一种红色长余辉纳米发光材料的制作方法
本发明涉及发光材料技术领域,更具体地,涉及一种红色长余辉纳米发光材料。
背景技术:
稀土长余辉发光材料是指一类能在吸收太阳光或灯光的能量后,将一部分能量存储起来,然后在一定激发下将存储能量缓慢的以可见光的形式释放出来。稀土长余辉发光材料被广泛应用于弱光照明、应急指示、建筑装饰及工艺美术等领域,近年来,在信息存储、高能射线探测等领域也有所拓展应用。可见光区的稀土长余辉发光材料主要分为蓝色、黄绿色和红色长余辉材料。纵观稀土掺杂红色长余辉的发展历程,其基质体系主要包含硫化物体系、钛酸盐体系、硫氧化物体系、硅酸盐体系,也有一些碱土金属氧化物的长余辉材料处于探索阶段。
然而,目前,红色长余辉纳米发光材料仍然较少,需要开发出新的性能优异的红色长余辉纳米发光材料。
并且,现有技术难以制备形貌稳定且均匀的长余辉纳米发光材料。因此,还需要开发出新的制备方法,该方法能够制备形貌稳定且均匀的红色长余辉纳米发光材料。
技术实现要素:
本发明为克服上述现有技术所述的缺陷,提供一种红色长余辉纳米发光材料,提供的红色长余辉纳米发光材料经高温热处理前后形貌稳定,其余辉时长可达3000s。
为解决上述技术问题,本发明采用的技术方案是:
一种红色长余辉纳米发光材料,如式(ⅰ)或式(ⅱ)所示:
la2o2co3:xeu3+(ⅰ);
la2o2co3:xeu3+,yho3+(ⅱ);
其中,0.5%≤x≤50%;1.0%≤y≤10%。
上述红色长余辉纳米发光材料的基质为la2o2co3,以eu3+为激活离子,或者,以eu3+和ho3+分别为激活离子和共激活离子;该红色长余辉纳米发光材料形貌稳定,其发光波长为600~650nm,余辉时长可达3000s。
本发明提供的红色长余辉纳米发光材料能够在弱光照明、应急指示、建筑装饰及工艺美术领域得到广泛的应用,具有极大的应用价值。
上述式(ⅰ)、式(ⅱ)中的x表示eu3+与材料中稀土离子总量的百分比;y表示ho3+与材料中稀土离子总量的百分比。式(ⅰ)中,稀土离子包括la3+和eu3+。式(ⅱ)中稀土离子包括la3+、eu3+和ho3+。
优选地,x满足0.5%≤x≤10%。更优选地,x的值为1%。x的值为1%时,红色长余辉纳米发光材料的余辉性能最优。
发明人研究发现,在掺杂eu3+的基础上,共掺杂少量ho3+后,纳米发光材料的余辉性能将得到提升。但是,如果ho3+的掺杂量太高,纳米发光材料的余辉性能将降低。因此,优选地,1.0%≤y≤5%。更优选地,y的值为1%。在eu3+的掺杂量为1%的基础上,掺杂1%的ho3+后,制得的纳米发光材料的余辉性能获得进一步提升。
优选地,所述红色长余辉纳米发光材料为纳米棒状。
本发明还保护上述红色长余辉纳米发光材料的制备方法,所述制备方法包括如下步骤:
s1.制备含la3+和掺杂离子的混合溶液;将混合溶液与尿素溶液混合,然后用氨水调节ph为碱性,得到反应溶液;所述掺杂离子仅为eu3+,或者,所述掺杂离子为eu3+和ho3+;
s2.将步骤s1.中的反应溶液在加热和搅拌条件下进行反应,经后处理,得到前驱体;
s3.将步骤s2.的前驱体在550~650℃条件下热处理1.5~2.5h,得到所述红色长余辉纳米发光材料。
上述制备方法通过沉淀法制备前驱体,然后在550~650℃条件下热处理1.5~2.5h,成功制备得到形貌稳定且均匀的棒状碳氧化物红色长余辉纳米发光材料。
所述尿素溶液的由15.015g尿素溶于100ml去离子水制得。所述氨水的浓度为25wt.%。所述热处理采用高温箱式炉,优选为马弗炉。热处理在空气氛围下进行。
优选地,步骤s3.中热处理的温度为600℃,时间为2h。
优选地,步骤s1.中混合溶液为la3+和掺杂离子的硝酸盐溶液。所述硝酸盐溶液可由对应的氧化物溶于稀硝酸制得。所述氧化物为la2o3和eu2o3,或者,所述氧化物为la2o3、eu2o3和ho2o3。
优选地,步骤s1.中用氨水调节ph的值为8~10。更优选地,步骤s1.中用氨水调节ph的值为9。
优选地,步骤s2.中反应的温度为85~95℃,反应的时间为1.5~2.5h。更优选地,步骤s2.中反应的温度为90℃,反应的时间为2h。将ph为9的反应溶液在90℃水浴条件下剧烈搅拌2h,然后自然冷却至室温,用去离子水和无水乙醇各离心洗涤三次,然后置于烘箱中90℃干燥12h。干燥后,稍加研磨,即可进行热处理。研磨可用玛瑙研钵。
所述搅拌,可以采用磁力搅拌器。步骤s2.中的反应采用可加热的磁力搅拌器。
与现有技术相比,本发明的有益效果是:
本发明提供的红色长余辉纳米发光材料的基质为la2o2co3,以eu3+为激活离子,或者,以eu3+和ho3+分别为激活离子和共激活离子。该红色长余辉纳米发光材料形貌稳定,其发光波长为600~650nm,余辉时长可达3000s,在弱光照明、应急指示、建筑装饰及工艺美术领域具有极大的应用价值。
另外,提供的制备方法通过沉淀法制备前驱体,然后在550~650℃条件下热处理1.5~2.5h,高温热处理前后形貌稳定,成功制备得到形貌稳定且均匀的红色长余辉纳米棒。
附图说明
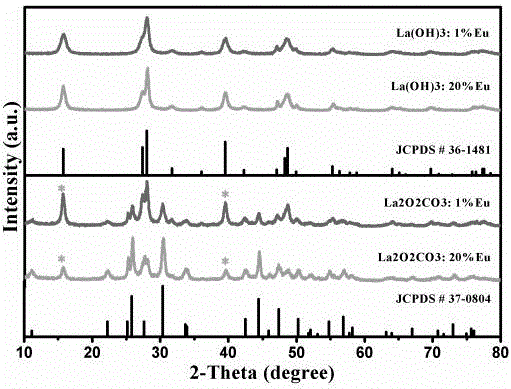
图1为本发明的实施例2和实施例4制备的样品的x射线衍射图。
图2为本发明的实施例2制备的样品透射电镜图。
图3为本发明的实施例1、2、3、4、5制备的样品的荧光激发光谱图和荧光发射光谱图。
图4为本发明的实施例1、2、3、4、5制备的样品的荧光寿命图。
图5为本发明的实施例1、2、3、4、5制备的样品的余辉衰减图。
图6为本发明的实施例2、6、7、8、9制备的样品的余辉衰减图。
图7为本发明的实施例2和实施例6制备的样品的长余辉衰减图。
图8为本发明的实施例6制备的样品的不同辐照时间的热释光图。
具体实施方式
下面结合具体实施方式对本发明作进一步的说明。
实施例中的原料均可通过市售得到;
除非特别说明,本发明采用的试剂、方法和设备为本技术领域常规试剂、方法和设备。
实施例1
称取氧化镧la2o3:1.621g溶于20ml稀硝酸中配制la(no3)3溶液,称取氧化铕eu2o3:0.000044g溶于20ml稀硝酸中配制eu(no3)3溶液,上述硝酸盐溶液各取15ml,混合,加入100ml去离子水,剧烈搅拌10min;制备0.25mol15.015g/100ml尿素溶液,加入上述溶液,剧烈搅拌20min;使用nh3•h2o(25wt.%)将上述混合溶液调ph=9,于90℃水浴剧烈搅拌2h,然后,自然冷却至室温,离心洗涤,去离子水和无水乙醇依次各洗涤三次;将洗涤离心后的样品于烘箱中90℃干燥12h,将干燥后的样品稍加研磨后,于600℃热处理2h;取出后即可得到组成为la2o2co3:0.5%eu3+的三价铕激活的长余辉纳米棒,该荧光粉在278nm光激发后发出612nm的红色长余辉,余辉衰减呈指数规律。
实施例2
本实施例与实施例1的区别在于,氧化镧la2o3为1.613g,氧化铕eu2o3为0.0002g;制得纳米发光材料的组成为la2o2co3:1%eu3+;
其他原料使用量及操作方法与实施例1相同。
实施例3
本实施例与实施例1的区别在于,氧化镧la2o3为1.466g,氧化铕eu2o3为0.018g;制得纳米发光材料的组成为la2o2co3:10%eu3+;
其他原料使用量及操作方法与实施例1相同。
实施例4
本实施例与实施例1的区别在于,氧化镧la2o3为1.303g,氧化铕eu2o3为0.070g;制得纳米发光材料的组成为la2o2co3:20%eu3+;
其他原料使用量及操作方法与实施例1相同。
实施例5
本实施例与实施例1的区别在于,氧化镧la2o3为0.815g,氧化铕eu2o3为0.440g;制得纳米发光材料的组成为la2o2co3:50%eu3+;
其他原料使用量及操作方法与实施例1相同。
实施例6
称取氧化镧la2o3:1.596g溶于20ml稀硝酸中配制la(no3)3溶液,称取氧化铕eu2o3:0.00044g溶于50ml稀硝酸中配制eu(no3)3溶液,称取氧化钬ho2o3:0.000472g溶于50ml稀硝酸中配制ho(no3)3溶液,上述硝酸盐溶液各取15ml,混合,加入100ml去离子水,剧烈搅拌10min,制备0.25mol15.015g/100ml尿素溶液,加入上述溶液,剧烈搅拌20min,使用nh3•h2o(25wt.%)将上述混合溶液调ph=9,于90℃水浴剧烈搅拌2h,然后,自然冷却至室温,离心洗涤,去离子水和无水乙醇依次各洗涤三次,将洗涤离心后的样品于烘箱中90℃干燥12h,将干燥后的样品稍加研磨后,于600℃热处理2h,取出后即可得到组成为la2o2co3:1%eu3+,1%ho3+的三价铕、钬共激活的长余辉纳米棒,该荧光粉在278nm光激发后发出612nm的红色长余辉,余辉衰减呈指数规律。
实施例7
本实施例与实施例1的区别在于,氧化镧la2o3为1.531g,氧化铕eu2o3为0.00044g,氧化钬ho2o3为0.0024g;制得纳米发光材料的组成为la2o2co3:1%eu3+,5%ho3+;
其他原料使用量及操作方法与实施例1相同。
实施例8
本实施例与实施例1的区别在于,氧化镧la2o3为1.482g,氧化铕eu2o3为0.00044g,氧化钬ho2o3为0.0038g;制得纳米发光材料的组成为la2o2co3:1%eu3+,8%ho3+;
其他原料使用量及操作方法与实施例1相同。
实施例9
本实施例与实施例1的区别在于,氧化镧la2o3为1.450g,氧化铕eu2o3为0.00044g,氧化钬ho2o3为0.0047g;制得纳米发光材料的组成为la2o2co3:1%eu3+,10%ho3+;
其他原料使用量及操作方法与实施例1相同。
结构表征与性能测试
x射线衍射检测采用bruker-d8advancex射线衍射仪;透射电镜图片检测采用捷克talosf200s场发射透射电子显微镜;激发光谱检测采用爱丁堡fls-980荧光光谱仪;发射光谱检测采用爱丁堡fls-980荧光光谱仪;余辉衰减光谱检测采用爱丁堡fls-980荧光光谱仪;热释光检测采用fj-427atlmeter(北京,中国)。
根据x射线衍射测试结果,如图1所示,从图中可以看出实施例2和实施例4制备的前驱体为la(oh)3,前驱体与标准卡片峰位一致,为纯相la(oh)3。600℃热处理2h后得到的产物la2o2co3,从与标准卡片的对比来看,存在微量的前驱体杂相,可以忽略不计。其他实施例的结果与实施例2和实施例4类似。
经透射电镜测试,如图2所示,实施例2制得的样品为纳米棒状。其他实施例与实施例2的样品的形貌一致。
根据荧光光谱,如图3所示,实施例1~5的单掺三价铕离子的荧光光谱图,表明该纳米发光材料的激发峰位位于278nm,发射峰位位于612nm,掺杂量为20%的样品荧光性能最优。一般来讲,微量共掺杂并不会改变荧光峰位,所以,后面实施例6~9讨论共掺杂时,无需再讨论共掺后的荧光谱图。
图4的荧光寿命图是在激发波长278nm,发射波长612nm下测得的,从图中可以看出,实施例1~5制得的纳米发光材料的荧光寿命长,故而进一步测试其余辉性能。采用实施例1~5的样品分别取0.083g于254nm的紫外灯下照射20min后于发射波长612nm监测下测试,得到图5,从图中可以看出,单掺杂1%的eu3+时的样品的余辉性能最优。
然后,采用实施例2、6~9的样品分别取0.083g于254nm的紫外灯下照射20min后于发射波长612nm监测下测试,得到图6;从图中可以看出,共掺杂1%的ho3+制得的样品最优。进一步对比实施例2和实施例6的余辉性能,如图7所示,其测试方法与图5和图6相同。从图7中可知,共掺后的性能大大提升。
采用实施例6的样品0.008g,测试前热处理300℃清空缺陷,用254nm的紫外灯在不同的辐照时间下得到的热释光图谱,得到图8。根据图8,可以看出辐照时间20min为最优的样品辐照时间,即能填满陷阱。
显然,本发明的上述实施例仅仅是为清楚地说明本发明所作的举例,而并非是对本发明的实施方式的限定。对于所属领域的普通技术人员来说,在上述说明的基础上还可以做出其它不同形式的变化或变动。这里无需也无法对所有的实施方式予以穷举。凡在本发明的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明权利要求的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!