生产环状碳酸酯的催化剂体系及相关方法与流程
本发明涉及一种生产环状碳酸酯的催化剂体系,以及利用所述催化剂体系制备环状碳酸酯的方法。
背景技术:
工业过程和燃料燃烧等产生的二氧化碳(co2)排放正在成为全世界的一个严重问题,因为这种排放被认为是气候变化的主要驱动因素。根据国际能源署(iea)发布的《2017年全球能源与二氧化碳现状报告》,全球与能源相关的二氧化碳排放量达到了历史最高的325亿吨。全球各地的立法者已经开始对大气中二氧化碳的无限制释放进行限制。在解决该问题的方法中,近年来,将co2化学转化为高价值化学品引起了相当大的关注。环状碳酸酯是其中之一,因为它们可以在温和的条件下通过将co2与环氧化物环加成而轻松生成。在涉及聚碳酸酯、乙二醇和聚酯的数十亿美元市场中,环状碳酸酯处于中心位置。但是,co2在动力学和热力学上是稳定的,因此需要大量的能量才能将co2转化为其他化学物质,包括环状碳酸酯。因此,一种有前途的方法是开发有效的催化剂,该催化剂允许在较低的温度下实现更高的co2转化率。
环状碳酸酯是由co2与环氧化合物发生环加成反应生成的。存在几种进行这种反应的催化剂体系,但是,这种体系中很少有能够在环境压力或中等压力(0.1至1mpa)下,使用不纯的co2进行操作。从气体混合物中捕获二氧化碳的能力是有利的,因为这可以很容易地应用于烟道气,因此对于环状碳酸酯的生产在商业上具有吸引力。所述催化剂的实例包括energyenvironsci.,2010,3,212-215中公开的双金属铝(salen)配合物,其将含有水气以及在co2中作为杂质的nox的co2,用于环状碳酸酯的生产。然而,由于有机骨架配位的铝原子的精细结构,所述配合物具有非常高的分子量,并且其制备涉及多个合成步骤。
us9,586,926b2公开了一种由co2碳化环氧化物来制备环状碳酸酯的方法。该方法通过均相催化剂体系进行,该体系包含选自ycl3、y2o3、y(no3)3、sccl2或lacl3的预催化剂和选自四丁基溴化铵、4-二甲基氨基吡啶或双(三苯基膦)亚胺氯化物的助催化剂,两者的摩尔比为1:1至1:2。但是,在环境条件下,使用不纯或稀释的co2时该系统的催化活性相对较低。
monteiro等(appliedcatalysisa:general(2017),544(25),46-54)公开了一种催化剂体系,该体系包括氯化1-甲基-3-(3-三甲氧基甲硅烷基丙基)咪唑鎓离子液体催化剂,以钛酸酯纳米管(tnt)或纳米线(tnw)为预催化剂,znbr2为助催化剂,用于合成环状碳酸酯。尽管催化剂对co2的选择性高,但该催化剂未应用于不纯的或稀释的co2的转化。
因此,需要开发新一代的催化剂体系,以在温和的条件下以高催化活性和高选择性,从co2,尤其是通过用稀释的和/或不纯的co2源,来制备环状碳酸酯。因此,本发明旨在提供一种催化剂体系,用于在温和条件下,使用纯的和不纯的co2,将co2和环氧化物转化为环状碳酸酯,该体系具有高催化活性且是有成本效益的。
技术实现要素:
在一个实施方案中,本发明提供了一种用于由二氧化碳(co2)和基于环氧的化合物生产环状碳酸酯的催化剂体系,该催化剂体系包含:
预催化剂,其选自具有式bixz的化合物;
具有式(i)的化合物
其中:
x和y各自独立地代表卤素;
z代表1至3的整数;
m代表1至4的整数;
r1、r2和r3独立地选自具有1至4个碳原子的烷基或具有1至4个碳原子的烷氧基;
r4、r5和r6独立地选自具有1至4个碳原子的烷基或芳基,或者r4、r5和r6一起形成杂芳基环;
以及
金属氧化物载体,其选自mgo、sio2、zno、al2o3或tio2。
在本发明的另一个实施方案中,本发明涉及一种制备催化剂体系的方法,其包括以下步骤:
(a)将具有式(iii)的硅烷化合物与基于胺的化合物,在有机溶剂中,于约100至150℃范围内的温度下,回流约12至72小时,其中硅烷化合物与基于胺的化合物的摩尔比在约5:1至约1:5的范围内,以获得化合物(i);
其中:
a代表卤素;
n代表1至4的整数;
r11、r12和r13独立地选自具有1至4个碳原子的烷基或具有1至4个碳原子的烷氧基。
(b)将含有由步骤(a)获得的在有机溶剂中的化合物(i)和选自mgo、sio2、zno、al2o3或tio2的金属氧化物载体的混合物,混合物悬浮在所述有机溶剂中,于约100至200℃范围内的温度下,回流约5至50小时,其中化合物(i)的浓度在约5至15%v/v的范围内,并且金属氧化物的浓度在约10至20%w/v的范围内;和
(c)使预催化剂与由步骤(b)获得的金属氧化物接触约1至5小时,其中预催化剂的浓度在金属氧化物的约1至约10重量%的范围内。
在本发明的另一个实施方案中,本发明涉及一种制备催化剂体系的方法,其包括以下步骤:
(a)将在有机溶剂中的基于胺的化合物和选自mgo、sio2、zno、al2o3或tio2的金属氧化物载体的混合物,混合物悬浮在所述有机溶剂中,于约100至200℃范围内的温度下,回流约5至50小时,其中基于胺的化合物的浓度在约5至15%v/v的范围内,并且金属氧化物的浓度在约10至20%w/v的范围内;和;
(b)将由步骤(a)获得的产物与烷基卤化物,在有机溶剂中,于约100至150℃范围内的温度下,混合约12至72小时,其中基于胺的化合物与烷基卤化物的摩尔比在约5:1至1:5的范围内;和
(c)使预催化剂与由步骤(b)获得的金属氧化物接触约1至5小时,其中预催化剂的浓度在金属氧化物的约1至10重量%的范围内。
在本发明的另一个实施方案中,本发明涉及一种生产环状碳酸酯的方法,其包括在催化剂体系的存在下,使基于环氧的化合物与二氧化碳反应,该催化剂体系包含:
预催化剂,其选自具有式bixz的化合物;
具有式(i)的化合物
其中:
x和y各自独立地代表卤素;
z代表1至3的整数;
m代表1至4的整数;
r1、r2和r3独立地选自具有1至4个碳原子的烷基或具有1至4个碳原子的烷氧基;
r4、r5和r6独立地选自具有1至4个碳原子的烷基或芳基,或者r4、r5和r6一起形成杂芳基环;
以及
金属氧化物载体,其选自mgo、sio2、zno、al2o3或tio2
其中所述方法如下进行:使用相对于基于环氧的化合物在约0.1至20摩尔%的范围内的催化剂体系浓度;在约1至100巴的范围内的二氧化碳的压力下;约10到200℃的范围内的温度;以及约1至8小时范围内的反应时间。
附图说明
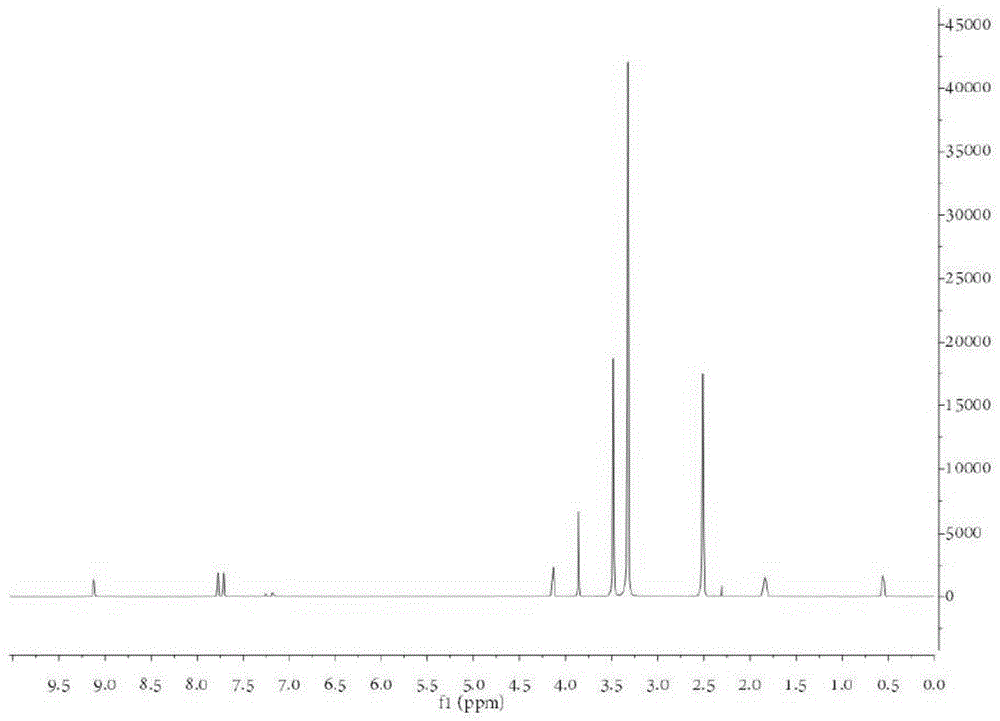
图1.1-甲基-3-(3-(三甲氧基甲硅烷基)丙基)-1h-咪唑-3-鎓溴化物的1h-nmr谱。
图2.1-甲基-3-(3-(三甲氧基甲硅烷基)丙基)-1h-咪唑-3-鎓碘化物的1h-nmr谱。
图3.负载在sio2上的1-甲基-3-(3-(三甲氧基甲硅烷基)丙基)-1h-咪唑-3-鎓碘化物的ft-ir谱。
图4.由本发明的催化剂获得的碳酸丙烯酯的1h-nmr谱。
具体实施方式
本发明提供一种具有成本效益和高催化活性的用于生产环状碳酸酯的催化剂体系,用于将co2和基于环氧的化合物转化为环状碳酸酯。此外,本发明提供一种制备环状碳酸酯的方法,该方法通过利用上述催化剂体系,在温和条件下使用纯的和不纯的co2的来制备环状碳酸酯。可以根据如下说明书阐明本发明的细节。
除非另有说明,本文所用的技术术语或科学术语具有如本领域具有普通技术的人员所理解的定义。
这里提到的设备、装置、方法或化学物质是指本领域技术人员通常操作或使用的设备、装置、方法或化学物质,除非另有明确说明它们是本发明中专门使用的设备、装置、方法或化学物质。
在权利要求书或说明书中,使用带有术语“包含”的单数或复数名词指“一个”,并且还指“一个或多个”、“至少一个”和“一个或一个以上”。
所公开和要求的所有组合物和/或工艺旨在包括本发明的的反应、操作、修饰方面,或改变任何参数而不进行与本发明明显不同的实验,以及尽管未特别提到权利要求,根据本技术领域的技术人员获得具有相同功用和结果的本发明的类似对象。因此,本发明的替代或类似对象,包括本领域技术人员可以清楚地看到的细微的修饰或改变,应视为在所附权利要求的本发明的范围、精神和概念内。
在整个本申请中,术语“约”用于表示本文中呈现的任何值都可能潜在地变化或偏离。这种变化或偏离可能是由于设备、计算中使用的方法的误差造成,或者是由于执行设备或方法的个人操作者造成。这些包括由于物理性质的变化而引起的变化或偏离。
以下是对本发明的详细描述,而无意于限制本发明的范围。
根据本发明的一个实施方案,本发明提供一种用于生产环状碳酸酯的催化剂体系,其包含:
预催化剂,其选自具有式bixz的化合物;
具有式(i)的化合物
其中:
x和y各自独立地代表卤素;
z代表1至3的整数;
m代表1至4的整数;
r1、r2和r3独立地选自具有1至4个碳原子的烷基或具有1至4个碳原子的烷氧基;
r4、r5和r6独立地选自具有1至4个碳原子的烷基或芳基,或者r4、r5和r6一起形成杂芳基环;
以及
金属氧化物载体,其选自mgo、sio2、zno、al2o3或tio2。
在另一个示例性的实施方案中,x是氯。
在一个优选的示例性的实施方案中,预催化剂是bicl3。
在另一个示例性的实施方案中,y选自氯、溴或碘。
在又一个示例性实施方案中,m是1。
在另一个示例性的实施方案中,r1、r2和r3独立地选自具有1至4个碳原子的烷氧基。优选地,r1、r2和r3是甲氧基。
在又一个示例性的实施方案中,r4、r5和r6独立地选自具有1至4个碳原子的烷基。优选地,r4、r5和r6是甲基。
在另一个示例性的实施方案中,r4、r5和r6一起形成具有式(ii)的杂芳基环
其中:
r7、r8、r9和r10独立地选自氢或具有1至4个碳原子的烷基。
在又一个示例性的实施方案中,r7、r8、r9和r10独立地选自氢或甲基。
在一个优选的示例性的实施方案中,r7、r9和r10是氢,并且r8是甲基。
在一个优选的示例性的实施方案中,具有式(i)的化合物选自1-甲基-3-(3-(三甲氧基甲硅烷基)丙基)-1h-咪唑-3-鎓或n,n,n-三甲基-3-(三甲氧基甲硅烷基)丙-1-胺鎓。
在一个优选的示例性的实施方案中,金属氧化物载体是sio2。
在本发明的另一个实施方案中,本发明涉及本发明的催化剂体系用于由co2和基于环氧的化合物生产环状碳酸酯的用途。
在本发明的另一个实施方案中,本发明涉及一种制备本发明的催化剂体系的方法,其包括以下步骤:
(a)将具有式(iii)的硅烷化合物与基于胺的化合物,在有机溶剂中,于100至150℃的温度下,回流12至72小时,其中硅烷化合物与基于胺的化合物的摩尔比在5:1至1:5的范围内,以获得化合物(i);
其中:
a代表卤素;
n代表1至4的整数;
r11、r12和r13独立地选自具有1至4个碳原子的烷基或具有1至4个碳原子的烷氧基。
(b)将含有由步骤(a)获得的在有机溶剂中的化合物(i)和选自mgo、sio2、zno、al2o3或tio2的金属氧化物载体的混合物,混合物悬浮在所述有机溶剂中,于约100至200℃范围内的温度下,回流约5至50小时,其中化合物(i)的浓度在约5至15%v/v的范围内,并且金属氧化物的浓度在约10至20%w/v的范围内;和
(c)使预催化剂与由步骤(b)获得的金属氧化物接触约1至5小时,其中预催化剂的浓度在金属氧化物的约1至10重量%的范围内。
在另一个示例性的实施方案中,a选自氯、溴或碘。
在另一个示例性实施方案中,n是1。
在另一个示例性的实施方案中,r11、r12和r13独立地选自具有1至4个碳原子的烷氧基。
在一个优选的示例性的实施方案中,r11、r12和r13是甲氧基。
在另一个示例性的实施方案中,基于胺的化合物是基于咪唑的化合物。
在一个优选的示例性的实施方案中,基于胺的化合物是n-甲基咪唑。
在另一个示例性的实施方案中,硅烷化合物与基于胺的化合物的摩尔比在约2:1至1:2的范围内。
在另一个示例性的实施方案中,有机溶剂是芳烃溶剂。
在一个优选的示例性的实施方案中,有机溶剂是甲苯。
在一个优选的示例性的实施方案中,金属氧化物载体是sio2。
在一个实施方案中,根据步骤(c),使预催化剂与从步骤(b)获得的金属氧化物接触可以通过,例如,浸浴法、浸渍法、固态反应法或类似方法进行,其中,例如研磨、研糊或球磨技术的固态反应法是优选的,因为操作简单并且提供优异的生产率。
在本发明的另一个实施方案中,本发明提供了一种用于制备本发明的催化剂体系的方法,其包括以下步骤:
(a)将在有机溶剂中的基于胺的化合物和选自mgo、sio2、zno、al2o3或tio2的金属氧化物载体的混合物,混合物悬浮在所述有机溶剂中,于约100至200℃范围内的温度下,回流约5至50小时,其中基于胺的化合物的浓度在约5至15%v/v的范围内,并且金属氧化物的浓度在约10至20%w/v的范围内;和;
(b)将由步骤(a)获得的产物与烷基卤化物,在有机溶剂中,于约100至150℃范围内的温度下,混合约12至72小时,其中基于胺的化合物与卤代烷烃的摩尔比在约5:1至1:5的范围内;和
(c)使预催化剂与由步骤(b)获得的金属氧化物接触约1至5小时,其中所述预催化剂的浓度在金属氧化物的约1至10重量%的范围内。
在另一个示例性的实施方案中,基于胺的化合物具有式(iv)。
其中:
q代表1至4的整数;
r14、r15和r16独立地选自具有1至4个碳原子的烷基或具有1至4个碳原子的烷氧基;
r17和r18独立地选自具有1至4个碳原子的烷基或芳基,或者r17和r18可以一起形成杂芳基环。
在一个优选的示例性的实施方案中,q是1。
在另一个示例性的实施方案中,r14、r15和r16独立地选自具有1至4个碳原子的烷氧基。优选地,r14、r15和r16是甲氧基。
在另一个示例性的实施方案中,r17和r18独立地选自具有1至4个碳原子的烷基。
在一个优选的示例性的实施方案中,r17是甲基。
在一个优选的示例性的实施方案中,r18是甲基。
在另一个示例性的实施方案中,其中有机溶剂选自芳烃溶剂。
在一个优选的示例性的实施方案中,有机溶剂是甲苯。
在一个优选的示例性的实施方案中,金属氧化物载体是sio2。
在一个实施方案中,根据步骤(c),使预催化剂与从步骤(b)获得的金属氧化物接触可以通过例如浸浴法、浸渍法、固态反应法或类似方法进行,其中,例如研磨、研糊或球磨技术的固态反应法是优选的,因为其操作简单并且提供优异的生产率。
在本发明的另一个实施方案中,本发明提供了一种由二氧化碳与基于环氧的化合物的反应生产环状碳酸酯的方法,该方法包括在催化剂体系的存在下,使基于环氧的化合物与二氧化碳反应,该催化剂体系包含:
预催化剂,其选自具有式bixz的化合物;
具有式(i)的化合物;和
其中:
x和y各自独立地代表卤素;
z代表1至3的整数;
m代表1至4的整数;
r1、r2和r3独立地选自具有1至4个碳原子的烷基或具有1至4个碳原子的烷氧基;
r4、r5和r6独立地选自具有1至4个碳原子或芳基,或者r4、r5和r6一起形成杂芳基环;
以及
金属氧化物载体,其选自mgo、sio2、zno、al2o3或tio2;
其中所述方法如下进行:使用相对于基于环氧的化合物在0.1至20摩尔%的范围内的催化剂体系浓度;在1至100巴的范围内的二氧化碳的压力下;10到200℃的范围内的温度;以及1至8小时的范围内的反应时间。
在一个优选的示例性的实施方案中,催化剂体系的浓度相对于基于环氧的化合物在约0.5至10摩尔%的范围内。
在一个优选的示例性的实施方案中,二氧化碳的压力在约1至10巴的范围内。
在一个优选的示例性的实施方案中,温度在约60至120℃的范围内。
在一个优选的示例性的实施方案中,反应时间在约2至4小时的范围内。
在另一个示例性的实施方案中,x是氯。
在一个优选的示例性的实施方案中,预催化剂是bicl3。
在另一个示例性的实施方案中,y选自氯、溴或碘。
在另一个示例性实施方案中,m是1。
在另一个示例性的实施方案中,r1、r2和r3独立地选自具有1至4个碳原子的烷氧基。优选地,r1、r2和r3是甲氧基。
在另一个示例性的实施方案中,r4、r5和r6独立地选自具有1至4个碳原子的烷基。优选地,r4、r5和r6是甲基。
在另一个示例性的实施方案中,r4、r5和r6一起形成具有式(ii)的杂芳基环
其中:r7、r8、r9和r10独立地选自氢或具有1至4个碳原子的烷基。
在另一个示例性的实施方案中,r7、r8、r9和r10独立地选自氢或甲基。
在一个优选的示例性的实施方案中,r7、r9和r10是氢,并且r8是甲基。
在一个优选的示例性的实施方案中,具有式(i)的化合物选自1-甲基-3-(3-(三甲氧基甲硅烷基)丙基)-1h-咪唑-3-鎓或n,n,n-三甲基-3-(三甲氧基甲硅烷基)丙-1-胺鎓。
在一个优选的示例性的实施方案中,金属氧化物载体是sio2。
此后,示出本发明的示例,而没有任何意图限制本发明的任何范围。
实施例
化学品和消耗品
所有化学品均购自商业渠道,按收到使用。前过渡金属卤化物在手套箱内存储和处理。不含金属的化合物存储在化学柜中,无需进一步预防措施即可使用。用于制备多相铋催化剂的二氧化硅载体,使用前在烘箱中在约150℃进行热处理,以去除痕量水分。纯的和稀释的co2(空气中50%浓度的co2)接收在金属瓶中,并通过调节器计量。
1-甲基-3-(3-(三甲氧基甲硅烷基)丙基)-1h-咪唑-3-鎓溴化物的合成
将n-甲基咪唑(约5ml)和(3-溴丙基)三甲氧基硅烷(约11.65ml)在干燥甲苯中的混合物在惰性气氛中回流约48小时。所得混合物用乙醚洗涤数次,并在真空下干燥约24小时。所得的n-3-(3-三甲氧基甲硅烷基丙基)-3-甲基咪唑鎓溴化物为棕色粘稠液体。如图1所示,在dmso-d6中通过1h-nmr确定该产物的结构。
1-甲基-3-(3-(三甲氧基甲硅烷基)丙基)-1h-咪唑-3-鎓碘化物的合成
将n-甲基咪唑(约5ml)和(3-碘丙基)三甲氧基硅烷(约11.65ml)在干燥甲苯中的混合物在惰性气氛中回流约48小时。所得混合物用乙醚洗涤数次,并在真空下干燥约24小时。所得的n-3-(3-三甲氧基甲硅烷基丙基)-3-甲基咪唑鎓碘化物为棕色粘稠液体。如图2所示,在dmso-d6中通过1h-nmr确定该产物的结构。
用离子液体对sio2进行官能化
sio2用1-甲基-3-(3-(三甲氧基甲硅烷基)丙基)-1h-咪唑-3-鎓碘化物进行官能化。将二氧化硅(约4g)悬浮在甲苯(约25ml)中,然后加入溶解在甲苯中的n-3-(3-三甲氧基甲硅烷基丙基)-3-甲基咪唑鎓碘化物(约2.5ml)。将混合物在约110℃下搅拌约48小时后,让二氧化硅沉淀下来。通过离心分离上清液,用甲苯洗涤改性二氧化硅若干次,然后在真空中干燥约24小时。
制得的催化剂的ft-ir谱(图3)显示了一些信号,如从2890至3151cm-1区域和1450至1630cm-1区域的信号中看出,这些信号对应表面上带有芳香性的有机物质的存在。通过以下元素分析确定了负载在载体上的有机物质的存在:c(14.05%);h(2.65%);n(3.83%)。
此外,sio2用n,n,n-三甲基-3-(三甲氧基甲硅烷基)丙-1-胺鎓碘化物进行官能化,然后用碘代甲烷进行胺的季铵化。将sio2(约4g)悬浮于甲苯(约25ml)中。然后加入溶于甲苯中的n-3-(3-三甲氧基甲硅烷基丙基)-3-二甲胺(约2.5ml)。将混合物在约110℃下搅拌约48小时后,使二氧化硅沉淀。通过离心分离上清液,用甲苯洗涤改性二氧化硅若干次,然后在真空中干燥约24小时。所得的官能化二氧化硅进一步与碘代甲烷在dmf中在黑暗条件下反应约2小时,得到季铵盐。此步骤重复两次,以确保季铵化胺的形成。通过离心分离上清液,用甲苯洗涤改性二氧化硅若干次,然后在真空中干燥约24小时。
预催化剂负载于改性sio2上
负载预催化剂在手套箱中进行。相对于二氧化硅的重量,约5重量%和10重量%的预催化剂的金属负载在官能化载体上。通过手动机械研磨工艺将bicl3(作为预催化剂)与载体一起研磨。将改性的二氧化硅载体和预催化剂的混合物在手套箱中研磨约2小时,并直接使用。
使用高压釜以纯的和稀释的co2合成环状碳酸酯
使用本发明的催化剂合成环状碳酸酯以测试其催化活性。在惰性气氛下(手套箱)在75ml高压釜中进行合成。将本发明的催化剂(约0.5至1g)和环氧丙烷(约5ml,71.4mmol)加入带有磁力搅拌棒的高压釜中。通过添加10巴的co2(使用约50%和约100%浓度的co2)来引发反应。将高压釜置于约60℃和约80℃的油浴中,并以约500rpm搅拌。约3小时后,容器可以在水浴中冷却至室温。收集粗制的反应混合物样品用于1hnmr。为了分离纯碳酸丙烯酯,将反应混合物过滤,并通过旋转蒸发仪蒸发未反应的环氧丙烷。从反应中得到的碳酸丙烯酯在cdcl3中通过1h-nmr确定(图4)。
从表1中,在约60℃和80℃和10巴co2压力下,使用纯的co2或约50%浓度的稀释的co2,对多相催化剂进行了初步研究。所研究的催化剂为负载在sio2上的n,n,n-三甲基-3-(三甲氧基甲硅烷基)丙-1-胺鎓碘化物和bicl3(sinme2-mei-bicl3),以及负载在sio2上的1-甲基-3-(3-(三甲氧基甲硅烷基)丙基)-1h-咪唑-3-鎓碘化物和bicl3(si-imi-bicl3)。在使用含金属的催化剂进行催化活性测试之前,研究了不含金属的官能化载体(条目1至4)的催化效率。值得注意的是,不含金属的官能化载体在将co2环加成环氧丙烷中显示出中等至好的催化活性。但是,bicl3的存在改善了所有条目的催化活性。与不存在金属相比,金属存在下的催化活性提高了约10至20%。条目5和6之间的比较表明,金属负载可以降低至约5重量%,催化活性没有任何最小的差异。在约80℃和约10巴的反应条件下,使用纯co2与10%sinme2-mei-bicl3的金属负载(条目9)提供最高的反应产率和在约3小时内co2转化完成。将温度降低至约60℃,使用纯的或稀释的co2(约50%浓度),导致转化率略降低。当在约80℃下操作时,使用稀释的co2导致反应产率降低,然而,在约60℃下,使用纯的co2和稀释的co2的反应产率相似。催化运行后回收sinme2-mei-bicl3,并再使用了额外两个连续循环。结果表明,该催化剂是可回收的,伴有催化活性的逐渐损失。这可能是由于催化剂在空气的暴露和/或当回收催化剂时发生的材料损失。
表1催化剂的催化活性
根据sinme2-mei-bicl3和si-imi-bicl3的良好的催化活性,使用稀释的co2(约50%浓度),通过改变温度和压力进行进一步的研究和优化,如表2所示。
表2中的数据显示,对于si-nme2-mei-bicl3,温度从约80℃升高到约120℃,使得产率增加,然而,在约80℃、和在约100至120℃之间,co2的转化率差别不大。压力对催化性能产生小的影响,因为表2中的数据表明,即使在约5巴的co2压力下也可以实现co2几乎完全的转化。表4(条目5)显示,约60℃和约7巴的co2压力,是以高收率(93%)实现co2转化的最温和条件。当使用si-imi-bicl3时,最有趣的观察是co2转化通常不会随温度增加而增加,而通常会降低,特别是当温度从约100℃升高到120℃时。这可能是因为在升高温度时,环氧丙烷的气化会减少底物与催化剂之间的接触。该观察得到以下事实的支持,在约5巴的co2压力下,当温度从约100℃升高到120℃时,转化率降低(表2,条目23和24),因为这条件有利环氧丙烷的蒸发。
表2制得的催化剂体系在几种反应条件下的催化活性
如本技术领域的技术人员容易理解的那样,上述描述是对本发明原理的实施方案的说明。该描述无意限制本发明的范围或应用,因为在不脱离如所附权利要求所限定的本发明的精神的前提下,本发明可以进行修改、变化和改变。
本发明最佳方式
本发明的最佳方式如本发明的说明书中所提供。
- 还没有人留言评论。精彩留言会获得点赞!