不存在SDA的沸石间转化和金属封装的制作方法
本发明涉及其中具有封装金属的沸石及其制备方法。更具体地,经由没有sda的直接水热合成通过沸石间转化制备沸石。
背景
沸石是具有良好限定晶体结构的有序微孔铝硅酸盐。分子大小的空隙允许沸石催化具有独特的反应性和选择性的化学反应。用于在沸石内封装金属簇的合成方案可以扩大催化化学的多样性,其通过微孔固体的性能使得这种多样性成为可能,微孔固体的性能是基于它们的分子大小和形状选择反应物、过渡态和产物以及保护活性位点免受作为毒物的更大的物质通过滴定活性位点。在不同空隙尺寸和几何形状的沸石内封装金属簇的通用方法可用于为特定催化应用定制或选择沸石结构;该方法包括在合成期间离子交换、初始润湿和引入金属前体。参见例如,gallezot,p.,合成后改性i(post-synthesismodificationi),2002年,第257页。
小孔沸石和中孔沸石内的孔隙通过从水性介质中离子交换阻止合成后的封装方法,其需要迁移不能通过这种沸石中的小孔扩散的溶剂化金属-含氧低聚物。最近,开发了利用配位稳定的金属前体来防止金属前体作为胶体氢氧化物在高ph和水热沸石结晶所需的温度下过早沉淀的封装方法。这些方法使得在lta内成功封装pt、pd、rh、ir、re和ag簇,以及在gis和sod内成功封装pt、pd、ru和rh簇。一些沸石需要甚至分解配体稳定的金属前体的合成温度。在这种情况下,强制封装是通过首先将金属簇置于在较温和条件下形成的沸石内(母体结构),然后使样品经受将该母体沸石转化为预期骨架(子结构)的条件,同时保持封装。这些方法使得在ana内成功封装pt和ru簇。
mfi(zsm-5)是中孔富含二氧化硅(高硅)的沸石,其通常需要高的结晶温度(423-473k)和ph(>11)用于其无模板合成。通过包括使用配体稳定的金属前体的直接水热合成以及合成后交换的方法,除了在一价或二价阳离子的情况下,这种材料中的封装仍然是达不到的。
如果更有效和更容易的金属封装方法可用于高硅沸石,将对行业是有益的。
发明概述
提供了一种通过以下方法将金属封装在沸石中的方法:
(a)将金属前体插入较低骨架密度(fd)沸石中,和
(b)在不存在有机结构导向剂(sda)的情况下,将较低骨架密度(fd)沸石转化为具有较高骨架密度值的沸石。该转化通常通过直接水热合成进行。
在一个实施方案中,所述金属为pt、rh、ru或cu。在另一个实施方案中,所述具有较高骨架密度值的沸石是zsm-5、ssz-35、zsm-12或菱沸石。
在一个实施方案中,所述具有较高骨架密度值的沸石是si/al比为10或更高的高硅沸石。
在其它因素中,本发明人已经发现,用于在高硅沸石例如zsm-5、ssz-35、zsm-12或菱沸石中封装金属簇的普通策略是可行的,其通过利用低骨架密度母体结构沸石(例如bea或fau)的沸石间转化为沸石子体结构而不需要有机结构导向剂(sda)。还发现了在高硅骨架(例如zsm-5)的空隙空间内选择性封装金属簇(pt、ru、rh、cu)的催化结果。
这些沸石间转化为在微孔固体内封装簇提供了普通和方便的途径,其中可以通过合成后交换或在水热结晶期间在大孔母体沸石结构内实现前体的成功放置。在其微孔空隙内包含金属簇的这种母体结构然后可以重结晶而封装入具有较高骨架密度的子体结构(例如mfi(zsm-5))没有损失,例如mfi(zsm-5),对于这样的沸石更直接的封装方法是不可用或不切实际的。转化也在不使用sda时发生。整个过程是高效和容易的。
附图简要说明
图1a显示了由(i)bea(si/al=37.5)和fau(si/al=40)母体沸石通过(a)直接、(b)模板辅助(使用tpabr)和(c)晶种辅助(使用mfi晶种)转化合成产物的x射线衍射图。所述合成是在423k,naoh/sio2=0.35(由bea),naoh/sio2=0.5(由fau)和h2o/sio2=65(由bea),h2o/sio2=95(由fau)下进行(表1)。
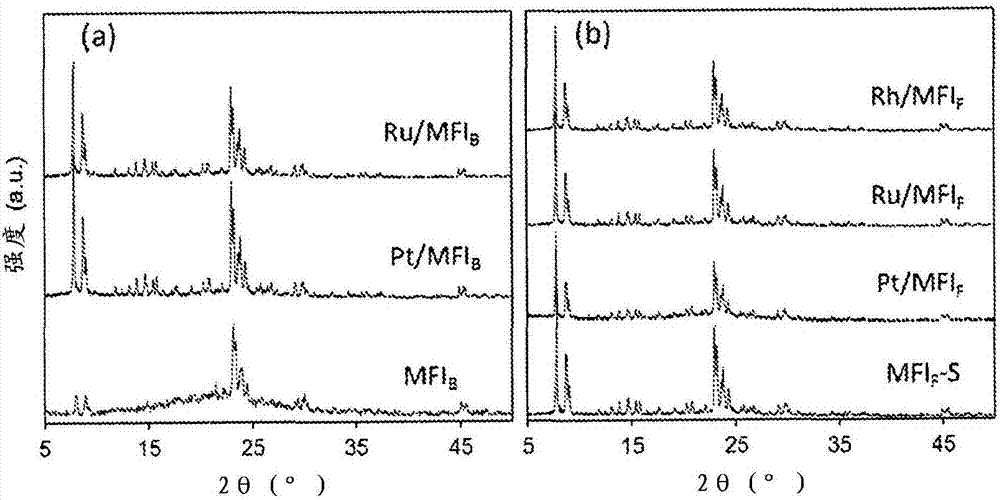
图2显示了通过作为母体沸石的包含金属簇的(a)bea和(b)fau的沸石间转化合成的mfi产物的x射线衍射图。在423k,由bea的摩尔组成为0.35naoh:1.0sio2:0.0133al2o3:65.0h2o没有晶种和由fau的摩尔组成为0.50naoh:1.0sio2:0.0125al2o3:95.0h2o带有10重量%mfi晶种下进行合成(表1)。
图3显示了(a)含有pt簇的母体bea和(b)通过离子交换方法合成的含有pt簇的母体fau的tem图像和金属簇尺寸分布,和(c)通过初始润湿浸渍法合成分散在sio2上的pt簇的tem图像和金属簇尺寸分布。
图4显示了由含pt簇的作为母体材料的(a)bea和(b)fau通过沸石间转化合成含mfi样品的pt的tem图像金属簇尺寸分布。
图5a显示了(a)通过ru/bea的沸石间转化合成的含mfi的ru和(b)通过含fau作为母体沸石的ru的沸石间转化合成的含mfi的ru和(c)通过含fau作为母体沸石的rh的沸石间转化合成的含mfi的rh的tem图像。
图6示出了在宽范围的合成条件下限制金属簇在mfi内的封装的合成因子的示意图。mi表示使用的合成方法(参见表5)。
发明详述
本发明提供了一种在沸石内封装金属的方法。沸石产物是比被转化的沸石具有更高骨架密度值的沸石。例如,包含金属或金属前体的bea(fd15.3)或fau(fd13.3)沸石可以转化成保存金属封装的mfi(fd18.4)、stf(fd16.9)、mtw(fd18.2)或cha(fd15.1)沸石。骨架密度(fd)定义为t原子/标准立方米(atoms/nm3),其中t代表沸石骨架中的si或al原子。骨架密度(fd)值可以是基于理论的全二氧化硅骨架结构的绝对值或归一化值。或者也可以用相对值,相对值将一致反映更高或更低的骨架密度值。转化在不需要有机sda下发生,同时仍保留金属封装。发现在将12-环结构(例如bea和fau)转化为8或10环结构(例如mfi、mtw、stf和cha)中特别的适用。然而,不管环结构如何,转化是从较低骨架密度沸石到较高骨架密度沸石。
转化为封装金属的高硅沸石是特别适用的,其中si/al比为10或更高,优选22,或甚至25或更高。而且,在水溶液中具有水合双层的金属前体通常大于产物或子体沸石的孔径,使得金属或金属簇不能使用常规方法插入产物沸石中,所述常规方法例如离子交换或浸渍。
本方法包括将金属前体插入较低骨架密度沸石中。然后在不存在结构化导向剂(sda)的情况下将较低骨架密度沸石转化为较高骨架沸石。在本方法中不需要有机sda的存在。通常,在转化之前或期间将子体沸石或产物沸石的晶种加入到母体或较低fd沸石中。然而,如果较低fd沸石和较高fd沸石具有共同的结构基序(commonstructuralmotif),则甚至不需要晶种。因为那时复合结构单元(compositebuildingunit)基本上相似,并且可以容易地实现更高密度的子体骨架。
不存在有机结构导向剂(sda)意味着合成不含可溶性sda。本合成不需要像在常规合成中那样使用sda试剂。因此在合成中没有可溶性sda。虽然可以使用沸石的晶种,即所述的晶种是原样制备的材料,外部添加,但是已经发现,可以与晶种相关的sda捕获在沸石的内部,并且不能离开沸石以影响合成。换句话说,新沸石不是由晶种释放sda成核的。没有从晶种释放sda,并且合成保持没有可溶性sda。
已发现转化通过直接水热合成进行。转化通常在碱性溶液中进行,例如ph>7,一直到13。水热合成的温度可以是任何合适的温度,并且可以高于较低fd沸石的结晶温度。
在实施本方法时,条件和组分的平衡可以提供改进的结果。例如,naoh含量与转化方法中使用的时间和温度平衡。在转化中,通常,二氧化硅和氧化铝由源沸石(例如,fau)和任何晶种提供。naoh/sio2的比值通常在0.25至1.00的范围,h2o/sio2的比值通常大于50。在一个实施方案中转化的时间在约1至约80小时的范围,在一个实施方案中,温度为高于较低fd沸石的结晶温度,其通常可以大于85℃,在85-130℃的范围内,或甚至更高,例如大于130℃,例如在从130-160℃的范围内。
合适的较低骨架密度沸石的实例包括y型沸石、bea和fau。合适的较高骨架密度沸石的实例包括mfi(zsm-5)、cha(菱沸石)、stf(ssz-35)和mtw(zsm-12)。
本方法可以应用于任何不能通过离子交换或浸渍直接进入子体沸石孔中的金属的封装。可以通过本发明成功地封装而不使用sda的诸多金属的实例是pt、rh、ru或cu,以及co、fe、v、ir、pd、re、ag和au。金属通常作为金属前体加入到较低fd沸石中。金属前体可以是胺或乙二胺络合物。金属前体也可以是配位的金属(ligatedmetal)。
在本方法的一个实施方案中,所用的较低fd沸石是bea或fau,金属是pt、rh或ru,较高fd沸石是zsm-5(mfi)。
在本方法的一个实施方案中,所用的较低fd沸石是y型沸石。它也可以是fau。金属是封装的铜,较高fd沸石是菱沸石、ssz-35或zsm-12。通过本方法将铜封装到菱沸石中在有益的应用和简单性方面具有特别的优点。
还已经发现,本方法中通过不使用sda,可以更好地控制金属在子体沸石或产物沸石内的位置,例如菱沸石、ssz-35和zsm-12,在一个非常有利的位置。所述位置涉及到卓越的催化优势。本方法考虑到更好地控制金属的放置。当制造封装铜的菱沸石时,这是特别有用的。
因此,通常通过本发明的简单方法可以实现将金属簇例如pt、ru、rh、cu,封装入高硅、较高fd的沸石如zsm-5、ssz-35、zsm-12或菱沸石中,本发明的方法包括将金属前体阳离子交换入母体沸石(bea、fau),用h2还原它们以形成金属簇,并将这些沸石在水热条件下转化成较高密度的子骨架。这些转化可能需要用于转化fau母体沸石的较高fd沸石的晶种,并且可保留被封装的簇。晶种也可以与bea一起用作母体或较低fd沸石,但当转化为mfi(zsm-5)时可能不需要。通常在bea和fau沸石中形成尺寸(例如,1.3-1.7nm)和表面清洁且可接近表面的均衡的簇,其尺寸在转变成zsm-5时基本上保持不变。通过直接水热合成法封装到zsm-5中发现是不成功的,因为金属前体在mfi合成所需的ph和温度下过早沉淀。在水热合成中金属前体和作为矿化剂的f-(代替oh-)的延迟引入提高了封装选择性,但是它们仍然低于通过本发明(沸石间转化)实现的封装选择性。
当前体可以通过交换引入到可以转化为具有更高骨架密度的目标子体沸石的沸石中时,无论是自发地还是通过使用晶种进行转化,本发明的这些转化为封装金属提供了普通和稳健的策略。
提供以下实施例目的是为进一步说明本发明。这些实施例仅是说明性的,而不意味着限制。
实施例1-4
实施例1-4中使用的材料包括热解sio2(cab-o-sil,hs-5,310m2g-1)、naoh(99.995%,sigmaaldrich)、fau(cbv780,zeolyst,h-fau,si/al=40)、bea(cp811e-75,zeolyst,h-bea,si/al=37.5)、四丙基溴化铵(tpabr,98%,sigmaaldrich)、naalo2(无水,riedel-de,haen,technical)、al(no3)3·9h2o(>98%,stremchemical)、nh4f(>98%,fluka)、原硅酸四乙酯(teos;98%,sigmaaldrich)、[pt(nh3)4](no3)2(99.99%,alfaaesar)、[rh(nh2ch2ch2nh2)3]cl3·3h2o(≥99.5%,aldrich)、rucl3(45-55重量%ru,sigmaaldrich)、ludoxas-30胶体二氧化硅(30重量%悬浮于h2o,sigmaaldrich)、[ru(nh3)6]cl3(98%,aldrich)、甲苯(≥99.9%,aldrich)、1,3,5-三甲基苯(98%,aldrich)、1,3,5-三异丙基苯(98%,aldrich)、he(99.999%,praxair)、air(99.999%,praxair)、0.5%o2/he(99.999%,praxair)、9%h2/he(99.999%,praxair)和h2(99.999%,praxair),按原始样品使用。
zsm-5晶种
在典型的合成中,将649g水、740g1moll-3naoh(bakerreagent)、98g四丙基溴化铵(kodakchemicals)加入到872gludoxas-30胶态sio2(dupont)中。然后将合成混合物转移到哈氏合金(hastelloy)衬里的不锈钢高压釜(3.8l)中,进行压力测试并在旋转(78rpm)下在对流烘箱中于423k下保持4天。4天后,将高压釜冷却,通过过滤并用去离子水洗涤,直到冲洗液的ph达到7-8来收集所得固体。得到的产物是结晶mfi(si/al
实施例1
在典型的合成中,将(0.5-1.0g)沸石bea(si/al=37.5)或fau(si/al=40)加入到naoh水溶液中,向其中加入具有表1所列的摩尔组成的mfi晶种或结构导向剂(tpabr)以制备最终混合物。将这些混合物置于密封的聚丙烯容器(nalgene,125cm3)中,并在环境温度下通过剧烈磁力搅拌(400rpm)均质1小时。然后将混合物转移到特氟隆衬里的不锈钢高压釜中并在静态条件于423k保持24-40小时。通过多孔圆盘布氏漏斗(chemglass,150ml,f)过滤,并用去离子水(电阻率17.9mω)洗涤,直到冲洗液的ph达到7-8以收集所得固体,并在对流烘箱中在373k下加热样品过夜,且所得产物的固体收率定义为
收率(%)=产物(g)/(母体沸石(g)+晶种(g))×100(1)
然后将所得产物在空气(1.67cm3g-1s-1)中以0.03ks-1的速率加热至623k,并在该温度下保持3小时。处理后的样品,当由bea合成时表示为mfib、mfib-t、mfib-s,当由fau合成时表示为mfif、mfif-t、mfif-s,分别表示以直接、模板辅助和晶种辅助沸石间转换方式进行。
表1样品a的初始合成摩尔组成、产物相态和收率
a在423k时,对于bea转化naoh/sio2=0.35,h2o/sio2=65,对于fau转化naoh/sio2=0.5,h2o/sio2=95
b晶种(重量%)=晶种材料(g)/母体沸石(g)×100
c初始合成摩尔组成不包括晶种中sio2的量
d收率(%)=产物(g)/母体沸石(g)×100
实施例2
由[pt(nh3)4](no3)2、[rh(nh2ch2ch2nh2)3]cl3·3h2o或[ru(nh3)6]cl3的水溶液(h2o:沸石的质量比为10:1,至实现
实施例3
通过使用m/bea样品作为母体沸石,根据本发明m/bea(m=pt、ru)的沸石间转化来实现金属簇在mfi内的封装。将m/bea(m=pt、ru)样品(0.5-1.0g)加入到naoh水溶液中以制备具有表1所列的摩尔组成的混合物。将这些混合物置于密封的聚丙烯容器(nalgene,125cm3)中,并在环境温度下通过剧烈磁力搅拌(400rpm)均质1小时。然后将混合物转移到特氟隆衬里的不锈钢高压釜中并在静态条件于所需结晶温度下(423k)下保持30小时。通过多孔圆盘布氏漏斗(chemglass,150ml,f)过滤,并用去离子水洗涤,直到冲洗液的ph达到7-8以收集所得固体。然后在对流烘箱环境空气中于373k下整夜加热样品,并于空气(1.67cm3g-1s-1)中以0.03ks-1的速率加热至673k,保持3小时;然后将金属前体暴露于9%h2/he流(1.67cm3g-1s-1)中,并以0.03ks-1加热至623k并保持2小时。在该处理之后,在暴露于环境空气之前,将样品在室温下在0.5%o2/he流(1.67cm3g-1s-1)中钝化1小时。处理后得到的样品表示为m/mfi(m=pt、ru),其由m/bea母体沸石通过沸石间转化合成。
实施例4
金属簇在mfi内的封装也通过使用m/fau样品作为母体沸石的m/fau(m=pt、ru、rh)沸石间转化来实现。将m/fau(m=pt、ru、rh)样品(0.5-1.0g)与10重量%mfi晶种(基于母体fau的重量%)一起加入到naoh水溶液中以制备具有表1中所列的摩尔组成的混合物。所有后续合成和处理步骤与通过m/bea样品的沸石间转化合成的m/mfib样品所述的相同。处理后得到的样品表示为m/mfif(m=pt、ru、rh),其由m/fau母体沸石通过沸石间转化合成。
实施例1-4的结构表征
通过粉晶x-射线衍射图(cukα辐射λ=0.15418nm,40kv,40ma,brukerd8advance)证明了产物沸石的身份和相纯度以及不存在大的金属簇。以2秒的扫描时间在0.02°间隔下测量2θ值为5-50°的衍射图。通过电感耦合等离子体原子发射光谱法(irisintrepid光谱仪;galbraith实验室)测量样品的si、al、na和金属(pt、ru或rh)含量。通过使用容积法的h2化学吸附吸收测定金属簇的分散度。在冷却至298k前,将样品在流动h2(1.67cm3s-1g-1)中以0.03ks-1加热至623k并保持1小时,然后在623k下抽空1小时以除去任何弱吸附的氢。在298k和5-50kpa的h2下在包含样品的金属上测量氢化学吸附吸收。使用1:1h:m表面(m=pt、ru、rh)吸附化学计量,外推至零压力,从总h2吸收和不可逆h2吸收之间的差异确定分散度。透射电子显微镜(tem)图像用在120kv下操作的philips/feitecnai12显微镜拍摄。在tem分析之前,将样品悬浮在乙醇中并分散在由400目cu网格(tedpellainc.)支撑的超薄碳/多孔碳膜上。通过对每个样品测量超过300个簇来确定金属簇的尺寸分布。使用下式计算表面平均簇直径dtem
dtem=σnidi3/σnidi2(2)
其中ni是具有直径di的晶体的数目。tem衍生的尺寸分布也用于计算金属簇的分散指数(di)。di值由表面平均直径(dtem;公式2)除以数均直径(dn=σnidi/σni)[30]给出。
分散指数(di)=dtem/dn=(σnidi3/σnidi2)/(σnidi/σni)(3)
该参数是金属簇的簇尺寸异质性的度量,其中单位值反映单峰簇且值小于1.5时表示相对均匀的尺寸分布。
实施例2-4的催化速率测量
在用热解sio2(cab-o-sil,hs-5,310m2g-1)稀释的催化剂样品上,使用具有活塞流的石英管式动力学反应器测量甲苯、1,3,5-三甲基苯(1,3,5-tmb)和1,3,5-三异丙基苯(1,3,5-tipb)的氢化速率。通过在稀释剂/催化剂质量比为10的情况下均匀混合、造粒和筛分颗粒以保留直径为0.18-0.25mm的聚集体来实现稀释。然后将这些颗粒(5-25mg)与相似尺寸的酸洗石英颗粒(fluka,酸纯化的,1.0g,0.18-0.25mm)混合。这种稀释用于避免颗粒或床浓度和温度梯度。
在测量氢化速率之前,通过在h2流(1.67cm3g-1s-1)中0.03ks-1下加热至623k并保持1小时以处理预还原和钝化的样品。用0.35kpa甲苯或0.26kpa1,3,5-tmb或0.15kpa1,3,5-tipb和100kpah2在473k下测量芳烃氢化速率。对于mfi(0.53×0.56nm),甲苯(动力学直径为0.59nm),而不是用于的1,3,5-tmb(动力学直径为0.74nm)以及对于bea(
当具有较低骨架密度和较大孔径的沸石可用于初始含有金属前体或簇,特别是更高价金属前体时,沸石间转化可提供用于在沸石空隙内封装金属簇的替代合成路线。然后可将这些材料随后转化为具有较高骨架密度和较小孔径的沸石,同时将封装的物质保留在沸石空隙内。
沸石间转化可以将具有较低骨架密度的结构转化为在热力学上倾向于更稳定的具有较高骨架密度的结构。这些转化可以避免昂贵的有机模板和/或减少结晶时间。这在制备具有较高si/al比,例如大于10或甚至50的较高骨架密度沸石时将是特别有价值的。它们还可以提供用于将簇封装在那些沸石中的更普通路线,否则将需要合成温度,这会导致在水热合成期间金属前体的分解,甚至对于含有保护配体的前体的分解。热力学通常允许增加沸石骨架密度(fd;这里报道为t原子/标准立方米)的转化,但不是所有这些方法在合成子体结构所需的水热条件下是动力学可达到的。
bea(fd15.3)和fau(fd13.3)可以在naoh水溶液中于一定温度下重结晶为具有更高的骨架密度的沸石,所述温度高于在水热条件下(360-400k)引起它们自己各自从无定形二氧化硅-氧化铝前体结晶的温度。在mfi晶种的存在或不存在下,在423k(摩尔组成,表1)的自生压力下,使用naoh水溶液,从bea成功地合成结晶mfi(fd18.4)样品。因此,我们得出结论,这种转化可以自发地发生,没有显着的动力学阻碍,甚至在没有mfi种子或有机结构导向剂(sda)的情况下。
母体bea和子体mfi的骨架结构和复合结构单元(cbu)包括共同mor结构基序,虽然fau和mfi缺少所述共同cbu。因此,看来似乎存在于bea中并且形成mfi需要的cbu在转化为mfi期间在bea衍生的中间体内基本上保持完整。该cbu可以帮助mfi的局部成核,并且在这样做时,减少动力学障碍,因此在甚至没有晶种时允许进行bea转化为mfi。因此,含有mor作为共同结构单元的bea至mfi转化(x射线衍射图1(i))在动力学上可行。这种共同的cbu可以充当用于使子体结构成核的动力学介质,这表明含有共同cbu单元的沸石能够克服动力学障碍,所述动力学障碍阻碍它们在沸石的热力学趋势所决定的相互转化的方向,从而阻碍形成具有较大骨架密度的结构。
在母体沸石和产物沸石之间存在共同cbu或在不存在共同cbu的情况下,合成的产物晶种有助于mfi晶体的成核,并且这样做能使从母体沸石形成的中间介质比从无定形二氧化硅和氧化铝凝胶更有效,这导致显著缩短的合成时间。结果,这些方案可以提供替代路线来合成一些沸石;这些路线可以缩短结晶时间并降低与有机部分相关的成本和环境影响。
实施例5-8
本节中的实施例描述了在bea和fau母体沸石内的pt、ru和rh金属簇的合成、结构表征和催化性能,目的是将这些材料随后用于转化为mfi。在[pt(nh3)4](no3)2、[ru(nh3)6]cl3或[rh(nh2ch2ch2nh2)3]cl3·3h2o水溶液中于353k下(使用实施例2中所述的步骤)通过与pt、ru和rh前体的离子交换合成包含金属的bea和fau(分别为m/bea和m/fau;m=pt、ru、rh)。
在流动空气中623k下持续3小时和在h2中573k下持续2小时交换和热处理之后分散在bea和fau沸石上的pt簇的tem图像显示在图3中。这些图像显示了在bea(dtem=1.6nm;下表2,使用式2计算)和fau(dtem=1.7nm;表2)中存在小的pt簇。这些簇在尺寸上分布狭窄(对于bea和fau分别为di=1.07和1.03;表2,来自式3)并且驻留在整个沸石晶体中。使用h2的金属表面化学吸附滴定给出pt部分的分散度,对于pt/bea为0.88和对于pt/fau为0.78(表2);当簇是球形的并且具有pt金属的堆积密度时,这些值分别对应于平均簇直径(dchem)为1.3和1.4nm。相比之下,在同样负载下且通过具有相同金属前体中孔sio2初始润湿浸渍制备的pt簇比pt/bea和pt/fau样品具有更大(dtem=2.4nm,dchem=1.8nm;表2)和更宽分布(di=1.96;表2),这表明在小沸石空隙内的限制抑制了烧结并伴随簇尺寸分布的拓宽。在这些沸石样品中化学吸附衍生的pt簇直径(1.3-1.4nm)很适合通过tem(1.4-1.7nm)测量,这表明可通过显微镜检测的簇包含通过h2滴定剂的化学吸附可接近的清洁表面,并且在合成期间存在的配体通过使用的热处理完全除去。类似地,分散在bea中的ru簇以及fau中的ru和rh簇显示dtem值分别为1.4、1.7和1.5nm,di值分别为1.08、1.16和1.09,dchem值分别为1.4、1.5和1.3nm,也与存在小的、均匀的和清洁的金属簇一起分散在整个bea和fau母体沸石中。
表2分散于sio2bea和mfi上金属簇的金属负载、分散度、平均尺寸和分散指数
a通过电感耦合等离子体发射光谱分析
b从h2化学吸附估计的金属分散度
c从由h2化学吸附测量获得的金属分散体估计的平均簇直径
d从tem分析估计表面加权平均簇直径(dtem),dtem=∑nidi3/∑nidi2
实施例5
沸石中的小孔允许它们基于其分子尺寸筛分反应物和产物。在位于可接近和不可接近位置内的位点处的小反应物和大反应物的相对反应速率可用于评估位于沸石空隙内的金属表面积的分数。使用甲苯和1,3,5-tipb反应物(动力学直径分别为0.59nm和0.84nm)的氢化速率来确认在母体bea
通过首先测量在分散于sio2上的非约束簇的小反应物(甲苯)和大反应物(1,3,5-tipb)氢化速率(χsio2=r甲苯/r1,3,5-tipb)来确定封装选择性;该速率比反映了在没有扩散约束的情况下这两种反应物分子的相对反应性。然后在金属-沸石样品(χ沸石)上这种比值的类似测量用来确定封装选择性参数(φ=χ沸石/χsio2),其反映样品中所有簇的表面积与在沸石晶体外部(完全可接近的)位置处的簇的表面积的比值。因此,封装选择性是在微孔网络内包含活性表面的程度的严格指示,所述微孔网络中甲苯(但不是1,3,5-tipb)可以进入。这种封装选择性参数对于无阻碍接近反应物(例如在外部沸石表面处的那些反应物)的簇而言接近1。相比之下,远大于1(
甲苯和1,3,5-tipb氢化反应导致在所有样品上各自排他地形成甲基环己烷和(顺式-和反式)1,3,5-三异丙基环己烷。下表3显示了分散在bea(m/bea)、fau(m/fau)和sio2(m/sio2)上的pt、ru、rh簇的芳烃氢化转换率。pt/bea和pt/fau上的甲苯加氢转换率与pt/sio2上的非常相似(表3),这与不存在簇尺寸效应或不存在对甲苯反应的扩散约束一致。相比之下,在pt/bea和pt/fau上得1,3,5-tipb转换率比在pt/sio2上的要低得多(分别为44和38的因子,表3),这表明1,3,5-tipb无法进入bea和fau样本中的大多数簇。因此,在pt/bea和pt/fau上的甲苯与1,3,5-tipb氢化转换率(分别为180和160的因子)比在pt/sio2(4.4)上的要高很多,这使得pt/bea和pt/fau的封装选择性参数(φ)分别为40.9和36.4(表3)。对于bea和fau母体沸石中的ru簇,封装选择性参数(表3)分别为14.3和15.4,对于fau样品中的rh簇,封装选择性参数为21.8。这些大的封装选择性值证实,当样品使用本文报道的交换和还原方法制备时,所有这些金属的簇优先存在于bea或fau沸石的空隙结构内。因此,这些材料非常适合于评估封装的金属簇是否可以(i)干扰fau或bea向mfi的转化和/或(ii)在沸石间转化期间保留。应进一步注意的是在金属配合物简单沉积在二氧化硅载体上的运行中,不提供封装保护。因此,表3中的选择性项为1.0。
表3含有bea、fau和sio2样品的金属在芳烃氢化中的催化性能a
a用0.35kpa甲苯/0.15kpa1,3,5-tipb和100kpa氢气在473k下进行氢化
b反应转换率定义为每秒每摩尔表面金属原子转化的反应物的摩尔数
cχj=r甲苯/r1,3,5-tipb,j=沸石、sio2
dφ=χ沸石/χsio2
实施例6
包含金属簇的m/bea和m/fau(m=pt、ru、rh)沸石在此用作形成mfi的前体材料,使用实施例3和4中描述的水热方法,并且在不存在金属簇时显示为成功的。所得样品在本文中表示为m/mfib(衍生自m/bea)和m/mfif(衍生自m/fau)。晶种和sda都不用于m/bea至m/mfi沸石间转化;在m/fau至m/mfi转化中使用mfi晶种(而不是sda),以避免静电和范德华相互作用,其在水热相互转换方案期间可能导致sda物质从晶体内mfi空隙中移出簇。
bea和fau沸石在母体沸石中成功地转化为具有或不具有封装簇的mfi(x-射线衍射图;没有金属,图1;有金属,图2)。随后在流动空气中673k下处理3小时,然后在623k的h2流中处理2小时没有引起mfi结晶度的可检测的变化。在623k下h2处理2小时(0.64-1.23重量%金属;表2)后,x射线衍射图在m/mfi(m=pt、rh、ru;图2)中也没有显示任何关于金属或氧化物相的线,这与mfi子体结构中不存在大的金属晶体一致。
还原和钝化m/mfi样品(m=pt、ru、rh)的tem图像检测到尺寸均匀的小簇(图4中的pt/mfib以及pt/mfif,图5中的ru/mfib、ru/mfif和rh/mfif)。从tem测量(表2)获得的表面平均的平均簇直径和di值,对于pt/mfib(对比母体pt/bea中的1.6nm和1.07)为1.7nm和1.41,对于pt/mfif为1.0nm和1.09(对比母体pt/fau中的1.7nm和1.03)。母体沸石的di值略大于相应的产物沸石,这表明在沸石间转化期间不发生显着的烧结或聚结。在pt/mfi上的h2化学吸附测量给出的平均簇直径,当从pt/bea(dchem=1.3)制备时为1.4,当从pt/fau(dchem=1.1)衍生时为1.5。这些化学吸附衍生的平均簇直径与来自tem的表面平均簇直径(0.8-1.6;表2)很好地吻合,这表明没有来自合成混合物的残余物沉积,并且在合成后处理期间没有除去。相反,pt/sio2显示dtem值为2.4nm、dchem值为1.8nm、di值为1.96;这些尺寸和分散度显著大于分散在母体(pt/bea和pt/fau)和产物(pt/mfi)沸石样品上的簇,这表明限制环境对于合成小的且均匀的金属簇是必要的。
类似地,di值(1.09-1.16对比母体样品的1.08-1.16;表2)、tem衍生的表面平均簇直径(1.3-1.5对比母体样品的1.4-1.5);(表2)和对于ru/mfib、ru/mfif和rh/mfif的化学吸附衍生的平均簇直径(1.2-1.5对比母体样品的1.1-1.2;表2)也与在mfi空隙内小的、均匀的和清洁的金属簇以及在从母体bea或fau材料的转变期间的保持封装一致。
实施例7
使用甲苯和1,3,5-tmb(动力学直径为0.59nm和0.74nm)的氢化速率来评估在mfi产物沸石(
1,3,5-tmb氢化反应导致在所有催化剂上排他地形成(顺式和反式)1,3,5-三甲基环己烷。表4显示了分散在sio2(m/sio2)和mfi(m/mfib和m/mfif)上的金属簇(m=pt、ru、rh)上的这些芳烃的氢化转换率。pt/mfib和pt/mfif上的甲苯加氢转换率比pt/sio2样品低一些(分别为1.2和2.7的因子,表4),这可能是因为通过mfi孔
表4含有mfi和sio2样品的金属在芳烃氢化中的催化性能a
a用0.35kpa甲苯/0.26kpa1,3,5-tipb和100kpa氢气在473k下进行氢化
b反应转换率定义为每秒每摩尔表面金属原子转化的反应物的摩尔数
cχj=r甲苯/r1,3,5-tmb,j=mfi、sio2
dφ=χ沸石/χsio2
m/mfi样品的高封装选择性值(8-23;表4)还表明,对于所有这些样品,超过88%的金属表面积包含在甲苯而不是1,3,5-tmb可接近的位置中。这些数据与tem衍生的和化学吸附衍生的平均簇直径和尺寸均匀性一起表明,最初存在于bea或fau空隙内的大多数金属簇在转化期间保留在沸石孔内,保持封装在所得的mfi样品中,其可以基于分子大小选择反应物并且仅允许小于mfi孔径尺寸的反应物进入活性位点。
实施例8
封装簇的成功合成首先需要在通过金属前体与oh-物质在所需的高ph下的反应形成不溶性胶体氢氧化物之前,mfi在碱性介质中成功的成核和生长。我们在这里寻求促进成核和生长,同时抑制金属前体的过早沉淀的合成条件。图6中显示了如何可以实现这些目的合成策略和条件的示意图。在下文中,我们根据图6所示的区域(例如,oh-或f-是否用作矿化剂)检查这些合成参数,目的是控制沸石成核和前体沉淀的相对速率。这些合成条件所用的额外细节,连同所形成的催化材料的封装选择性的值在表5中给出。这些金属-沸石材料的封装选择性由甲苯和1,3,5-tmb的氢化速率报告,如通过bea或fau沸石间转化成mfi制备的材料所做的那样。
在区域iii(图6)中,合成在高ph和温度(ph12,433k)下进行,这有利于mfi的快速成核和结晶,而且从金属前体快速形成不溶性氢氧化物。在这些条件下形成的产物的封装选择性参数接近1,这表明加在沸石外面的团簇占主导。sda部分(四丙基溴化铵)的使用也促进快速和选择性mfi的结晶,但是这样的物质可以填充晶体内空隙,从而阻止金属前体的封装,即使是溶剂化的单体。在这种情况下,产物的选择性参数再次接近1(对于使用pt(nh3)4(no3)2前体的pt为0.85),这表明所形成的簇不在晶体内mfi空隙间驻留。结果,在合成期间必须避免sda物质(或它们的浓度保持非常低),同时还维持不利于形成金属前体的胶体氢氧化物的条件。使用最少量的sda填充晶体内空隙,同时保持低温(383k)但高oh-浓度(ph12.9)没有导致mfi结构的结晶(m4,表5),并且所形成的无定形结构不提供任何接触约束。
考虑较低的温度和oh-浓度。在区域i(403-523k,7-11,图6)的低温和ph下,金属前体是稳定的,但是组装成mfi骨架的硅酸盐物质也是稳定的,这导致在这种情况下合成时间非常长,在一些报告的案件中大约几个月。
虽然封装可以通过直接水热合成方法实现,水热合成方法是通过在沸石合成后期引入金属前体或通过使用f-而不是使用oh-作为矿化剂来降低ph,但是使用延迟添加前体的方法导致适中的封装选择性以及使用氟化物合成的方法导致的低封装产率。然而,与本发明方法一致的沸石间转化方案避免了在直接水热合成中的封装挑战,并且这样做提供了将金属簇封装在mfi晶体空隙内的具有高选择性和金属含量的通用方法。这样的方法仅需要所述能够交换进入比mfi或其它子体沸石的空隙大的母体沸石。母体沸石表现出较低的骨架密度(这里为bea或fau),其随后转化为子体沸石(这里为mfi),其对于交换或更直接的封装方法是不可行的。似乎合理推断的是,在水热条件下无论它们是自发发生还是通过使用动力学助剂(例如晶种或有机结构导向剂),用于保留金属簇的所述沸石间转化方法通常可以扩展到在水性介质中的具有阳离子络合物的任何金属,以及增加沸石骨架密度的任何转变。
基于上述实施例1-8,可以得出这样的结论,在mfi空隙内是实现金属簇(pt、ru、rh)的成功封装是借助于包含bea或fau沸石的金属沸石间转化,其通过控制加入金属前体的时机的低温水热合成,和在氟化物介质中的直接水热合成。与直接水热合成方法相比,沸石间转化提供了以更少的时间、更低成本合成沸石的机会并且代表更经济和环境意识的方法,并且在合成期间这样做是通过帮助所需产物沸石的成核并避免使用有机结构。这些沸石间转化方法还导致金属簇在mfi沸石内的成功封装,而通过开发包括与配体稳定的金属前体的直接水热合成和合成后交换的方案,对封装是不可行的。当组合x-射线衍射、电子显微镜和h2化学吸附测量时,证实了母体沸石向mfi的转化和小的、均匀的和清洁的金属簇的存在。在分散于mfi和sio2上的金属簇上的甲苯和1,3,5-tmb的氢化相对速率表明,沸石样品中的金属簇主要存在于mfi空隙内,其中它们仅能被较小的甲苯反应物接近。我们期望所开发的沸石间转化方法,其可用于具有/不具有封装金属簇的mfi的合成,可以进一步延伸到不同骨架、空隙环境和骨架组成的沸石,并且延伸到封装具有催化重要性的其它金属、金属氧化物和金属硫化物的簇。
表5通过直接水热合成法合成的含金属mfi的合成方法和封装选择性
a相对于sio2报告金属前体的摩尔组成
bt=按天计所需的合成时间
cφ=χ沸石/χsio2,χj=r甲苯/r1,3,5-tmb,j=mfi、sio2
实施例9-12
在实施例9-12中,使用以下材料:
naoh(99.994%,sigmaaldrich),fau(cbv780,zeolyst,h-fau,si/al=40),cu(acac)2(>99.99%,sigmaaldrich)和[cu(nh3)so4h2o(98%,sigmaaldrich)按原始样品使用。在本实施例中,使用前述用于cha(菱沸石)1,stf(ssz-35)2和mtw(zsm-12)3沸石的合成方法制备用作晶种的材料。
(1)zones,s.i.美国专利us8007763b2,2011年8月30日;(2)musilova-pavlackova,s.;zones,s.i.;cejka,j.top.catal.2010,53,273;(3)jones,a.j.;zones,s.i.;iglesia,e.j.phys.chem.c2014,118.17787。
实施例9
cha、stf和mtw沸石的合成通过fau作为母体沸石的沸石间转化来实现。将fau(0.5-1.0g)加入到naoh水溶液中以达到xnaoh:1.0sio2:95h2o(x=0.50、0.68、0.85)的摩尔组成,向其中加入具有下述表6中所列的摩尔组成的10重量%(基于母体fau的重量%)晶种(cha、stf或mtw)以制备最终混合物。将这些混合物置于密封的聚丙烯容器(nalgene,125cm3)中,并在环境温度下通过剧烈磁力搅拌(400rpm;ikarctbasic)均质1小时。然后将混合物转移到特氟隆衬里的不锈钢高压釜中并在静态条件于所需结晶温度下(423、428或433k)下保持40小时。通过多孔圆盘布氏漏斗(chemglass,150ml,f)过滤,并用去离子水(电阻率17.9mω)洗涤,直到冲洗液的ph达到7-8以收集所得固体。在对流烘箱中在373k下加热样品过夜,然后将样品在管式炉中于流动干燥空气(1.67cm3g-1s-1)中以0.03ks-1的速率加热至873k,并在该温度下保持3小时。经过处理后得到的样品表示为chaf、stff、mtwf,其分别使用cha、stf(ssz-35)和mtw(zsm-12)的晶种通过fau的沸石间转化合成。
表6使用cha、stf和mtw晶种进行fau转化的初始合成摩尔组成a,b
a对于所有合成h2o/sio2=95且合成时间=40h
b初始合成摩尔组成排出晶种中sio2的量
c晶种(重量%)=晶种(g)/母体沸石(g)×100
dam.=无定形
e收率(%)=产物(g)/母体沸石(g)×100
实施例10
通过在353k磁力搅拌(400rpm;ikarctbasic)24小时下从cu(acac)2的水溶液(10:1h2o的质量比;沸石,实现
实施例11
将cu/fau(0.5-1.0g)与10重量%的cha晶种(基于母体fau的重量%)一起加入naoh水溶液(摩尔组成为0.68naoh:1.0sio2:95h2o)制备合成混合物,通过沸石间转化实现cu在cha中的封装。将这些混合物置于密封的聚丙烯容器(nalgene,125cm3)中,并在环境温度下通过剧烈磁力搅拌(400rpm;ikarctbasic)均质1小时。然后将混合物转移到特氟隆衬里的不锈钢高压釜中并在静态条件于所需结晶温度下(423k)下保持40小时。通过多孔圆盘布氏漏斗(chemglass,150ml,f)过滤,并用去离子水(电阻率17.9mω)洗涤,直到冲洗液的ph达到7-8以收集所得固体。在对流烘箱中在373k下整夜加热样品,得到的样品表示为cu/chacuf,其通过cu/fau的沸石间转化合成。
实施例12
在合成凝胶中添加cu前体通过作为母体材料的fau沸石间转化实现cu在cha中的封装。将fau(0.5-1.0g)与10重量%的cha晶种(基于母体fau的重量%)和[cu(nh3)4]so4h2o(
上述说明书、实施例和数据提供了本发明组合物的制造和使用的完整描述。由于在不脱离本发明的实质和范围的情况下可以进行本发明的许多实施例,因此本发明在于所附的权利要求。
- 还没有人留言评论。精彩留言会获得点赞!