧光粉的制备方法,包括以下步骤:
a)按照化学组成称取钡源化合物、铕源化合物、硅源化合物、铝源化合物、锶源化合物、柠檬酸、乙醇、聚乙二醇和水混合搅拌,得到溶胶;
b)将所述溶胶加热,得到凝胶;
c)将所述凝胶在空气条件下预烧后,再在氨气条件下1100~1300℃烧结10~24h,得到具有式(III)、式(IV)或式(V)所示化学式的适用于近紫外/蓝光激发的白光LED用绿/黄色荧光粉。
本发明首先按照化学组成称取钡源化合物、铕源化合物、硅源化合物、铝源化合物、锶源化合物、柠檬酸、乙醇、聚乙二醇和水混合搅拌,得到溶胶。
(1)当所述适用于近紫外/蓝光激发的白光LED用绿/黄色荧光粉的化学式为式(III)时,所述溶胶具体按照如下方法进行制备:
将钡源化合物和铕源化合物溶解于水中,得到混合溶液;
向所述混合溶液中加入柠檬酸进行搅拌,接着加入无水乙醇、硅源化合物以及聚乙二醇混合搅拌,得到溶胶。
(2)当所述适用于近紫外/蓝光激发的白光LED用绿/黄色荧光粉的化学式为式(IV)时,所述溶胶具体按照如下方法进行制备:
将钡源化合物、铝源化合物和铕源化合物溶解于水中,得到混合溶液;
向所述混合溶液中加入柠檬酸进行搅拌,接着加入无水乙醇、硅源化合物以及聚乙二醇混合搅拌,得到溶胶。
(3)当所述适用于近紫外/蓝光激发的白光LED用绿/黄色荧光粉的化学式为式(V)时,所述溶胶具体按照如下方法进行制备:
将钡源化合物、锶源化合物和铕源化合物溶解于水中,得到混合溶液;
向所述混合溶液中加入柠檬酸进行搅拌,接着加入无水乙醇、硅源化合物以及聚乙二醇混合搅拌,得到溶胶。
在上述(1)、(2)和(3)的情况中,所述钡源化合物选自钡的硝酸盐、钡的醋酸盐或钡的氯化物,优选为钡的硝酸盐;所述铕源化合物选自铕的硝酸盐、铕的醋酸盐或铕的氯化物,优选为铕的硝酸盐;所述铝源化合物选自铝的硝酸盐、铝的醋酸盐或铝的氯化物,优选为铝的硝酸盐;所述锶源化合物选自锶的硝酸盐、锶的醋酸盐或锶的氯化物,优选为锶的硝酸盐。所述硅源化合物选自正硅酸四乙酯。
所述柠檬酸与所述混合溶液中金属离子的摩尔比优选为2:1。
所述聚乙二醇的数均分子量为两万,所述聚乙二醇在所述溶胶中的浓度为0.01mol/L。
将所述溶胶进行加热,得到凝胶。在本发明中,优选将所述溶胶置于烘箱中进行静置加热,所述加热的温度优选为70~85℃,更优选为80~85℃所述加热的时间优选为12~24h,更优选为18~20小时。
将所述凝胶在空气条件下预烧后,再在氨气条件下进行烧结,得到具有式(III)、式(IV)或式(V)所示化学式的适用于近紫外/蓝光激发的白光LED用绿/黄色荧光粉。
在本发明中,所述预烧的温度为400~600℃,优选为500℃;所述预烧的时间为4~8小时,优选为5~7小时。
所述烧结的温度为1100~1300℃,优选为1150~1250℃,更优选为1200℃;所述烧结的时间为10~24小时,优选为12~15小时。
烧结时,所述氨气的流量为0.1~1.5L/min,优选为0.5~1.0L/min。
在本发明中,所述预烧和烧结都在常压下进行。
本发明提供的制备方法操作步骤简单,反应烧结温度较低,烧结时间短,所使用的原料成本低廉,可重复性强,可以实现批量制备,减少了工作量和能耗。避免了传统制备方法所需的高温、高压、长时间烧结等条件。另外,本发明提供的荧光材料激发范围更广,不仅与蓝光LED芯片相匹配,更可以与近紫外光LED芯片匹配,并且具有高的热稳定性和化学稳定性改善材料在近紫外/蓝光区的吸收强度,更好地与商业LED芯片匹配,并导致光谱红移,有效地改善WLEDs的显色指数与色温,实现“暖白光”照明的要求。
本方案中通过阳离子/阴离子部分取代,如用N部分取代O,或用Sr取代Ba,Al取代Si,可以提高荧光材料的热稳定性,改善材料在近紫外/蓝光区的吸收强度,更好地与商业LED芯片匹配,并导致光谱红移,有效地改善WLEDs的显色指数与色温,实现“暖白光”照明的要求。
为了进一步理解本发明,下面结合实施例对本发明提供的适用于近紫外/蓝光激发的白光LED用绿/黄色荧光材料及其制备方法进行说明,本发明的保护范围不受以下实施例的限制。
实施例1:Ba2.91Si6O15:0.09Eu2+
(1)称取0.7605g Ba(NO3)2,浓度为1mol/L的90μL Eu(NO3)3溶液,加入去离子水至溶液体积为10mL,搅拌至固体物完全溶解,加入1.2608g柠檬酸,继续搅拌30分钟,加入30mL无水乙醇,搅拌均匀后加入1.341mL TEOS搅拌均匀,最后加入数均分子量为20000的PEG 8g,室温快速搅拌2小时,形成透明溶胶。
(2)将溶胶转移至陶瓷平底蒸发皿中,置于80℃烘箱中静置24小时形成凝胶,将凝胶在400℃的马弗炉中预烧5h;将得到产物简单研磨后转移至刚玉坩埚中,在800℃煅烧6h,得前驱体。
(3)将步骤(2)中制备得到的前驱体置于管式炉中,通入氨气,控制流量在1.0L/min,升温至1250℃,常压下烧结10h;烧结结束后,冷却至室温,取出样品得到最终产物。
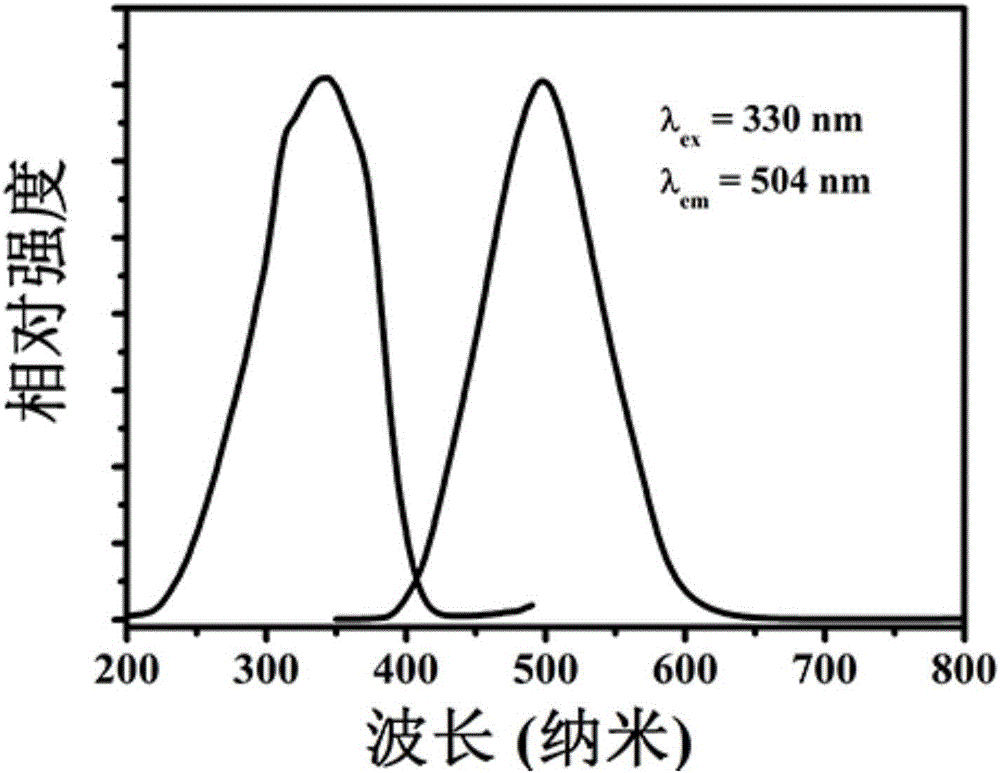
图1为实施例1制备的荧光材料的XRD图谱,粉体为BaSi2O5的单相。图2为实施例1制备的荧光材料的激发、发射光谱图,图2显示实施例1可以被近紫外光(365-380nm)有效地激发,发出明亮的蓝绿光,峰值波长为504nm。
实施例2:Ba2.91Si6O15-zNδ:0.09Eu2+
(1)称取0.7605g Ba(NO3)2,浓度为1mol/L的90μL Eu(NO3)3溶液,加入去离子水至溶液体积为10mL,搅拌至固体物完全溶解,加入1.2608g柠檬酸,继续搅拌30分钟,加入30mL无水乙醇,搅拌均匀后加入1.341mL TEOS搅拌均匀,最后加入数均分子量为20000的PEG 8g,室温快速搅拌2小时,形成透明溶胶。
(2)将溶胶转移至陶瓷平底蒸发皿中,置于85℃烘箱中静置12小时形成凝胶,将凝胶在500℃的马弗炉中预烧7h;将得到产物简单研磨后转移至刚玉坩埚中,得灰黑色前驱体。
(3)将步骤(2)中制备得到的前驱体置于管式炉中,通入氨气,控制流量在1.0L/min,升温至1200℃,常压下烧结12h;烧结结束后,冷却至室温,取出样品得到最终产物。
图1为实施例2制备的荧光材料的XRD图谱,粉体为Ba2Si2O5与Ba3Si6O12N2的中间相。图3为实施例2制备的荧光材料的激发、发射光谱,图3显示实施2不仅可以被(近)紫外光激发还可以被蓝光有效激发,发出明亮的绿色和蓝色。较之实施例1,通过1200℃氨气氮化作用,氮的掺杂,发射光谱从504nm红移至520nm,对近紫外-蓝光区的吸收增强,热稳定性明显提高。图8是本发明实施例1~8制备的荧光材料的热猝灭曲线及与商业Ba2SiO4:Eu2+绿粉、YAG:Ce3+黄粉的热衰减对比图。实施例2制备的荧光材料与商业Ba2SiO4:Eu2+绿粉的热衰减对比曲线如图8c所示。
实施例3:Ba2.91Si6-xAlxO15-zNδ:0.09Eu2+,x=0.03荧光粉
(1)称取0.7605g Ba(NO3)2、0.0113g Al(NO3)3·9H2O、浓度为1mol/L的90μL Eu(NO3)3溶液,加入去离子水至溶液体积为10mL,搅拌至固体物完全溶解,加入1.2860g柠檬酸,继续搅拌30分钟,加入30mL无水乙醇,搅拌均匀后加入1.328mL TEOS搅拌均匀,最后加入数均分子量为20000的PEG 8g,室温快速搅拌2小时,形成透明溶胶。
(2)将溶胶转移至陶瓷平底蒸发皿中,置于75℃烘箱中静置20小时形成凝胶,将凝胶在600℃的马弗炉中预烧4h;将得到产物简单研磨后转移至刚玉坩埚中,得灰黑色前驱体。
(3)将步骤(2)中制备得到的前驱体置于管式炉中,通入氨气,升温至1150℃,控制流量在0.5L/min,常压下烧结12h;烧结结束后,冷却至室温,取出样品得到最终产物。
实施例4:Ba2.91Si6-xAlxO15-zNδ:0.09Eu2+,x=0.12
(1)称取0.7605g Ba(NO3)2、0.045g Al(NO3)3·9H2O、浓度为1mol/L的90μL Eu(NO3)3溶液,加入去离子水至溶液体积为10mL,搅拌至固体物完全溶解,加入1.2860g柠檬酸,继续搅拌30分钟,加入30mL无水乙醇,搅拌均匀后加入1.328mL TEOS搅拌均匀,最后加入数均分子量为20000的PEG 8g,室温快速搅拌2小时,形成透明溶胶。
(2)将溶胶转移至陶瓷平底蒸发皿中,置于80℃烘箱中静置24小时形成凝胶,将凝胶在500℃的马弗炉中预烧4h;将得到产物简单研磨后转移至刚玉坩埚中,得灰黑色前驱体。
(3)将步骤(2)中制备得到的前驱体置于管式炉中,通入氨气,升温至1200℃,控制流量在0.5L/min,常压下烧结12h;烧结结束后,冷却至室温,取出样品得到最终产物。
实施例5:按照Ba2.91Si6-xAlxO15-zNδ:0.09Eu2+,x=0.3荧光粉
(1)称取0.7605g Ba(NO3)2、0.1125g Al(NO3)3·9H2O、浓度为1mol/L的90μL Eu(NO3)3溶液,加入去离子水至溶液体积为10mL,搅拌至固体物完全溶解,加入1.2860g柠檬酸,继续搅拌30分钟,加入30mL无水乙醇,搅拌均匀后加入1.328mL TEOS搅拌均匀,最后加入数均分子量为20000的PEG 8g,室温快速搅拌2小时,形成透明溶胶。
(2)将溶胶转移至陶瓷平底蒸发皿中,置于80℃烘箱中静置24小时形成凝胶,将凝胶在500℃的马弗炉中预烧4h;将得到产物简单研磨后转移至刚玉坩埚中,得灰黑色前驱体。
(3)将步骤(2)中制备得到的前驱体置于管式炉中,通入氨气,升温至1200℃,控制流量在0.5L/min,常压下烧结12h;烧结结束后,冷却至室温,取出样品得到最终产物。
图4为实施例3、4、5制备的荧光材料的XRD图谱,粉体为Ba3Si6O12N2的单相。图5显示实施例3、4、5可以同时被近紫外及蓝光有效激发,发出明亮的绿光,较之实施例1、实施例2,通过Al-N共掺杂(摩尔百分比为1-30%)使激发光谱在蓝光区的吸收增强,发射光谱从504nm(实施例1)红移至520nm,热稳定性有所提高。图8是本发明实施例1~8制备的荧光材料的热猝灭曲线及与商业Ba2SiO4:Eu2+绿粉、YAG:Ce3+黄粉的热衰减对比图。图8a显示实施例3、4、5制备的荧光材料的热衰减曲线。图8c显示实施例3与商业Ba2SiO4:Eu2+绿粉的热衰减对比。
实施例6:按照(Ba0.97-ySry)3Si6O15-zNδ:0.09Eu2+,y=0.1荧光粉
(1)称取0.3685g Ba(NO3)2、0.0635g Sr(NO3)2、浓度为1mol/L的90μL Eu(NO3)3溶液,加入去离子水至溶液体积为10mL,搅拌至固体物完全溶解,加入1.608g柠檬酸,继续搅拌30分钟,加入30mL无水乙醇,搅拌均匀后加入1.341mL TEOS搅拌均匀,最后加入数均分子量为20000的PEG 8g,室温快速搅拌2小时,形成透明溶胶。
(2)将溶胶转移至陶瓷平底蒸发皿中,置于70℃烘箱中静置24小时形成凝胶,将凝胶在500℃的马弗炉中预烧4h;将得到产物简单研磨后转移至刚玉坩埚中,得灰黑色前驱体。
(3)将步骤(2)中制备得到的前驱体置于管式炉中,通入氨气,升温至1200℃,控制流量在0.6L/min,常压下烧结10h;烧结结束后,冷却至室温,取出样品得到最终产物。
实施例7:按照(Ba0.97-ySry)3Si6O15-zNδ:0.09Eu2+,y=0.3荧光粉
(1)称取0.3685g Ba(NO3)2、0.1905g Sr(NO3)2、浓度为1mol/L的90μL Eu(NO3)3溶液,加入去离子水至溶液体积为10mL,搅拌至固体物完全溶解,加入1.608g柠檬酸,继续搅拌30分钟,加入30mL无水乙醇,搅拌均匀后加入1.341mL TEOS搅拌均匀,最后加入数均分子量为20000的PEG 8g,室温快速搅拌2小时,形成透明溶胶。
(2)将溶胶转移至陶瓷平底蒸发皿中,置于80℃烘箱中静置24小时形成凝胶,将凝胶在500℃的马弗炉中预烧4h;将得到产物简单研磨后转移至刚玉坩埚中,得灰黑色前驱体。
(3)将步骤(2)中制备得到的前驱体置于管式炉中,通入氨气,升温至1150℃,控制流量在0.5L/min,常压下烧结24h;烧结结束后,冷却至室温,取出样品得到最终产物。
实施例8:按照(Ba0.97-ySry)3Si6O15-zNδ:0.09Eu2+,y=0.5荧光粉
(1)称取0.3685g Ba(NO3)2、0.3174g Sr(NO3)2、浓度为1mol/L的90μL Eu(NO3)3溶液,加入去离子水至溶液体积为10mL,搅拌至固体物完全溶解,加入1.608g柠檬酸,继续搅拌30分钟,加入30mL无水乙醇,搅拌均匀后加入1.341mL TEOS搅拌均匀,最后加入数均分子量为20000的PEG 8g,室温快速搅拌2小时,形成透明溶胶。
(2)将溶胶转移至陶瓷平底蒸发皿中,置于85℃烘箱中静置12小时形成凝胶,将凝胶在600℃的马弗炉中预烧4h;将得到产物简单研磨后转移至刚玉坩埚中,得灰黑色前驱体。
(3)将步骤(2)中制备得到的前驱体置于管式炉中,通入氨气,升温至1200℃,控制流量在0.5L/min,常压下烧结12h;烧结结束后,冷却至室温,取出样品得到最终产物。
图6为实施例6、7、8制备的荧光材料的XRD图谱,由图6显示,粉体为Ba3Si6O12N2的单相。图7为实施例6、7、8制备的荧光材料的激发、发射光谱。由图7可知,荧光材料可以同时被近紫外及蓝光有效激发,发出明亮的黄光,较之实施例1、2、3,通过Sr-N共掺杂使激发光谱在蓝光区的吸收增强,发射光谱从504nm(实施例1)红移至556nm,热稳定性有所提高。图8b显示实施例6、7、8的热衰减曲线。图8d显示实施例7与商业YAG:Ce3+黄色荧光粉的热衰减对比。
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!