一种测定石斛鲜条中有机氯和拟除虫菊酯农药残留的方法与流程
本发明属于农药残留领域的检测方法,具体涉及一种同时测定石斛鲜条中有机氯类和拟除虫菊酯类农药残留的方法。
背景技术:
石斛味甘、性微寒,具有益胃生津、滋阴清热的功效,作为名贵的中药材和保健品,越来越受到消费者的青睐和追捧。为了增加产量,农药已广泛应用于石斛生产中,有机氯类和拟除虫菊酯类农药因广谱、高效、价廉而被广泛使用。但这两类农药对人体神经中枢和消化系统毒害较大,并且会在人体肝脏中蓄积,长期过量使用后对人体健康危害较大,因此,对石斛中有机氯类和拟除虫菊酯类农药残留进行检测并有效控制具有十分重要的意义。
针对农药残留,欧盟、美国、日本等国家都制定了严格的农药残留限量标准,我国主要施行GB 2763《食品安全国家标准 食品中农药最大残留限量》标准,目前也有一些严于国家标准的石斛地方标准限量发布实施,如:DB53/T 683《地理标志产品 芒市石斛》、DB53/T 589《地理标志产品 龙陵紫皮石斛》、DB34/T 486《霍山石斛》等,这就意味着对石斛产品的质量安全指标要求更高、更严。
查阅相关文献,目前对石斛鲜条农药残留检测方法还没有系统的研究报道。现有的研究主要集中在对茶叶、中草药、保健食品等方面。阎正、封棣等采用固相萃取-毛细管气相色谱法开展了中草药中13种有机氯农药的残留量的研究;訾伟旗,杜斌采用毛细管气相色谱法测定了中药材中拟除虫菊酯类农药等,这些方法均采用石油醚、丙酮或其两者的混合溶剂来进行农残的提取,对于干燥和颜色浅的物质来说,这种方法还是可行有效,但对于水分和多糖含量高、粘度大及颜色深的石斛鲜条来说,这种方法不能很好的把残留在其上面的农药充分提取出来。
为了解决以上问题,本发明的目的是建立一种同时测定石斛鲜条中有机氯类和拟除虫菊酯类农药残留的方法。经反复实验确证,该方法简便、准确、快速、实用,各项技术指标满足检测要求,能够很好的应用于石斛鲜条农药残留的测定。
技术实现要素:
本发明提供了一种测定石斛鲜条中有机氯和拟除虫菊酯农药残留的方法,目的是解决目前石斛鲜条农残测定前处理繁琐、种类单一、操作复杂、结果不准确等问题。石斛鲜条经切碎、乙腈提取、Florisil 固相萃取柱净化后,采用气相色谱ECD检测检测,外标法定量。该方法简便、快速、灵敏、准确,能够满足石斛鲜条中有机氯类和拟除虫菊酯类农药残留的定性和定量分析。
为了实现上述发明目的,通过大量研究,本发明提供的技术方案是:
一种同时测定石斛鲜条中有机氯类和拟除虫菊酯类农药残留的方法,该检测方法由两部分组成:
第一部分是标准曲线的建立。即用已知浓度的有机氯类和拟除虫菊酯类农药进气相色谱仪检测分析,以溶液质量浓度(C)为横坐标(x),峰面积(A)为纵坐标(y)绘制标准曲线,进行定量分析。具体实施步骤为:
1、依次吸取浓度均为100 ug/ml的六六六和滴滴涕混标、三氯杀螨醇、联苯菊酯、甲氰菊酯、氯菊酯、高效氯氰菊酯、氯氰菊酯、顺式氰戊菊酯、氟氰戊菊酯、氰戊菊酯、溴氰菊酯农药标准溶液各500μl于50 ml棕色容量瓶中,用色谱纯的正己烷定容至刻度,配制成浓度为1.0μg/ml的有机氯类和拟除虫菊酯类农药混合标准储备液。
2、用吸量管吸取步骤1中的混合标准储备液,用正己烷逐级稀释,配制成浓度为0.008μg/ml、0.02μg/ml、0.08μg/ml、0.1μg/ml、0.2μg/ml、0.4μg/ml的混合标准工作液。
3、色谱条件设置:色谱柱:30 m×0.32mm×0.25μm的 HP-5毛细管色谱柱;进样口温度:200℃;ECD检测器温度:320℃;升温程序:150℃,保持2min,以6℃/min升至250℃,保持4 min,以1℃/min升至270℃,保持3 min;进样量:1μl,分流比10:1。
4、按照步骤3所述色谱条件,准确吸取1.0μl 步骤2所述有机氯类和拟除虫菊酯类农药混合标准工作液,以各农药组分的色谱峰保留时间定性,以溶液质量浓度(C)为横坐标(x),峰面积(A)为纵坐标(y)绘制标准曲线进行定量。
第二部分是石斛鲜条中有机氯类和拟除虫菊酯类农药残留的提取和检测
1、石斛鲜条农残的提取:将石斛鲜条切碎成长度不超过2mm的小块状,制成试样。称取10.0 g置于100ml具塞比色管中,加入50 ml乙腈,盖紧塞子,超声提取30分钟,迅速用滤纸将滤液过滤到50ml具塞量筒中,收集滤液40ml,加入5g氯化钠,盖上塞子,激烈震荡1分钟,静置,使乙腈和水相分层,然后吸取10.0ml乙腈溶液于10ml比色管中,在氮吹仪上通氮气蒸发近干,加入2.0ml正己烷,超声溶解,待净化。
2、试样的净化:取规格为500mg、6ml的Florisil固相萃取柱一支,装入1g碳粉,用5.0ml丙酮与正己烷体积比1:4的混合溶液淋洗,再用5.0ml正己烷淋洗,当溶剂液面到达柱吸附层表面时,立即倒入步骤(1)所得的样品溶液,用10ml比色管接收洗脱液,用5ml丙酮与正己烷体积比1:4的混合溶液清洗样品管,洗液倒入固相萃取柱上,重复一次;待固相萃取柱上的洗液滴干时将盛有淋洗液的比色管置于氮吹仪上,在水浴温度50℃下,氮吹浓缩至1~2 ml,最后用正已烷准确定容至5.0ml,旋涡混合器上混匀,用0.22μm的滤膜过滤后供气相色谱检测。
3、测定:待仪器稳定后,按照第一部分3所述色谱操作条件,准确吸取1.0μl步骤2所述试样净化液注入色谱仪中,以色谱峰保留时间定性,峰面积代入第一部分步骤4中相应的校准曲线,计算含量。
4、计算公式:
W:样品中某种农药残留含量,单位:mg/kg;
C:待测试样中某种农药浓度,单位:ug/ml,
m:称取样品质量,单位:g。
测定石斛鲜条中有机氯类和拟除虫菊酯类农药残留的色谱条件与第一部分步骤(3)中的条件相同。
5、方法验证
(1)线性范围:测定结果表明,在上述色谱分离条件下,18种有机氯类和拟除虫菊酯类农药得到很好的分离,在0.008~0.4μg/ml浓度范围内,线性关系良好,其相关系数均大于0.996,可以作为定量分析的依据,详见表1。
(2)方法的检出限、精密度和加标回收率
取不含农药的石斛样品,精确添加0.05mg/kg和0.5mg/kg的混合标准溶液,按3的步骤处理,在同一分析条件下重复测定5次,获得的色谱图按信噪比3倍(S/N=3)条件计算方法的检出限,结果:18种农残的回收率在75.3%~109.2%之间,相对标准偏差在2.9%~8.6%之间,检出限在0.001mg/kg~0.01mg/kg之间(见表2),符合石斛鲜条多农药残留分析的相关技术要求。
表2 18种农药精密度和回收率试验结果
6、上述方案中有关内容解释
(1)所用标准物质六六六和滴滴涕混标、三氯杀螨醇、联苯菊酯、甲氰菊酯、氯菊酯、高效氯氰菊酯、氯氰菊酯、顺式氰戊菊酯、氟氰戊菊酯、氰戊菊酯、溴氰菊酯均购于农业部环境保护科研检测所,浓度均为100 ug/ml。
(2)所用试剂乙腈、丙酮、正己烷均为色谱纯,购于天津市光复精细化工研究所,氯化钠为分析纯购于天津市风船化学试剂科技有限公司。
(3)所用材料500mg/6ml的弗罗里硅土florisil固相萃取柱购于美国安捷伦科技有限公司,石斛鲜条由本地石斛协会提供。
(4)所用仪器设备:配有ECD电子捕获检测器的GC-2010Plus气相色谱仪购于日本岛津公司,MTN-2800W氮吹浓缩装置购于天津奥特赛恩斯仪器有限公司,KYT-JT-B均质器购于北京同德创业科技有限公司。
(5)对于含2个及2个以上异构体的农药,以各异构体的峰面积之和计算该农药的测定值。
本发明的优点和有益效果
虽然液相色谱-质谱联用仪、气相色谱-质谱联用仪具有较高的准确性和较低的检出限,在农残检测领域已有广泛运用,但是其价格昂贵、操作要求高,而气相色谱仪相对其价格适中、操作简便,作为实验室农残检测常用仪器,目前许多县级检测部门都有配置。
本发明提出的石斛鲜条中有机氯类和拟除虫菊酯类农药残留的测定方法,在上述条件下各农药组分间分离效果好、经加标回收和精密度计算,测定结果准确可靠,各项指标均满足农药多残留分析的要求,能够很好的运用于石斛鲜条中多组分有机氯类和拟除虫菊酯类农药残留的定性、定量分析。
与现行许多国家标准方法相比,本发明前处理操作简单,提取效率高,净化过程使用的洗脱剂少,对环境污染小,农药损失率低,实用性强,具有很好的推广使用价值。
附图说明
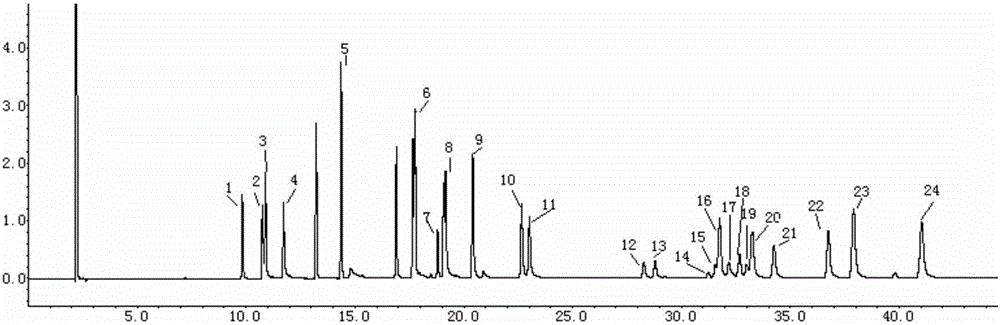
图1为本发明实施实例中18种有机氯类和拟除虫菊酯类农药混合标准溶液色谱图;
图中,浓度均为0.2ug/ml,编号依次为:1为α-六六六;2为β-六六六;3为γ-六六六;4为δ-六六六;5为三氯杀螨醇;6为p,p'-DDE ;7为p,p'-DDD;8为o,p'-DDT;9为p,p'-DDT;10为联苯菊酯;11为甲氰菊酯;12、13为氯菊酯;.14、15、16为高效氯氰菊酯;17、18为氯氰菊酯;19、20为顺式氰戊菊酯;21为氟氰戊菊酯;22、23为氰戊菊酯;24为溴氰菊酯。
具体实施方式
下面结合附图对本发明作进一步详细说明,但本发明不仅仅局限于以下实施实例。
所述方法由两部分组成,第一部分是建立标准曲线:
依次吸取浓度均为100 ug/ml的六六六和滴滴涕混标、三氯杀螨醇、联苯菊酯、甲氰菊酯、氯菊酯、高效氯氰菊酯、氯氰菊酯、顺式氰戊菊酯、氟氰戊菊酯、氰戊菊酯、溴氰菊酯农药标准溶液各500μl于50 ml棕色容量瓶中,用色谱纯的正己烷定容至刻度,配制成浓度为1.0μg/ml的有机氯类和拟除虫菊酯类农药混合标准储备液,放于2-4℃的冰箱中备用。用吸量管吸取混合标准储备液,用正己烷逐级稀释,配制成浓度为0.008μg/ml、0.02μg/ml、0.08μg/ml、0.1μg/ml、0.2μg/ml、0.4μg/ml的混合标准工作液(现用现配)。在下述色谱条件下,色谱柱:30 m×0.32mm×0.25μm的 HP-5毛细管色谱柱;进样口温度:200℃;ECD检测器温度:320℃;升温程序:150℃,保持2min,以6℃/min升至250℃,保持4 min,以1℃/min升至270℃,保持3 min;进样量:1μl,分流比10:1,载气为高纯氮气。准确吸取1.0μl 农药混合标准工作液,进气相色谱分析检测,以溶液质量浓度(C)为横坐标(x),峰面积(A)为纵坐标(y)绘制各农药组分标准曲线。
第二部分石斛鲜条中有机氯类和拟除虫菊酯类农残的提取和检测:
称取切碎长度不超过2mm的石斛鲜条10.0 g于100ml具塞比色管中,加入50 ml乙腈,盖紧塞子,超声提取30分钟,迅速用滤纸将滤液过滤到50ml具塞量筒中,收集滤液40ml,加入5g氯化钠,盖上塞子,激烈震荡1分钟,静置,使乙腈和水相分层,然后吸取10.0ml乙腈溶液于10ml比色管中,在氮吹仪上通氮气蒸发近干,加入2.0ml正己烷,超声溶解,将溶解液倒入事先用5.0ml丙酮与正己烷体积比为1:4的混合溶液淋洗,再用5.0ml正己烷淋洗过的Florisil固相萃取柱上,下面用10ml比色管接收洗脱液,当样液快到达柱吸附层表面时,用5ml丙酮与正己烷体积比1:4的混合溶液清洗样品管,洗液倒入固相萃取柱上,重复一次;待固相萃取柱上的洗液滴干时将盛有淋洗液的比色管置于氮吹仪上,在水浴温度50℃下,氮吹浓缩至1~2 ml,用正已烷准确定容至5.0ml,旋涡混合器上混匀,用0.22μm的滤膜过滤后制成待测样。
在上述色谱条件下,准确吸取1.0μl待测液注入气相色谱仪分析,得到样品色谱图,通过保留时间定性后,将峰面积代入第一部分相应农药校准曲线,计算出农药浓度值(对于含2个及2个以上异构体的农药,以各异构体的峰面积之和计算该农药的测定值),再将浓度值代入定量计算公式,求出石斛鲜条农残含量。
计算公式:
W:样品中某种农药残留含量(单位:mg/kg);C:待测试样中某种农药浓度(单位:ug/ml);m:称取样品质量(单位:g)。
5、方法验证
(1)线性范围:测定结果表明,在上述色谱分离条件下,18种农药得到很好的分离,在0.008~0.4μg/ml浓度范围内线性关系良好,其相关系数均大于0.996,可以作为定量分析的依据,详见表1。
表1 18种农药保留时间、回归方程和相关系数
(2)方法的检出限、精密度和加标回收率
取不含农药的石斛样品,精确添加0.05mg/kg和0.5mg/kg的混合标准溶液,按3的步骤处理,在同一分析条件下重复测定5次,获得的色谱图按信噪比3倍(S/N=3)条件计算方法的检出限,结果:18种农残的回收率在75.3%~109.2%之间,相对标准偏差在2.9%~8.6%之间,检出限在0.001mg/kg~0.01mg/kg之间(见表2),符合石斛鲜条多农药残留分析的相关技术要求。
表2 18种农药精密度和回收率试验结果
上述实施实例中所用农残标准物质浓度均为100 ug/ml,购于农业部环境保护科研检测所;所用试剂色谱纯乙腈、色谱纯丙酮、色谱纯正己烷购于天津市光复精细化工研究所,分析纯氯化钠购于天津市风船化学试剂科技有限公司,所用材料500mg/6ml的 florisil固相萃取柱购于美国安捷伦科技有限公司;石斛鲜条由当地石斛协会提供;所用仪器设备配有ECD电子捕获检测器的GC-2010Plus气相色谱仪购于日本岛津公司,MTN-2800W氮吹浓缩装置购于天津奥特赛恩斯仪器有限公司,KYT-JT-B均质器购于北京同德创业科技有限公司。
- 还没有人留言评论。精彩留言会获得点赞!