一种改善铈锆材料中锆偏聚问题的方法与流程
本发明涉及一种改善铈锆材料中锆偏聚问题的方法,属于有色金属材料领域。
背景技术:
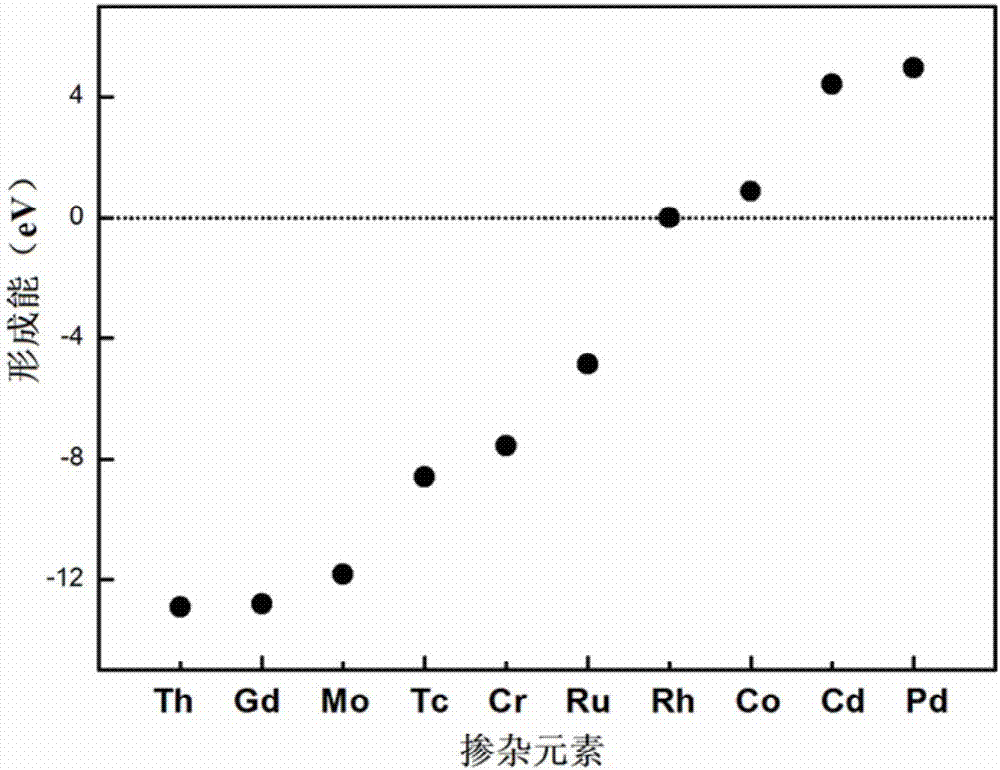
:CeO2-Zr基材料具有典型的立方萤石(CaF2)结构,在室温到其熔点范围内均保持立方萤石结构,具有非常好的稳定性。其本身所具有的氧化还原能力使得CeO2具有优秀的催化性能。同时,CeO2具有储氧能力(OxygenStorageCapacity),能够快速地形成和填充氧空位。这使得CeO2被广泛应用于处理机动车尾气NOx、CO和碳氢化合物的三效催化剂中。另外CeO2还能够作为催化剂,对化石燃料进行重整从而获得大量的氢气。不仅如此,同样由于其灵活的得失氧能力使得CeO2成为一种有前途的中温固体氧化物燃料电池的电解质材料。此外,CeO2还被广泛地应用于WGS(WaterGasShift)反应,而且由于其本身结构的结构特点和所具有的较高的硬度等特点,它还是一种重要的工业磨料和抛光材料。总之,CeO2是一种非常有用的稀土材料,在国民经济中有着大量的应用。在氧化铈材料中掺入锆,可以提高材料的催化效率,但是锆在提高铈基材料得失氧能力的同时,又带来了新的问题。其中最突出的就是材料的不均匀性。掺杂元素Zr的偏析对于材料的电化学性能、热力学稳定性以及力学性能都有相当大的负面影响。作为尾气催化材料的重要组成部分,其安全性需要严格的把关。因此该问题亟需要研究与改善。氧化铈在不同退火温度处理后,晶粒尺寸不同,在一定程度上影响到了其得失氧的能力,这是氧化铈材料薄膜化过程中可能遇到的问题,同时,氧化铈材料薄膜化后的机械强度是否达到生产要求,以及掺杂后体系产生的偏析与高温下的稳定性,都直接关系到生产应用。技术实现要素:基于现有技术存在的不足,本发明的目的在于提供一种改善铈锆材料中锆偏聚问题的方法,利用计算机模拟技术进行高效的材料性能改善与提高。为实现上述目的,本发明采用以下技术方案:一种改善铈锆材料中锆偏聚问题的方法,包括以下步骤:(1)稳定性筛选:在寻常的CeO2-Zr材料中引入第二种掺杂元素M,利用第一性原理计算方法,首先建立具有萤石结构的二氧化铈晶胞,弛豫优化后得到的结构晶胞参数为a=5.48,以此为基础建立2*2*2超胞、共96个原子的体系,选择体心位置进行掺杂,从原子尺度模拟不同掺杂元素以及掺杂方式对氧化铈的热力学结构稳定性的影响,寻找能稳定铈基材料的最佳掺杂元素;(2)改善Zr偏聚性能筛选:在现有文献基础上选择不同的元素组合,尝试双掺杂组合,模拟掺杂后材料的结构稳定性、M与Zr之间的相互作用强度、材料力学性能,以期筛选出性能表现优良的组合;(3)力学性能筛选:建立表面体系模拟薄膜状材料,通过第一性原理拉伸实验获得材料应力应变曲线,观察其随掺杂元素的改变情况并考察掺杂元素对铈基材料力学性能的影响,得到满足预期需要的性能指标的双掺杂体系。本发明使用双掺杂体系,引入第二种掺杂元素M,由于M与体系中Zr原子间存在相互吸引,因此在Zr的偏聚扩散中有效的钉扎,阻碍Zr的偏聚,从而改善材料的不均匀性。双掺杂体系还可以提高材料的力学性能,这对于材料的薄膜化十分有意义。本发明借助第一性原理计算方法,对13种元素(Mn、Mo、Pd、Rh、Ru、Tc、Th、Gd、As、Bi、Cd、Co、Cr)进行初步热稳定性测试,筛选出能维持稳定结构的元素后,进行性能测试,包括能否改善铈锆材料中锆偏聚问题,能否满足实际生产中力学性能要求等。本发明的优点在于:本发明利用第一性原理计算方法从原子尺度模拟不同掺杂元素对铈基材料的结构稳定性和化学性能的影响,尝试多种不同元素的组合形式;可以有效地预测、筛选能稳定材料结构、提高材料性能的掺杂元素,结果对实验更具有指导意义,同时也预测了各种掺杂后形成的新体系材料热稳定性。模拟材料的力学性能,对铈基材料实际制备的薄膜化有很重要的意义。本发明通过对铈锆材料的双掺杂模拟计算,结果表明M=Mn、Mo、Rh、Ru、Gd、Cd、Cr时的双掺杂体系完全满足预期需要的性能指标,认为可以有效的解决铈锆材料中锆偏聚等不均匀性所导致的性能指标达不到使用要求的问题。并且同时,在10%形变量范围内,力学性能表现得更加优越。附图说明图1为双掺杂体系CeO2-M-Zr模型晶体结构。图2为双掺杂体系CeO2-M-Zr形成能。图3为双掺杂体系M-Zr原子团耦合能。具体实施方式下面结合附图和实施例对本发明作详细说明,但并不意味着对本发明保护范围的限制。第一性原理计算是指不借助任何经验参数或者最大限度地进行“非经验性”处理,在量子力学原理基础上,通过直接求解组成物质的粒子运动的薛定谔方程而得到物质的性质。由于通过直接求解薛定谔方程得到材料的电子结构,使材料的计算从宏观和原子层次深入到电子层次,从本质上探讨的许多特性,因而相比其他材料的模拟方法,材料的第一性原理计算是一种真正意义上的预测,能够在新材料尚未制造出来之前准确预测材料的性质,从而指导材料的创新设计。对于固体这样一个含有巨大粒子数的多粒子系统,精确求解量子力学方程实际上是不可能的,必须采用一些近似和简化;通过绝热近似将原子核与电子的运动分开;通过Hartree-fock自洽场方法或者密度泛函理论将多电子问题简化为单电子问题,该近似又称为独立粒子近似;通过周期边界条件以及将固体抽象成为具有平移周期性的理想晶体。将求解巨大数目电子的运动问题转化为求解原胞内电子的运动问题。通过第一原理计算可以在最短时间内,尝试多种掺杂形式,而不需受实验条件限制,根据计算结果对实验进行指导与预测,从而提高实验的效率与项目推进进度。本发明中使用的模拟工具是VASP软件。实施例具体计算方法与结果如下:1、稳定性筛选:目前计算过的元素M包括:Mn、Mo、Pd、Rh、Ru、Tc、Th、Gd、As、Bi、Cd、Co、Cr。首先建立2*2*2CeO2晶胞,ENCUT设置为500ev,K点设置为4*4*4,进行M元素与Zr的掺杂,最终结构如图1所示。对于掺杂,首先应该判断的是掺杂后的体系是否稳定,此处使用体系形成能作为判断标准,双掺体系能量与M原子、Zr原子能量之和减去单独的氧化铈、单独铈原子能量,所得即为掺杂后体系的形成能,能量越负,表示体系越稳定,计算公式如下:ΔGf=E(CeO2-M-Zr)+E(M)+E(Zr)-E(CeO2)-2E(Ce),结果如图2所示。2、改善Zr偏聚性能筛选:本实验方法是通过掺杂元素M与Zr的相互作用来进行材料优化。因此对掺杂元素的功效作用判断标准是M-Zr原子团耦合能,单独氧化铈能量与双掺体系能量,减去单独掺杂M和单独掺杂Zr的能量,所得即为M-Zr原子团耦合能,能量越负,表示体系越稳定,计算公式如下:ΔGf=E(CeO2)+E(CeO2-M-Zr)-E(CeO2-M)-E(CeO2-Zr),结果如图3所示。根据上述两项结果,我们的筛选标准为既要能保持体系稳定又要M-Zr原子间相互作用强大的元素,结果如表1所示。表1形成能与耦合能计算结果形成能(ev)M-Zr原子团耦合能(ev)CeO2-Th-Zr(-12.92)CeO2-Cr-Zr(-1.01)CeO2-Gd-Zr(-12.81)CeO2-Mn-Zr(-0.86)CeO2-Mn-Zr(-11.97)CeO2-Co-Zr(-0.70)CeO2-Mo-Zr(-11.82)CeO2-Mo-Zr(-0.61)CeO2-Tc-Zr(-8.59)CeO2-Pd-Zr(-0.56)CeO2-Cr-Zr(-7.57)CeO2-Tc-Zr(-0.32)CeO2-Ru-Zr(-4.86)CeO2-Rh-Zr(-0.25)CeO2-Rh-Zr(-0.0053)CeO2-Cd-Zr(-0.15)CeO2-Bi-Zr(+0.54)CeO2-Bi-Zr(-0.12)CeO2-Co-Zr(+0.88)CeO2-Gd-Zr(-0.08)CeO2-Cd-Zr(+4.44)CeO2-Ru-Zr(-0.08)CeO2-Pd-Zr(+4.97)CeO2-Th-Zr(-0.03)3、力学性能筛选:该部分计算是针对铈锆材料作为汽车尾气催化剂,喷涂薄膜等使用方式时,对其机械强度有一定的要求。利用第一性原理计算方法,建立2*2*2CeO2-M-Zr晶胞,ENCUT设置为500ev,K点设置为4*4*4,ISIF设置为2,弛豫优化结构后,对体系分别沿Y轴、Z轴施加形变,形变量依次为2%、4%、6%、8%、10%、12%、14%、16%,计算体系在上述形变量下对应的受力,通过计算,结果如表2、表3所示。表2沿Y轴方向应力应变(Gpa)表3沿Z轴方向应力应变(Gpa)根据以上结果,通过对铈锆材料的双掺杂模拟计算,结果表明M=Mn、Mo、Rh、Ru、Gd、Cd、Cr时的双掺杂体系完全满足预期需要的性能指标,认为可以有效的解决铈锆材料中锆偏聚等不均匀性所导致的性能指标达不到使用要求的问题。并且,在10%形变量范围内(工业使用中一般允许材料的变形范围即为10%),力学性能表现得更加优越。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1