磁性颗粒粒径分选方法与流程
本发明涉及分离技术领域,特别是涉及一种磁性颗粒粒径分选方法。
背景技术:
磁性颗粒在众多领域被广泛应用。但是,现在并没有针对磁性颗粒的有效的分选方法。另外,目前,为了得到合适粒径的磁性颗粒,常常采用后续处理的方法对制备的产品进行粒径分选,比较常用的方法是透析法。
现有市场上出售的透析膜规格以截留分子量来划分,截留分子量可以从数百到数百万不等,理论上可以使粒径为十几个纳米的材料透过膜孔。但是,透析法难以分选出不同粒径范围的纳米材料。由于透析膜是允许粒径小的分子透出,分子量大的颗粒被留在透析袋内,而且受透析膜的规格限制,粒径大于十几纳米的材料很难透过膜孔。这样使得分选出不同粒径的纳米磁性材料变得很困难。另外,透析法分选不同粒径的磁性材料,需要并用多种规格的透析膜,透析膜的价格通常很高。而且采用透析的分选方法,过程繁琐,效率低、速度慢,处理量小。
技术实现要素:
基于此,有必要提供一种效率较高的磁性颗粒粒径分选方法。
一种磁性颗粒粒径分选方法,包括如下步骤:
S1,制备分选管:
将磁性吸附介质按照预设的高径比填充在层析柱中得分选管;
S2,磁性吸附:
将所述分选管移至磁场内,使所述分选管内的磁性吸附介质磁化;
将含待分选磁性颗粒的悬浮液上样于所述分选管内,使所述待分选磁性颗粒吸附在已磁化的磁性吸附介质上;
S3,洗脱:
上样结束后,将吸附有所述待分选磁性颗粒的分选管移出磁场,采用洗脱液将所述分选管内的所述待分选磁性颗粒洗脱下来,收集产品。
在其中一个实施例中,所述含待分选磁性颗粒的悬浮液是通过共沉淀法、水热法或热分解法制备而成。
在其中一个实施例中,所述磁性颗粒的悬浮液为葡聚糖@Fe3O4纳米颗粒的悬浮液。
在其中一个实施例中,在步骤S1中,所述磁性吸附介质选自铁磁性微球、顺磁性微球或软磁性微球。
在其中一个实施例中,在步骤S1中,所述磁性吸附介质选自铁珠、钢珠或硅钢珠。
在其中一个实施例中,在步骤S1中,所述磁性吸附介质的粒径为0.1mm-5mm。
在其中一个实施例中,在步骤S1中,所述高径比为100:5-80。
在其中一个实施例中,在步骤S1中,所述层析柱的材质为金属或塑料;所述层析柱的形状为圆柱体、三棱柱体或四棱柱体。
在其中一个实施例中,在步骤S1之前,还包括对所述磁性吸附介质和所述层析柱的内壁进行预处理的步骤,其步骤如下:
根据所述待分选磁性颗粒的悬浮液的亲水性或疏水性,相应地分别预先对所述磁性吸附介质和所述层析柱的内壁进行亲水性或疏水性处理。
在其中一个实施例中,在步骤S2中,所述磁场通过永磁铁产生。
本发明具有如下有益效果:
本发明针对磁性颗粒(尤其是磁性纳米颗粒)和磁性吸附介质可以被磁化的特点,设计了一种磁性颗粒粒径分选方法,包括如下步骤:将磁性吸附介质按照预设的高径比填充在层析柱中得分选管,分选管移至磁场内以使磁性吸附介质磁化,将待分选磁性颗粒的悬浮液上样,并使待分选磁性颗粒吸附在已磁化的磁性吸附介质上,再将分选管移出磁场,采用洗脱液洗脱,收集磁性颗粒。该磁性颗粒粒径分选方法通过磁性吸附分选出磁性颗粒,并且磁性吸附介质填充时具有一定的孔径通道,进而分选出不同粒径范围的磁性颗粒。由于分选后的磁性颗粒粒径分布范围变窄,磁性颗粒产品整体的分散性和稳定性会显著提高。本发明的磁性颗粒粒径分选方法分选过程简单,处理量较大,且效率高。
本发明的磁性颗粒粒径分选方法通过对层析柱和磁性吸附介质进一步进行亲水性或疏水性处理,增加磁性颗粒对磁性吸附介质的表面的浸润性或吸附性,并优化磁性吸附介质的种类、粒径范围及填充的高径比和磁场强度等,能够进一步降低悬浮液中磁性颗粒的粒径分布范围,提高悬浮液中磁性颗粒的分散性和稳定性。
附图说明
图1为一实施方式的磁性颗粒粒径分选方法示意图;
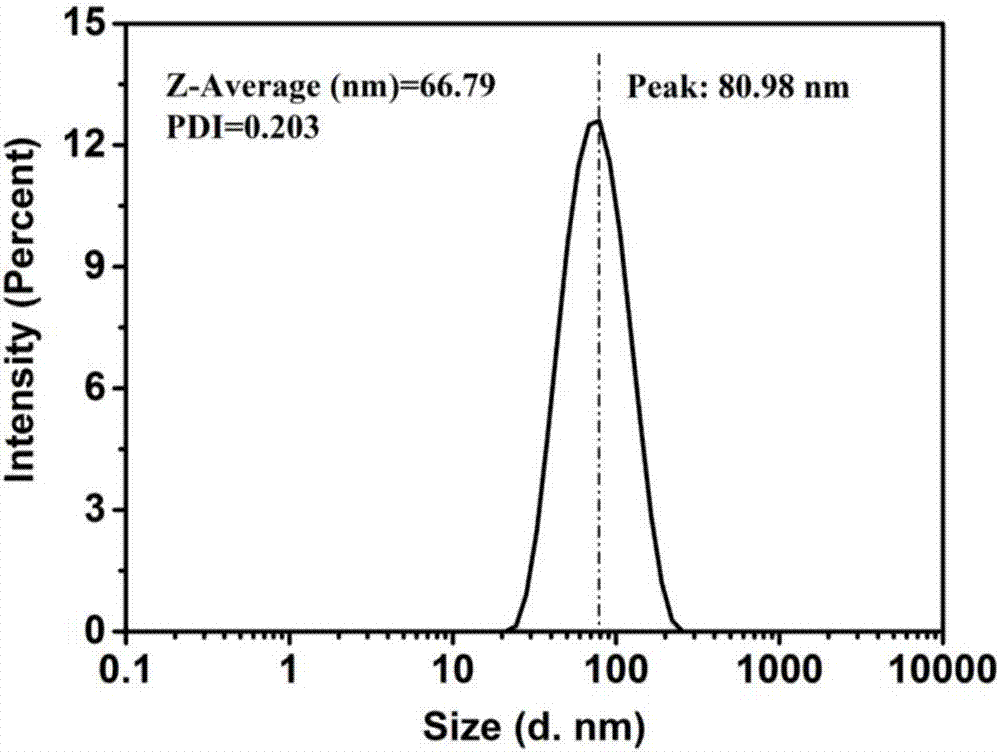
图2为实施例1制备获得的葡聚糖@Fe3O4纳米颗粒经粒径分选前的粒径分布图;
图3为实施例1制备获得的葡聚糖@Fe3O4纳米颗粒经粒径分选后的粒径分布图;
图4为实施例2制备获得的葡聚糖@Fe3O4纳米颗粒未经粒径分选方法处理的第一天的粒径分布图;
图5为实施例2制备获得的葡聚糖@Fe3O4纳米颗粒未经粒径分选方法处理放置一周后的粒径分布图;
图6为实施例2制备获得的葡聚糖@Fe3O4纳米颗粒未经粒径分选方法处理放置一个月后的粒径分布图;
图7为实施例2制备获得的葡聚糖@Fe3O4纳米颗粒经粒径分选(磁性吸附介质填充高度:35mm)的第一天的粒径分布图;
图8为实施例2制备获得的葡聚糖@Fe3O4纳米颗粒经粒径分选(磁性吸附介质填充高度:35mm)后的一周后的粒径分布图;
图9为实施例2制备获得的葡聚糖@Fe3O4纳米颗粒经粒径分选(磁性吸附介质填充高度:35mm)后的一个月后的粒径分布图;
图10为实施例2制备获得的葡聚糖@Fe3O4纳米颗粒经粒径分选(磁性吸附介质填充高度:55mm)的第一天的粒径分布图;
图11为实施例2制备获得的葡聚糖@Fe3O4纳米颗粒经粒径分选(磁性吸附介质填充高度:55mm)后的一周后的粒径分布图;
图12为实施例2制备获得的葡聚糖@Fe3O4纳米颗粒经粒径分选(磁性吸附介质填充高度:55mm)后的一个月后的粒径分布图;
图13为实施例2制备获得的葡聚糖@Fe3O4纳米颗粒经粒径分选(磁性吸附介质填充高度:100mm)的第一天的粒径分布图;
图14为实施例2制备获得的葡聚糖@Fe3O4纳米颗粒经粒径分选(磁性吸附介质填充高度:100mm)后的一周后的粒径分布图;
图15为实施例2制备获得的葡聚糖@Fe3O4纳米颗粒经粒径分选(磁性吸附介质填充高度:100mm)后的一个月后的粒径分布图。
具体实施方式
为了便于理解本发明,下面将参照相关附图对本发明进行更全面的描述。
请结合图1,一实施方式的磁性颗粒分选方法10,包括如下步骤:
S1,制备分选管:
将磁性吸附介质按照预设的高径比填充在层析柱中得分选管。该高径比指的是磁性吸附介质在层析柱中的填充高度与横截面直径的比值。
在本实施方式中,优选地,磁性吸附介质选自铁磁性微球、顺磁性微球或软磁性微球,例如铁珠、钢珠或硅钢珠,饱和磁化强度大,在磁场中被磁化后,与待分选的磁性颗粒的作用力大。磁性吸附介质的粒径为0.1mm-5mm,以控制产品中的磁性纳米颗粒的粒径分布范围。
可以理解,在其他实施例中,当对磁性颗粒的粒径范围没有要求时,磁性吸附介质的粒径范围可以不受限制,只要能够满足吸附量和流速的要求,进行适应性调整即可。
在本实施方式中,层析柱的材质为塑料管,该塑料管为圆柱形的两端开口的中空塑料管,且样品流出端呈逐渐收窄的结构。可以理解,在其他实施方式中,层析柱的材质也可以为金属材质等,其整体形状可以为三棱柱体或四棱柱体等,只要能够保证磁性吸附介质在层析柱能够满足粒径分选所需的高径比,满足待分选磁性颗粒的吸附面积、悬浮液的流速及洗脱液的流速要求即可。
在本实施方式中,优选地,磁性吸附介质的粒径为0.1mm-5mm,高径比为100:5-80,防止悬浮液中待分选磁性颗粒不能完全吸附的损失,同时满足洗脱液的靠重力下降的速度,控制洗脱时间。
在本实施方式中,优选地,对所述磁性吸附介质和所述层析柱的内壁提前进行预处理,其步骤如下:
根据待分选磁性颗粒的悬浮液的亲水性或疏水性,相应地分别预先对磁性吸附介质和层析柱的内壁进行对应的亲水性或疏水性处理,例如涂覆亲水性涂料、表面活性剂、油漆等,以加强悬浮液与层析柱的内壁及磁性吸附介质的相互浸润,提高吸附效率和吸附量。另外,该预处理的步骤也可以分别预先对磁性吸附介质和层析柱的内壁刻蚀处理,提高与待分选磁性颗粒的悬浮液的亲润性、接触面积和吸附作用力。
在其他实施方式,当层析柱的材质为塑料时,也可以预先对其内壁进行电晕处理,以增加表面活性,提高待分选磁性颗粒在磁性吸附介质的吸附作用。
S3,磁性吸附:
制备待分选磁性颗粒的悬浮液。具体地,待分选磁性颗粒的悬浮液可通过共沉淀法、水热法或热分解法制备而成。该待分选磁性颗粒的悬浮液,例如可以为通过共沉淀法制备的葡聚糖@(包裹)Fe3O4纳米颗粒的悬浮液等,其分散剂可以为有机溶剂或水等。
将分选管移至磁场内,使磁性吸附介质磁化。该磁场优选通过永磁铁产生,磁场强度大,且稳定性高。
将含待分选磁性颗粒的悬浮液上样于分选管内,使待分选的磁性颗粒吸附在已磁化的磁性吸附介质上。
S4,洗脱:
将吸附有待分选磁性颗粒的分选管移出磁场,采用洗脱液将已吸附在磁性吸附介质上的待分选磁性颗粒洗脱下来。该洗脱液可以对应悬浮液采用的分散剂。
在本实施方式中,该磁性颗粒分选方法可以通过调整填充磁性吸附介质的使用量、高径比及粒径大小、调整所施加的磁场大小以及调整上样量等,来改善所分选出来的磁性颗粒的粒径分布范围、分散性和稳定性。同时,可以根据流出的悬浮液或者洗脱液的颜色或紫外吸光度值监控磁性吸附过程和洗脱过程,以获得目标产品,即分选出的分散性好、稳定性高及粒径分布范围窄的磁性颗粒的悬浮液。
本实施方式的磁性颗粒分选方法10通过磁性吸附可分选出磁性颗粒,并且磁性吸附介质的填充时具有一定的孔径通道,进而也可以分选出不同粒径范围的磁性颗粒。由于粒径分布范围变窄,磁性颗粒产品整体的分散性和稳定性会显著提高。
下面结合具体实施例,对本发明做进一步说明。
实施例1分散性效果测试
本实施例采用共沉淀法制备葡聚糖@Fe3O4纳米颗粒,并对其进行分选,其步骤如下:
(1)取100mL去离子水,加热到50℃,通入氮气以去除水中的游离氧,用1000r/min的速度进行搅拌。
(2)加入8g葡聚糖,在机械搅拌下溶解完全。
(3)加入6mmol FeCl3.6H2O和3mmol FeCl2.4H2O,搅拌10min。
(4)逐滴加入氨水,直到溶液变成黑色。颜色不再变化后,停止滴加。
(5)升温至60-75℃,继续搅拌反应1小时。反应停止后,冷却到室温,用盐酸调节溶液pH至中性,即得葡聚糖@Fe3O4纳米颗粒的悬浮液。
(6)制备分选管。选取层析柱,该层析柱为圆柱体并具有逐渐收窄的尖端以用于液体的流出,两端开口。该层析柱的总高度为135mm,其内直径为9.0mm,收窄后的出口的直径为1.5mm,逐渐收窄的尖端的高度为40mm。该层析柱的内壁通过表面活性剂聚乙二醇涂覆处理,选用磁性吸附介质为钢珠,粒径为1.5±0.3mm,填充高度为55mm,得分选管。
(7)将分选管移至磁场。再将步骤(5)制备获得的葡聚糖@Fe3O4纳米颗粒的悬浮液上样于步骤(6)中的分选管中,即将所制备的葡聚糖@Fe3O4纳米颗粒的悬浮液从分选管的入口滴入,直到分选管的出口端有黄褐色液体流出为止,表明钢珠表面已吸附了最大量的葡聚糖@Fe3O4纳米颗粒。然后将分选管移出磁场,用去离子水将吸附在钢珠表面的葡聚糖@Fe3O4纳米颗粒洗脱出来,得到分选后的葡聚糖@Fe3O4纳米颗粒的悬浮液。
图2和图3为Fe3O4纳米颗粒的悬浮液通过分选管分选前后的粒径分布图。由图2和图3可知,Fe3O4纳米颗粒的悬浮液分选前,悬浮液中Fe3O4纳米颗粒的平均粒径为66.79nm,多分散系数PDI为0.203;分选后,分选出来产品中Fe3O4纳米颗粒的平均粒径为71.17nm,多分散系数PDI为0.095,磁珠分散性得以极大提高(PDI越小,表明磁珠的分散性越好,PDI≤0.05时,磁珠呈单分散状态)。
实施例2稳定性效果测试
本实施例参照实施例1的方法重新制备一批葡聚糖@Fe3O4纳米颗粒的悬浮液,验证产品中的葡聚糖@Fe3O4纳米颗粒分选前后的稳定性。
(1)制备不同填充高度的分选管。
准备A、B、C三根层析柱,其尺寸与实施例1中相同,对应依次填充钢珠(粒径与实施例1相同),填充高度依次为35mm、55mm和100mm。其中A、C层析柱内的钢珠是经过聚乙二醇处理的,而B层析柱内的钢珠是经过聚乙烯吡咯烷酮(K13-19)处理的。
(2)将三根分选管置于磁场中,再把重新参照实施例1制备的葡聚糖@Fe3O4纳米颗粒的悬浮液逐滴加入分选管内,直到分选管的出口端有黄褐色液体流出。
(3)将三根分选管撤出磁场,分别向三根分选管中滴加去离子水以冲洗出吸附在管内钢珠表面的的葡聚糖@Fe3O4磁性颗粒,直到从分选管的出口端流出的是清澈的去离子水为止。收集由三根分选管中冲洗出来的葡聚糖@Fe3O4磁性颗粒的悬浮液,以备下一步检测用。
(4)对悬浮液中的Fe3O4磁性颗粒进行粒度表征。为对比起见,再表征一组未经过分选方法处理的原悬浮液中的Fe3O4纳米颗粒的粒度变化。
为验证分选出来的磁性颗粒产品的稳定性,对上述四组的样品在保存一周和一个月后再进行粒径分布测试。具体结果见表1以及图4至图12。
表1实施例2的悬浮液中的Fe3O4磁性颗粒粒径分布情况及其稳定性测试
通过对比表1和图4至图15,可以看出,经过分选方法处理的悬浮液中的Fe3O4磁性颗粒粒径分布范围变窄且均匀,分散性提高,且至少一个月内具有非常好的稳定性。
以上所述实施例的各技术特征可以进行任意的组合,为使描述简洁,未对上述实施例中的各个技术特征所有可能的组合都进行描述,然而,只要这些技术特征的组合不存在矛盾,都应当认为是本说明书记载的范围。
以上所述实施例仅表达了本发明的几种实施方式,其描述较为具体和详细,但并不能因此而理解为对发明专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。因此,本发明专利的保护范围应以所附权利要求为准。
- 还没有人留言评论。精彩留言会获得点赞!