一种DNA银纳米簇及其细胞原位的合成方法和应用与流程
本发明涉及一种利用细胞原位合成制备的dna银纳米簇用于干细胞增殖和分化过程中端粒酶的长期监控,属于材料合成及生物成像与传感技术领域。
背景技术:
端粒酶(telomerase)是负责端粒延长的一种酶,端粒缩短的细胞复制能力受限,端粒酶能以自身rna为模版延长端粒长度,从而保持染色体稳定性和细胞活性,在细胞增殖及分化过程中起重要作用。间充质干细胞是一种具有多向分化潜能的干细胞,常用于临床上治疗中枢神经系统受损,相较于骨髓源和脂肪源干细胞,人脐带间充质干细胞易取材且不受伦理法制限制,在医学上的应用更为广泛。然而,影响人脐带间充质干细胞增殖和分化潜力的重要的端粒酶在上述过程中的表达,却未曾有实时原位监测的方法。
银纳米簇具有光稳定性好,毒性小,尺寸小等特性,常应用于生物标记。另外一些特定的碱基,金属离子和生物分子会影响银纳米簇的荧光,使其增强或者猝灭。因而可以利用不同寡核苷酸修饰的银纳米簇作为多功能荧光探针,用来高灵敏检测细胞内功能分子如酶、葡萄糖、蛋白质、dna等。
然而化学方法合成的dna-银纳米簇虽用途广泛生物相容性较好,但仍易被免疫系统通过肾清除排出体外,从而限制了探针在生物体内的停留时间,无法长期实时监控端粒酶等其他功能分子的活性。
本发明公开的一种新的诱导细胞原位合成的方法来制备dna银纳米簇可用于细胞内端粒酶的长期监控。因生物方法原位合成的dna银纳米簇可以不受限于细胞免疫排斥的影响,极高的生物相容性大大提高了探针在生物体内的滞留时间,使得探针可以长期留在细胞内,对细胞在生长分化过程中的状态进行实时动态成像。还可以作为一种纳米荧光探针来识别和标记细胞内的功能分子,为活体荧光实时动态成像及细胞疾病的早期诊断和治疗提供了新的思路和方法。
技术实现要素:
本发明的目的在于提供了一种利用细胞原位合成制备dna银纳米簇,用于干细胞增殖和分化过程中端粒酶的长期监控;干细胞中谷胱甘肽(gsh)含量显著高于普通细胞,gsh是一种含有巯基的三肽,是动物细胞中关键的抗氧化剂,真核细胞通过其可逆的氧化还原反应以维持细胞的活性氧平衡。当干细胞输入大量dna和银离子,细胞内部会通过gsh自发地还原组装银纳米颗粒,形成dna为模板的银纳米簇。所述dna银纳米簇具有生物相容好,在体时间长和检测灵敏度高的优点。
本发明提供了一种细胞内源dna银纳米簇的原位合成方法,所述方法包括以下步骤:
(1)上调细胞内谷胱甘肽(gsh)的水平:
培养细胞,待细胞覆盖率达75~85%左右时,加入琉辛酸(α-lipoicacid)进行共孵育。
步骤(1)中,所述细胞选自人骨髓间充质干细胞、人脐带间充质干细胞、人胚胎间充质干细胞;优选地,为人脐带间充质干细胞。
步骤(1)中,培养细胞的培养基的组分为sciencell公司提供的混有hmscs生长因子的高糖/f12(dmem/f12)培养液(货号7501)。
步骤(1)中,优选地,待细胞覆盖率达80%时,加入琉辛酸。
步骤(1)中,加入琉辛酸前,所述琉辛酸的浓度为4~8μm;优选地,为5μm。
步骤(1)中,加入琉辛酸后,3ml培养液中,含104个细胞,所述琉辛酸的浓度为5×10-5μm。
步骤(1)中,所述琉辛酸的作用为诱导刺激细胞内还原型的谷胱甘肽含量上调。
步骤(1)中,所述共孵育的温度为37℃。
步骤(1)中,所述共孵育的时间为12-36h;优选地,为24h。
步骤(1)中,所述共孵育优选地在5%co2的条件下进行。
步骤(1)中,培养结束后,缓慢倾去培养液并用新鲜培养液替换,其目的是终止诱导、让细胞恢复正常生长环境。
(2)检测细胞内gsh含量:
离心收集细胞,冻融破碎细胞,释放细胞中的gsh,用试剂盒测定gsh的含量。
步骤(2)中,收集细胞前还包括用pbs洗涤细胞的步骤。
步骤(2)中,收集细胞进行冻融破碎前,还包括加入蛋白去除试剂的步骤。
步骤(2)中,所述冻融破碎细胞的方法为,利用液氮和37℃水浴对样品进行两次快速的冻融破碎细胞。
步骤(2)中,所述测定gsh的试剂盒是dtnb试剂盒。
步骤(2)中,将所测定的诱导后的细胞内gsh含量的结果与空白细胞内的gsh含量测定结果对照,如果上调后的细胞内的gsh含量高于空白细胞内的gsh含量的15%,则说明上调成功。或者,如果上调后的细胞内的gsh含量接近肝癌hepg2细胞内的gsh含量(71μg/gprotein),则说明细胞内的gsh含量适用于进行转染等步骤。
(3)转染dna:
取上述已上调gsh含量的细胞,加入新鲜细胞培养液和转染液进行培养,去除培养液和转染液。加入新鲜细胞培养液继续培养。
步骤(3)中,所述新鲜细胞培养液为sciencecell公司提供的混有hmscs生长因子的高糖/f12(dmem/f12)培养液(货号7501)。
步骤(3)中,加入新鲜细胞培养液和转染液后,培养的时间为2-4h;优选地,为4小时。
步骤(3)中,加入新鲜细胞培养液继续培养时间为2-4h;优选地,为2小时。
步骤(3)中,所述细胞、新鲜培养液、转染液的用量比为105个:3ml:88.75μl。
步骤(3)中,所述转染液的配方是,88.75μl不含抗生素和谷氨酰胺(glutamine)的dmem溶液,22.5μllipotap及3.75μgdna;所述dna序列是gcccccccc;即,所述转染液包括:88.75μl不含抗生素和glutamine的dmem溶液,22.5μllipotap及3.75μgdnagcccccccc。
步骤(3)中,所述加入转染液的手法是,在一洁净无菌离心管内依次加入上述成份后用枪轻轻吹打混匀,25℃孵育10分钟后再把上述混合物均匀加入到培养盒内,随后轻轻混匀。
(4)诱导细胞原位合成dna银纳米簇:
倒去培养液,加入agno3共孵育;然后用新鲜dmem替换,陈化后置于荧光显微镜下观察;另取部分细胞裂解收集dna银纳米簇,进行形貌观察和成分分析。
步骤(4)中,加入agno3前还包括用hepes缓冲液冲洗细胞的步骤。
步骤(4)中,所述agno3的浓度为0.005-0.1mm;优选地,为0.01m。
步骤(4)中,所述agno3的用量为105个细胞中加入100μl的0.01magno3。
步骤(4)中,所述共孵育的温度为30-37℃;优选地,为37℃。
步骤(4)中,所述共孵育的时间为5min-2h;优选地,为20min。
步骤(4)中,所述陈化时间为12h-48h;优选地,为24h。
步骤(4)中,裂解细胞的方法为,利用液氮和37℃水浴对细胞进行两次快速的冻融,破碎细胞。
步骤(4)中,所述收集dna银纳米簇的方法是,离心除去破碎的细胞膜,收集悬浊液,即是dna银纳米簇;优选地,在1500rpm/min下离心l0min除去破碎的细胞膜。
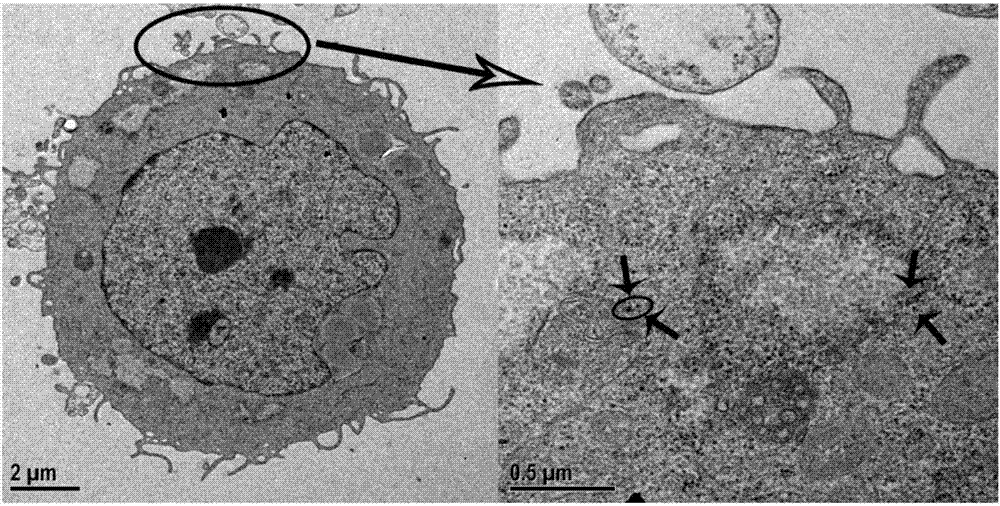
本发明还提供了由上述方法制备得到的dna银纳米簇,所述dna银纳米簇在经上述方法处理的msc细胞的树脂切片中富集在食物泡周围,为黑色簇状物;将细胞裂解,提取其中的dna银纳米簇,可以得知,所述dna银纳米簇为均匀分散的纳米簇,银纳米簇高度约为2.7nm,粒径约为3.1nm,晶面间距为0.24nm;在360nm和470nm附近有明显紫外吸收峰;并且使用488nm光源激发时有较强的610nm的荧光发射;x光电子能谱表征在366.4ev处有明显峰,对应于零价银的3d5/2。如图2~4是本发明一具体实施方式制备的dna银纳米簇。
本发明还提供了将所述dna银纳米簇用于端粒酶的长期监控的应用,尤其是用于干细胞增殖和分化过程中端粒酶的长期监控。
本发明还提供了一种利用上述dna银纳米簇原位监测人脐带间充质干细胞在增殖过程中的端粒酶活性变化的方法,所述方法包括以下步骤:
经上述方法诱导细胞原位合成dna银纳米簇,利用荧光共聚焦显微镜观察相应的样品,采用488nm波长进行激发,进行共荧光成像观察。并利用软件计算每个细胞内的平均荧光强度,对比得到每代间充质干细胞中的端粒酶相对活性。
其中,所述细胞为人脐带间充质干细胞;优选地,为第一代的人脐带间充质干细胞。
其中,所述荧光共聚焦显微镜的观察样品时,采用488nm波长进行激发,63x的物镜倍数进行共荧光成像观察。
其中,所述计算端粒酶相对活性的方法,指的是用每代细胞成像随机得到的50个细胞区域内荧光强度平均值,以第三代为基准取相对值即为每代间充质干细胞中的端粒酶相对活性。
本发明还提供了一种利用上述dna银纳米簇原位监测人脐带间充质干细胞诱导分化成神经干细胞过程的端粒酶活性变化的方法,所述方法包括以下步骤:
(a)将间充质干细胞以一定浓度接种于培养瓶,一段时间后更换成诱导液,在倒置显微镜下定期观察细胞的生长情况及其形态变化,并拍照纪录;诱导结束后,进行神经干细胞标记蛋白巢蛋白免疫荧光鉴定,计算其分化前后端粒酶活性的变化;
(b)将成功分化的神经干细胞继续传代,重复步骤(1)同样方法监控其生长情况和端粒酶活性变化。
步骤(a)中,所述接种的浓度为5×109l-1。
步骤(a)中,接种后,培养2-12小时,加入诱导液;优选地,培养8h,加入诱导液。
步骤(a)中,所述诱导液的配方为:含质量百分比2%的b27和60g/l的葡萄糖的dmem/f12培养基,另加20μg/l的表皮生长因子(egf)和20μg/l的碱性成纤维生长因子(fgf)。
步骤(a)中,加入诱导液的用量和细胞用量之间的关系为3ml/5×109个细胞。
步骤(a)中,加入诱导液诱导的时间为14-21天;优选地,为18天。
步骤(a)中,采用常规方法对神经干细胞标记蛋白巢蛋白,并进行免疫荧光鉴定。
在本发明的一个具体实施方式中,所述细胞原位合成的方法来制备dna银纳米簇的制备方法和用其长期监控干细胞内端粒酶的活性的方法,包括以下步骤:
(1’)细胞内源dna银纳米簇的原位合成
(a’)上调细胞内gsh水平:将细胞培养在25ml细胞培养瓶中,待细胞覆盖率达80%左右时加入琉辛酸(α-lipoicacid)在37摄氏度,5%co2的条件下共孵育24h后,缓慢倾去培养液并用新鲜培养液替换。
其中所述的加入琉辛酸(α-lipoicacid’)的体积是10μl,浓度是5μm。
(b’)检测细胞内gsh含量:用pbs洗涤细胞一次,离心收集细胞,吸尽上清。加入细胞沉淀体积3倍量的蛋白去除试剂,然后利用液氮和37℃水浴对样品进行两次快速的冻融破碎细胞。4℃或冰浴放置5分钟后,4℃,10000g离心10分钟。取上清用试剂盒测定谷胱甘肽的含量。
其中,所述的试剂盒是dtnb试剂盒。
(c’)转染dna:取上述已上调gsh含量的细胞,确保细胞盒中准确加入3毫升新鲜培养液,按照lipotap转染试剂盒加入转染夜。细胞培养箱内培养4小时后,去除含有lipotap-dna的培养液。加入3ml新鲜细胞培养液继续培养2h。
其中,所述的加入转染液的配方是,适量不含抗生素和glutamine的dmem溶液,22.5μllipotap及3.75μgdna,最终体积为115μl;所述的转染液的dna的序列是gcccccccc;所述的加入转染液的方法是,在一洁净无菌离心管内依次加入上述成份后用枪轻轻吹打混匀,25℃孵育10分钟后再把上述混合物均匀加入到培养盒内,随后轻轻混匀。
(d’)诱导细胞原位合成dna银纳米簇:缓慢倾倒掉培养液,用hepes缓冲液轻轻冲洗细胞两次,加入100μl的0.01magno3共孵育20min。之后用新鲜dmem替换溶液,陈化24h后置于荧光显微镜下观察。另取部分细胞裂解收集银纳米簇,进行形貌观察和成分分析。
其中,所述的裂解细胞的方法是利用液氮和37℃水浴对样品进行两次快速的冻融破碎细胞;所述的收集dna银纳米簇的方法是,1500rpm/min离心l0min除去破碎的细胞膜,收集悬浊液即是dna银纳米簇。
(2’)原位监测人脐带间充质干细胞在增殖过程中的端粒酶活性变化
端粒酶传感器的构建:将第一代的人脐带间充质干细胞经步骤(1)处理后,置于细胞培养箱中培养,每到传代时,取部分铺板。利用荧光共聚焦显微镜观察相应的样品,采用488nm波长进行激发,63x的物镜倍数进行共荧光成像观察。并利用软件计算每个细胞内的平均荧光强度,对比得到每代间充质干细胞中的端粒酶相对活性。
其中所述的计算端粒酶相对活性的方法,指的是用每代细胞成像随机得到的50个细胞区域内荧光强度平均值,以第三代为1取相对值即为每代间充质干细胞中的端粒酶相对活性。
另取每代间充质干细胞裂解并提取端粒酶使用elisa试剂盒检测每代细胞的端粒酶活性。
(3’)诱导人脐带间充质干细胞分化成神经干细胞并原位监控该过程的端粒酶活性变化
借助传感器监控端粒酶在干细胞分化过程中的表达:将每代间充质干细胞以一定浓度接种于25ml培养瓶,一段时间后更换成诱导液,在倒置显微镜下定期观察细胞的生长情况及其形态变化,并拍照纪录。诱导结束后,进行神经干细胞标记蛋白巢蛋白免疫荧光鉴定。取每代间充质干细胞诱导分化得到的神经干细胞重复步骤(3)计算其分化前后端粒酶活性的变化,另将成功分化的神经干细胞分别继续传代,重复同样方法监控其生长情况和端粒酶活性变化。
另取每代间充质干细胞分化后的神经干细胞裂解并提取端粒酶使用elisa试剂盒检测每代细胞的端粒酶活性
本发明的有益效果在于,本发明首先采用诱导细胞原位合成的方法来制备dna银纳米簇,因生物方法原位合成的dna银纳米簇不受限于细胞免疫排斥的影响,在生物体内的滞留时间长,可用于细胞内端粒酶的长期监控。原位合成dna银纳米簇的方法中采用药物诱导细胞的还原型谷胱甘肽(gsh)水平上调,然后输入大量的dna和银离子,诱导细胞自发地还原组装成银纳米颗粒,形成dna银纳米簇。由于本发明原位合成的dna银纳米簇的方法中使用的dna模版是富c序列,方便银纳米簇靠近端粒的重复序列(ttaggg)n,又因为银纳米簇靠近富g碱基序列时会有荧光增强的效应,因而可以用来监控端粒酶的活性。本发明使用该生物方法原位合成的dna银纳米簇可用于长期监控人脐带间充质干细胞增殖分化过程中端粒酶活性变化,不受限于细胞免疫排斥的影响,极高的生物相容性大大提高了探针在生物体内的滞留时间,使得探针可以长期留在细胞内实时动态成像干细胞在增殖分化过程中的状态。因该探针可以长期留在细胞内,实现对细胞在生长分化过程中的端粒酶活性进行实时动态成像,为活体荧光实时动态成像及细胞疾病的早期诊断和治疗提供了新的思路和方法。本发明方法还可以通过更换不同序列的寡核苷酸修饰银纳米簇作为多功能荧光探针,用来高灵敏检测细胞内其他功能分子如酶、葡萄糖、蛋白质、dna等,可广泛应用于生物标记及疾病监测等领域。
附图说明
图1是实施例1中上调msc细胞内的gsh后,用试剂盒检测细胞内gsh含量的结果。
图2是实施例1生物方法原位合成dna银纳米簇的msc细胞的树脂切片电镜照片。
图3是实施例1生物方法原位合成的dna银纳米簇的紫外-可见吸收光谱和荧光发射光谱。
图4是实施例1生物方法原位合成的dna银纳米簇的形貌表征图;其中,a为原子力图,b图中对应于a图中线标处银纳米簇高度,c为透射电镜图,d为高分辨射电镜图。
图5是实施例1生物方法原位合成的dna银纳米簇的x光电子能谱表征。
图6是实施例1生物方法原位合成的dna银纳米簇的用于msc细胞端粒酶活性成像的结果;其中,a为荧光成像图,b为软件计算得到的平均荧光强度,c为计算得到的相对端粒酶活性。
图7是实施例1中诱导第三代msc细胞分化成nsc细胞的结果;其中,a为细胞形貌变化,b为免疫荧光鉴定nsc细胞的结果。
图8是实施例1生物方法原位合成的dna银纳米簇的用于nsc细胞端粒酶活性成像的结果;其中,a为荧光成像图,b为软件计算得到的平均荧光强度,c为计算得到的相对端粒酶活性。
图9是实施例1用elisa试剂盒检测msc和nsc细胞端粒酶活性的结果。
具体实施方式
结合以下具体实施例和附图,对本发明作进一步的详细说明。实施本发明的过程、条件、实验方法等,除以下专门提及的内容之外,均为本领域的普遍知识和公知常识,本发明没有特别限制。
实施例1细胞内源dna银纳米簇的原位合成
(1)实施例上调细胞内gsh水平:
将细胞培养在25ml细胞培养瓶中,待细胞覆盖率达80%左右时加入10μl的5μm琉辛酸(α-lipoicacid)在37摄氏度,5%co2的条件下共孵育24h后,缓慢倾去培养液并用新鲜培养液替换。
用pbs将上述细胞洗涤一次,离心收集细胞,吸尽上清。加入细胞沉淀体积3倍量的蛋白去除试剂,然后利用液氮和37℃水浴对样品进行两次快速的冻融破碎细胞。4℃或冰浴放置5分钟后,4℃,10000g离心10分钟。取上清用dtnb试剂盒测定谷胱甘肽的含量。
(2)转染dna:
取上述已上调gsh含量的细胞,确保细胞盒中准确加入3毫升新鲜培养液,按照lipotap转染试剂盒加入转染夜。细胞培养箱内培养4小时后,去除含有lipotap-dna的培养液。加入3ml新鲜细胞培养液继续培养2h。加入转染液的配方是,适量不含抗生素和glutamine的dmem溶液,22.5μllipotap及3.75μgdna,最终体积为115μl;转染液的dna的序列是gcccccccc;加入转染液的手法是,在一洁净无菌离心管内依次加入上述成份后用枪轻轻吹打混匀,25℃孵育10分钟后再把上述混合物均匀加入到培养盒内,随后轻轻混匀。
(3)诱导细胞原位合成dna银纳米簇:
缓慢倾倒掉培养液,用hepes缓冲液轻轻冲洗细胞两次,加入100μl的0.01magno3共孵育20min。之后用新鲜dmem替换溶液,陈化24h后置于荧光显微镜下观察。另取部分细胞裂解收集银纳米簇,进行形貌观察和成分分析。裂解细胞的方法是利用液氮和37℃水浴对样品进行两次快速的冻融破碎细胞;所述的收集dna银纳米簇的方法是,1500rpm/min离心l0min除去破碎的细胞膜,收集悬浊液即是dna银纳米簇。
图1可以看到药物诱导后的间充质干细胞内的gsh含量大大提高。图2为原位合成dna银纳米簇的msc细胞树脂切片电镜图,可清晰的看到细胞内食物泡周围聚集有大量黑色簇状物,即为所述dna银纳米簇。将细胞裂解,提取其中的dna银纳米簇进行表征。图3为dna银纳米簇的光谱表征,银纳米簇在360nm和470nm附近有明显紫外吸收峰,并且使用488nm光源激发时有较强的610nm的荧光发射(图3)。图4为银纳米簇的形貌表征图,从结果可以看出成功合成均匀分散的纳米簇。原子力图可以看出银纳米簇高度约为2.7nm(图4a)。从透射电镜图可看出,粒径约为3.1nm,晶面间距为0.274nm(图4c&d)。进一步通过x光电子能谱表征可以看出,银纳米簇在366.4ev处有明显峰,对应于零价银的3d5/2,证明合成的银纳米簇是0价的(图5)。本发明成功使用生物方法诱导细胞原位合成了dna银纳米簇。
实施例2实施例1原位合成的dna银纳米簇探针用于长期监测干细胞增殖
分化过程中的端粒酶活性变化。
(1)原位监测人脐带间充质干细胞在增殖过程中的端粒酶活性变化:
将经实施例1处理后的第一代的人脐带间充质干细胞,置于细胞培养箱中培养。每到传代时,取部分铺板。利用荧光共聚焦显微镜观察相应的样品,采用488nm波长进行激发,63x的物镜倍数进行共荧光成像观察。并利用软件计算每个细胞内的平均荧光强度,对比得到每代间充质干细胞中的端粒酶相对活性。
计算端粒酶相对活性的方法,是用每代细胞成像随机得到的50个细胞区域内荧光强度平均值,以第三代为1取相对值即为每代间充质干细胞中的端粒酶相对活性。
(2)诱导人脐带间充质干细胞分化成神经干细胞并原位监控该过程的端粒酶活性变化
将每代间充质干细胞以一定浓度接种于25ml培养瓶,一段时间后更换成诱导液,在倒置显微镜下定期观察细胞的生长情况及其形态变化,并拍照纪录。诱导结束后,进行神经干细胞标记蛋白巢蛋白免疫荧光鉴定。取每代间充质干细胞诱导分化得到的神经干细胞重复步骤(3)计算其分化前后端粒酶活性的变化,另将成功分化的神经干细胞分别继续传代,重复同样方法监控其生长情况和端粒酶活性变化。
由图6可知,随着加入端粒酶抑制剂,银纳米簇的荧光强度大幅减弱,证明本发明中的原位合成的dna银纳米簇探针的荧光强度能灵敏响应端粒酶活性变化。银纳米簇的荧光强度在间充质干细胞传至第三代时最亮,随后迅速衰弱。这说明第三代间充质干细胞的端粒酶活性最高。而后面的诱导分化实验中也发现第三代间充质干细胞的分化潜能明显高于其他几代细胞,也从另一方面证明了第三代间充质干细胞的端粒酶活性较好。虽然干细胞的前五代都能成功诱导出神经干细胞,然而仅第三代间充质干细胞诱导出来的神经干细胞(图7)密度最大,在以后传代过程中的端粒酶表达也明显高于其他几代间充质干细胞分化出来的神经干细胞(图8),另外神经干细胞在分化出来后,其端粒酶活性明显低于间充质干细胞,并在传代过程中迅速衰退。
实施例3体外验证实验
为了评估本发明实施例1原位合成的dna银纳米簇探针,用于长期监测干细胞增殖分化过程中的端粒酶活性变化的准确性,分别取每代间充质干细胞裂解并提取端粒酶使用elisa试剂盒检测每代细胞的端粒酶活性。另取每代间充质干细胞分化后的神经干细胞裂解并提取端粒酶使用elisa试剂盒检测每代细胞的端粒酶活性。
图9a是实施例3验证实验所用到的一系列细胞。图9b是每代间充质干细胞裂解提取端粒酶,使用elisa试剂盒检测的结果。从图中可看到每代间充质干细胞的端粒酶活性变化趋势与实施例2使用dna银纳米簇探针监测的结果大致吻合。图9c是每代间充质干细胞分化出来的神经干细胞裂解所提取端粒酶,使用elisa试剂盒检测的结果。从图中可看到每代神经干细胞的端粒酶活性变化趋势与实施例2使用dna银纳米簇探针监测的结果大致吻合。证明本发明中的原位合成的dna银纳米簇探针可用于长期监测干细胞增殖分化过程中的端粒酶活性变化。
本发明的保护内容不局限于以上实施例。在不背离发明构思的精神和范围下,本领域技术人员能够想到的变化和优点都被包括在本发明中,并且以所附的权利要求书为保护范围。
<110>华东师范大学
<120>一种dna银纳米簇及其细胞原位的合成方法和应用
<160>1
<170>patentinversion3.3
<210>1
<211>9
<212>dna
<213>人工序列
<400>1
gcccccccc
- 还没有人留言评论。精彩留言会获得点赞!