一种阿昔替尼原料及其制剂中有关物质的分析方法与流程
本发明涉及化学药物分析方法
技术领域:
,尤其涉及一种阿昔替尼原料及其制剂中有关物质的分析方法。
背景技术:
:阿昔替尼(axitinib),化学名为n-甲基-2-({3-[(1e)-2-(吡啶-2-基)乙烯-1-基]-1h-吲唑-6-基}硫烷基)苯甲酰胺,其分子式为c22h18n4os,分子量为386.47,cas号为319460-85-0,其结构式如下:阿昔替尼是新的口服酪氨酸激酶抑制剂(tki),能有效并选择性抑制血管内皮生长因子受体vegfr-1、vegfr-2和vegfr-3,抑制血管和淋巴管的新生,抑制肿瘤的生长和转移,发挥抗肿瘤活性。阿昔替尼片原研厂家是美国辉瑞公司,首先于2012年1月在美国获得fda的上市批准,批准的制剂为阿昔替尼片,规格为1mg和5mg,商品名为inlyta,同年6月,辉瑞日本分公司生产的阿昔替尼片在日本批准上市。该药是自2005年来fda批准用于治疗转移或晚期肾细胞癌的第7种药物。该药为口服药丸,一日2次服用。通过阻断肿瘤生长过程中的蛋白激酶起到抑制肿块生长和癌症进展的作用。多项临床试验研究表明,阿昔替尼对接受过多种药物治疗无效的arcc患者显示出了临床活性。一项随机、开放、国际多中心的ⅲ期临床试验中,对于既往接受过治疗的晚期肾细胞癌患者,阿昔替尼与索拉非尼相比,显著延长无进展生存期,且显示出总的良好的安全性。为了保证药物的安全有效,需要对药物原料及其制剂中的有关物质进行研究、检测和监控。由于药品的合成工艺不同,药品的杂质谱也会发生变化,因此需要根据不同的合成工艺建立合适的分析方法,达到对阿昔替尼有关物质准确、有效的检测和监控。技术实现要素:基本
背景技术:
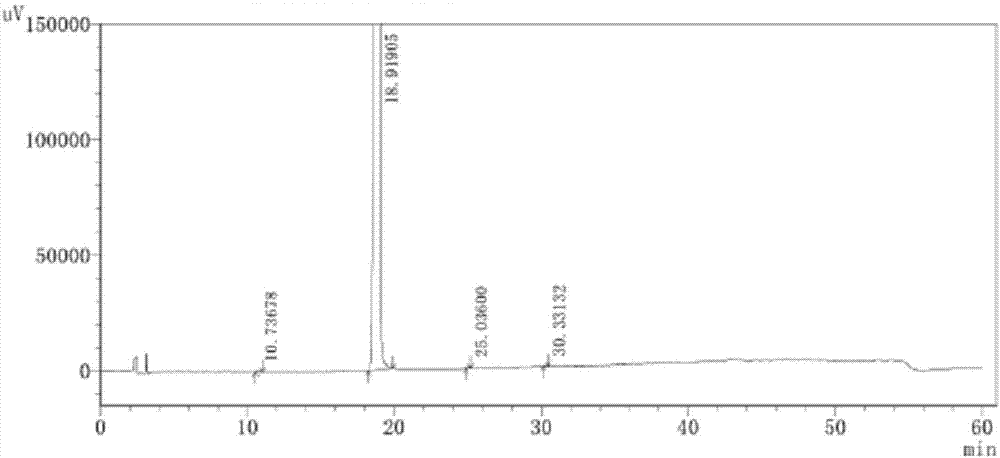
存在的技术问题,本发明提出了一种阿昔替尼原料及其制剂中有关物质的分析方法,本发明通过加校正因子的主成分自身对照法能快速、有效、准确监控阿昔替尼中的有关物质。本发明提出的一种阿昔替尼原料及其制剂中有关物质的分析方法,采用高效液相色谱法,其色谱条件包括:色谱柱为十八烷基硅烷键合硅胶色谱柱,以磷酸盐缓冲水溶液和乙腈的体积比为88-92:8-12为流动相a,以磷酸盐缓冲水溶液和乙腈的体积比为8-12:88-92为流动相b,检测波长为218-222nm,进行梯度洗脱;所述梯度洗脱过程为:0.01-10min内,流动相a和流动相b的体积比为80:20;10-40min内,流动相a和流动相b的体积比从80:20匀速渐变至30:70;40-50min内,流动相a和流动相b的体积比为30:70;50-50.01min内,流动相a和流动相b的体积比从30:70匀速渐变至80:20;50.01-60min内,流动相a和流动相b的体积比为80:20。优选地,色谱柱的长度为250mm,直径为4.6mm,填料粒径为5μm。优选地,在流动相a中,磷酸盐缓冲水溶液和乙腈的体积比可以为88.5:11.5、89:11、89.5:10.5、90:10、90.5:9.5、91:9或91.5:8.5。优选地,在流动相b中,磷酸盐缓冲水溶液和乙腈的体积比可以为8.5:91.5、9:91、9.5:90.5、10:90、10.5:89.5、11:89或11.5:88.5。优选地,磷酸盐缓冲水溶液的ph=3.2-3.4。优选地,磷酸盐缓冲水溶液的ph可以为3.21、3.22、3.23、3.24、3.25、3.26、3.27、3.28、3.29、3.3、3.31、3.32、3.33、3.34、3.35、3.36、3.37、3.38或3.39。优选地,在磷酸盐缓冲水溶液中,用磷酸调节ph=3.2-3.4。优选地,在磷酸盐缓冲水溶液中,磷酸盐的浓度为0.003-0.007mol/l。优选地,在磷酸盐缓冲水溶液中,磷酸盐的浓度可以为0.004、0.0045、0.005、0.0055、0.006或0.0065mol/l。优选地,在磷酸盐缓冲水溶液中,磷酸盐为磷酸二氢铵。优选地,流速为0.95-1.05ml/min。优选地,流速可以为0.96、0.97、0.98、0.99、1.0、1.01、1.02、1.03或1.04ml/min。优选地,柱温为25-33℃。优选地,柱温为26、26.5、27、27.5、28、28.5、29、29.5、30.5、31、31.5、32或32.5。优选地,进样量为5-50μl。优选地,进样量为6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30、31、32、33、34、35、36、37、38、39、40、41、42、43、44、45、46、47、48或49μl。优选地,所述有关物质为:上述杂质中,杂质4为起始原料,杂质7和杂质8为中间体,杂质1为工艺杂质和降解杂质,杂质2为工艺杂质,杂质3、5、6为降解杂质。本发明的具体步骤为:分别配制系统适用性溶液、对照溶液和供试品溶液并进样,通过加校正因子的主成分自身对照法计算供试品中各杂质的含量。上述系统适用性溶液为:分别取杂质1、2、3、4、5、6、7对照品约12.5mg,精密称定,置25ml棕色量瓶中,加甲醇适量使溶解并稀释至刻度,摇匀,作为杂质对照品贮备液一;另取杂质8对照品约12.5mg,精密称定,置25ml棕色量瓶中,加dmf适量使溶解,再加甲醇稀释至刻度,作为杂质对照品贮备液二;另取阿昔替尼对照品约25mg,精密称定,置50ml棕色量瓶中,加甲醇适量使溶解,精密量取杂质对照品贮备液一、杂质对照品贮备液二各0.5ml,置同一量瓶中,加甲醇稀释至刻度,摇匀,作为系统适用性溶液。上述供试品溶液为:取阿昔替尼供试品适量,精密称定,用甲醇溶解并定容得到含阿昔替尼浓度为0.5mg/ml的供试品溶液。上述对照溶液为:精密量取供试品溶液1.0ml于100ml量瓶中,用甲醇定容至刻度,摇匀得到对照溶液。本发明人对阿昔替尼、杂质1-8分别进行紫外吸收光谱扫描,结果见表1:表1阿昔替尼和各杂质的紫外吸收波长由表1可以看出,阿昔替尼及各杂质在220nm波长处虽然不是最大吸收,但是均有较大紫外吸收,综合比较后选择220nm波长作为本品有关物质检查波长。本发明人通过筛选合适流动性组分并优化各组分比例,以及筛选合适的其他色谱条件,对阿昔替尼以及上述8个杂质进行色谱分析,确定了本发明分析方法,通过对起始原料、反应中间体、工艺杂质、降解杂质、阿昔替尼的峰定位试验,干扰性试验和阿昔替尼的降解试验对本发明进行专属性验证,结果见表2和图1:表2专属性验证结果由表2和图1可以看出,各杂质峰之间、阿昔替尼主峰及其相邻杂质峰之间的分离度均大于1.5,阿昔替尼主峰的理论板数大于3000,本发明的专属性好。本发明人还选用市场上常用的制备阿昔替尼制剂的药用辅料和空白溶剂对本发明进行了研究,发现常用的药用辅料和空白溶剂对本发明没有干扰。本发明人对阿昔替尼和各杂质的检测限、定量限、线性、校正因子进行检测,结果见表3:表3阿昔替尼和各杂质的检测限、定量限、线性、校正因子试验结果上述报告限度是指超出此限度的杂质均应在检测报告中报告,并应报告具体的检测数据。由表3可以看出,本发明阿昔替尼和各杂质检测灵敏度均较高,其检测限均小于报告限度,并且各杂质在较低浓度范围内线性关系良好。本发明人配制供试品溶液,分别于配制后的0、2、4、8h进样并记录图谱,统计并计算供试品中阿昔替尼和各杂质的含量,计算得到供试品中杂质3、5和阿昔替尼的含量相对标准偏差rsd分别为8.61%,3.85%,0.49%,其它已知杂质均未检出,并统计新生杂质个数为0。结果表明,供试品溶液在8h内稳定。本发明人对各杂质进行回收率试验,对杂质3和杂质5进行重复性、中间精密度试验,结果见表4和表5:表4各杂质的回收率验证结果试验回收率80-120%回收率rsd≤10%杂质1106.755.20杂质2105.004.61杂质3103.763.44杂质4107.685.40杂质5101.054.32杂质6101.923.78杂质7108.555.66杂质892.473.42表5杂质3和杂质5的重复性、中间精密度试验结果试验重复性-峰面积rsd≤15%中间精密度-含量rsd≤20%杂质37.9814.91杂质512.2812.04由表4和表5可以看出,本发明的回收率试验结果均符合要求,本发明回收率高,杂质3和杂质5的重复性、中间精密度均符合要求,杂质3、杂质5的重复性、中间精密度均良好。本发明人取阿昔替尼配制供试品溶液,进样并记录图谱,按加校正因子的主成分自身对照法计算供试品中杂质1-8的含量,结果见表6和图2。表6阿昔替尼中各杂质的含量测定结果名称杂质1杂质2杂质3杂质4含量%未检出未检出0.032未检出名称杂质5杂质6杂质7杂质8含量%0.026未检出未检出未检出由表6和图2可以看出,阿昔替尼供试品中杂质3和5含量分别为0.032%和0.026%,其它已知杂质均未检出。本发明能快速、有效、准确监控阿昔替尼中的有关物质;本发明具有良好的专属性,各杂质峰之间、阿昔替尼主成分峰及其相邻杂质峰之间的分离度均大于1.5,阿昔替尼主峰的理论板数大于3000,杂质峰和主峰可以有效分离;本发明检测限、定量限均较小,本发明的灵敏度好;本发明的重复性好,中间精密度高好,回收率高,可以准确测量阿昔替尼原料和制剂中的有关物质;本发明通过加校正因子自身对照法对上述8个杂质进行定量分析,增加本发明有关物质检测的准确性。附图说明图1为系统适用性溶液色谱图。图2为阿昔替尼原料有关物质色谱图。图3为阿昔替尼制剂有关物质色谱图。具体实施方式下面,通过具体实施例对本发明的技术方案进行详细说明。实施例1高效液相色谱条件:十八烷基硅烷键合硅胶色谱柱(250×4.6mm,5μm),以磷酸调节ph=3.3的磷酸二氢铵缓冲水溶液和乙腈的体积比为90:10为流动相a,以磷酸调节ph=3.3的磷酸二氢铵缓冲水溶液和乙腈的体积比为10:90为流动相b,检测波长为220nm,流速为1.0ml/min,柱温为30℃,其中,磷酸二氢铵缓冲水溶液中磷酸二氢铵的浓度为0.005mol/l,进行梯度洗脱;所述梯度洗脱过程为:0.01-10min内,流动相a和流动相b的体积比为80:20;10-40min内,流动相a和流动相b的体积比从80:20匀速渐变至30:70;40-50min内,流动相a和流动相b的体积比为30:70;50-50.01min内,流动相a和流动相b的体积比从30:70匀速渐变至80:20;50.01-60min内,流动相a和流动相b的体积比为80:20。样品配制:系统适用性溶液:分别取杂质1、2、3、4、5、6、7对照品约12.5mg,精密称定,置25ml棕色量瓶中,加甲醇适量使溶解并稀释至刻度,摇匀,作为杂质对照品贮备液一;另取杂质8对照品约12.5mg,精密称定,置25ml棕色量瓶中,加dmf适量使溶解,再加甲醇稀释至刻度,作为杂质对照品贮备液二;另取阿昔替尼对照品约25mg,精密称定,置50ml棕色量瓶中,加甲醇适量使溶解,精密量取杂质对照品贮备液一、杂质对照品贮备液二各0.5ml,置同一量瓶中,加甲醇稀释至刻度,摇匀,作为系统适用性溶液。试验操作:取系统适用性溶液10μl进样,记录色谱图。典型色谱图见图1。实施例2高效液相色谱条件:十八烷基硅烷键合硅胶色谱柱(250×4.6mm,5μm),以磷酸调节ph=3.2的磷酸二氢铵缓冲水溶液和乙腈的体积比为92:8为流动相a,以磷酸调节ph=3.4的磷酸二氢铵缓冲水溶液和乙腈的体积比为8:92为流动相b,检测波长为218nm,流速为1.05ml/min,柱温为25℃,其中,磷酸二氢铵缓冲水溶液中磷酸二氢铵的浓度为0.007mol/l,进行梯度洗脱;所述梯度洗脱过程同实施例1。样品配制:系统适用性溶液:同实施例1。供试品溶液:取阿昔替尼供试品适量,精密称定,用甲醇溶解并定容得到含阿昔替尼浓度为0.5mg/ml的供试品溶液。对照溶液:精密量取供试品溶液1.0ml于100ml量瓶中,用甲醇定容至刻度,摇匀得到对照溶液。试验操作:取系统适用性溶液、供试品溶液、对照溶液各50μl进样,记录色谱图。实施例3高效液相色谱条件:十八烷基硅烷键合硅胶色谱柱(250×4.6mm,5μm),以磷酸调节ph=3.4的磷酸二氢铵缓冲水溶液和乙腈的体积比为88:12为流动相a,以磷酸调节ph=3.2的磷酸二氢铵缓冲水溶液和乙腈的体积比为12:88为流动相b,检测波长为222nm,流速为0.95ml/min,柱温为33℃,其中,磷酸二氢铵缓冲水溶液中磷酸二氢铵的浓度为0.003mol/l,进行梯度洗脱;所述梯度洗脱过程同实施例1。样品配制:系统适用性溶液:同实施例1。供试品溶液:同实施例2。对照溶液:同实施例2。试验操作:取系统适用性溶液、供试品溶液、对照溶液各5μl进样,记录色谱图。实施例4高效液相色谱条件:同实施例1。样品配制:系统适用性溶液:同实施例1。供试品溶液:同实施例2。对照溶液:同实施例2。试验操作:取系统适用性溶液、供试品溶液、对照溶液各10μl进样,记录色谱图。典型色谱图见图2。实施例5高效液相色谱条件:同实施例1。样品配制:系统适用性溶液:同实施例1。供试品溶液:取规格为5mg的阿昔替尼片10片,研碎混匀,取细粉适量(约相当于含阿昔替尼25mg),精密称定,置50ml量瓶中,用甲醇溶解并定容,过滤得到供试品溶液。对照溶液:精密量取供试品溶液1.0ml于100ml量瓶中,用甲醇定容至刻度,摇匀得到对照溶液。试验操作:取系统适用性溶液、供试品溶液、对照溶液各10μl进样,记录色谱图。典型色谱图见图3。以上所述,仅为本发明较佳的具体实施方式,但本发明的保护范围并不局限于此,任何熟悉本
技术领域:
的技术人员在本发明揭露的技术范围内,根据本发明的技术方案及其发明构思加以等同替换或改变,都应涵盖在本发明的保护范围之内。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1