一种检测流产组织DNA拷贝数变异和嵌合体的方法与流程
本发明涉及离体组织的基因信息分析领域,具体地,涉及一种检测流产组织dna拷贝数变异和嵌合体的方法。
背景技术:
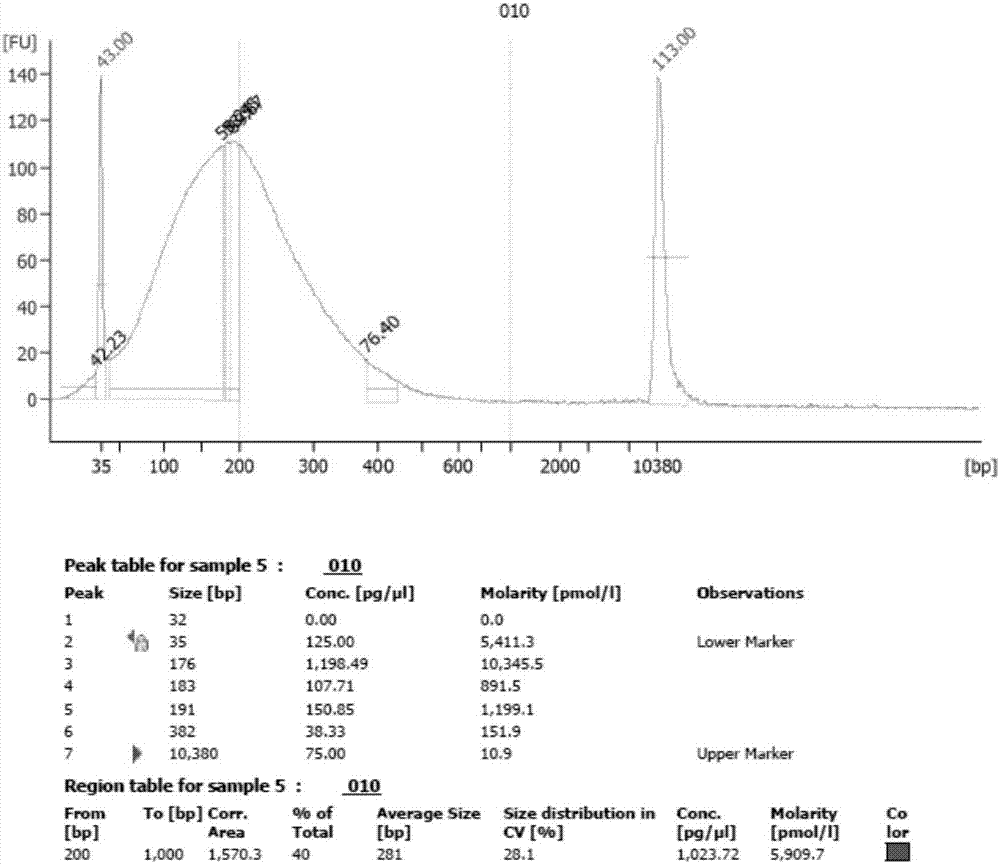
:流行病学证据显示基因的因素在流产发生中起重要作用。染色体异常例如三体,单体,多倍体是用传统方法检测出的常见致流产原因,为50~70%小于10周妊娠的流产做出了解释。由于传统方法的分辨率有限,-30~40%的流产胚胎的具有正常的检测结果即二倍体,没有更多发现用于解释流产的原因。染色体嵌合体在不同的样本中发生率不同。在羊水样本中约0.20%~0.25%,在绒毛组织样本中约0.8%~0.2%。由于染色体基因芯片用于检测嵌合体只能检出25~70%的嵌合,以及嵌合体的检测收样本采集的影响很大。所以在分析流产的原因时,嵌合体的发生率是被低估了。尤其是低比例的嵌合体。分子核型分析的方法,如多重荧光原位杂交(mfish),多重连接依赖探针扩增(mlpa)和实时定量酶连反应(qpcr),克服了传统核型分析的劣势,提供了比传统核型分析更好的分辨率。但是这些分子核型方法的缺点是通量低、分辨率有限,对于衡量全部染色体的变异情况能力有限。在目前现有检测方法中,g带染色体核型分析技术是检测染色体异常的“金标准”。g带核型分析是一种传统的染色体分析方法,在自发性流产原因的探索中占重要地位。但是细胞培养失败,母源细胞污染,染色体制备失败等因素在很大程度上限制了g带核型分析的应用。微阵列比较基因组杂交技术是一种新型高效的分子核型分析技术,其优势在于省去了细胞培养,一定程度上避免了母体细胞污染。可以直接在dna水平上进行分析并将分析结果比对数据库做出诊断。很大程度上又要依赖于细胞样品的质量。如果样本细胞量小或被污染,则很难或不能检测出结果。利用高通量测序技术对流产组织进行检测的方法相比传统方法具有明显优势。该方法最低起始量只需3ngdna样本,相比于微阵列比较基因组杂交技术对样本量要求少,对样本质量要求不如微阵列方法严格。检测的准确性、灵敏度及可靠性都大大提高。通过低深度测序,能够对流产组织dna的拷贝数变化进行准确检测,可以克服传统检测方法耗时长,需要细胞培养,分辨率低等缺点。目前,市场上基于高通量基因测序法检测流产组织的诊断产品都是基于illumina测序平台,没有针对iontorrent平台开发的配套的生物信息学算法。本发明旨在提供一套用于iontorrent测序平台检测流产组织中染色体dna拷贝数变异和嵌合体的算法。技术实现要素:本发明为了克服现有技术的上述不足,提供一种基于iontorrent测序平台检测流产组织dna拷贝数变异和嵌合体的方法。为了实现上述目的,本发明是通过以下技术方案予以实现的:一种基于iontorrent测序平台检测流产组织dna拷贝数变异的方法,包括数据质控、数据校正、确定拷贝数变异、结果展示、确定参考范围;所述确定拷贝数变异包括:(1)校正后的数据根据circularbinarysegmentation算法确定拷贝数变异数值,(2)根据隐马尔科夫模型hiddenmarkovmodel算法确定拷贝数变异数值,(3)根据z-score对区间内拷贝数变异显著性进行进一步统计;所述确定参考范围为:r值的参考范围在[-0.2,0.2]之间,z值的参考范围是[-3,3],对于r值大于0.2或小于-0.2,z值大于或小于3的区间,则提示该染色体区域存在拷贝数重复或缺失的情况。隐马尔科夫模型是生物信息学中比较流行的机器学习和模式识别方法,具有对模型中一些隐性参数识别优化的能力,可以随着训练深入提高识别精度,能够自适应的实现检测过程中的参数优化,进一步提高检测灵敏度。所述数据质控包括:(1)去除低质量的reads:包括大于q15的碱基比例不小于80%、reads长度不小于50bp;(2)去重:同一条reads被多次复制的当作一条reads;(3)比对reads唯一性:对于多次比对到基因组不同部位的reads,将其从结果中去除。所述数据校正包括:(1)基因组的端粒、着丝粒、卫星、微卫星区域进行掩盖;(2)将基因组切割成50kb一个的窗口,统计落在每一个窗口中的reads个数;(3)窗口内如果参考基因组的n碱基的比例大于10%,直接去除该窗口,否者对reads进行中位数校正;(4)对窗口内的reads根据每个窗口的gc百分数进行loess校正;(5)对窗口内的reads根据每个窗口的实际比对率进行lowess校正;(6)根据女性或者男性样本的基线数据分别根据性别进行校正。一种基于iontorrent测序平台检测流产组织嵌合体的存在和比例的方法,包括数据质控、数据校正、采用人工模拟染色体嵌合体配比样本log2ratio值和嵌合体比例之间的关系,具体如下:设立了7个梯度的模拟嵌合体比例,分别为12.5%,35%,47.5%,50%,62.5%,75%,87.5%。经过测序数据分析得出不同比例的嵌合体log2ratio值。然后将log2ratio值转化为标准化读长频率。做标准曲线。根据标准曲线从标准化读长频率可以推测出样本是否存在嵌合体以及嵌合体比例。与现有技术相比,本发明具有如下有益效果:本发明提供的基于iontorrent测序平台检测流产组织dna拷贝数变异和嵌合体的方法,可以用于流产原因分析,进一步具体地阐述基因因素在流产发生中的重要作用。附图说明图1为样本基因组dna打断后2100质控图。图2为样本基因组dna片段选择后质控图。图3为染色体具体区间拷贝数变异图。图4为染色体拷贝数变异的核型图。图5为采用人工模拟染色体嵌合体配比样本log2ratio值和嵌合体比例之间的关系做的标准曲线;上图为图a,下图为图b,其中a为按人工配的不同比例嵌合做拷贝数扩增的标准曲线,b为按人工配的不同比例嵌合做拷贝数缺失的标准曲线。具体实施方式下面结合说明书附图和具体实施例对本发明作出进一步地详细阐述,所述实施例只用于解释本发明,并非用于限定本发明的范围。下述实施例中所使用的试验方法如无特殊说明,均为常规方法;所使用的材料、试剂等,如无特殊说明,为可从商业途径得到的试剂和材料。实施例1一种基于iontorrent测序平台检测流产组织dna拷贝数变异和嵌合体的方法,具体步骤如下:1、全血基因组dna提取:按照dneasyblood&tissuekit(50)(厂家:qiagen,货号:69504)试剂盒操作说明书进行全血基因组dna提取。dna溶液可置于-20℃保存。2、dna打断:将从健康未孕女性混合血细胞中提取的基因组dna用covarism220超声波打断,设置10个重复,用qubit2.0和dsdnahsassaykit测定dna的浓度,agilent2100bioanalyzer测定dna片段长度分布,见图1。3、回收100~200bp长度的dna片段:往以上打断的基因组dna中加入相应比例的磁珠(ampurexpbeads),去除大片段和小片段,用超纯水洗脱,回收目的dna片段。用qubit2.0和dsdnahsassaykit测定dna的浓度,agilent2100bioanalyzer测定dna片段长度分布,见图2。4、文库构建(1)将末端补平试剂置于冰上溶解,根据实验样本数计算各反应体系的具体用量,并配置反应混合液。涡旋混匀,离心机低速离心数秒,使管壁和盖子上无明显液滴。每个样本所需的末端反应体系如下:试剂体积(μl)h2o9.55xendrepairbuffer10endrepairenzyme0.5总体积20磁珠纯化末端修复产物dna片段末端加接头2)将接头连接试剂置于冰上溶解。3)将p1母液及1~32号接头母液分别与无核酸酶水以1:5的比例稀释备用。4)按根据操作说明。根据实验样本数计算各反应体系的具体用量,并配置反应混合液。每个样本所需的末端反应体系如下:金属浴中25℃反应20min。5)磁珠纯化连接产物6)pcr扩增dna片段根据实验样本数计算各反应体系的具体用量,并配置反应混合液。每个样本所需的末端反应体系如下:试剂1x(μl)platinumpcrsupermixhighfidelity47.5libraryamplificationprimermix2.5总体积507)磁珠纯化pcr产物8)文库质检采用qubit2.0和2100bioanalyzer分别进行dna初步浓度和片段大小检测。5、按照ionpgmtmhiqtmot2reagents200(厂家:lifetechnologies,货号:a26428)和ionpgmtmhiqtmot2solutions200(厂家:lifetechnologies,货号:a26429)的试剂盒操作说明书进行模板制备和模板富集。6、按照ionpgmitmhiqtmsequencing200reagents(厂家:lifetechnologies,货号:a26431)、ionpgmtmhiqtmsequencing200solutions(厂家:lifetechnologies,货号:a26430)和ionpgmtmsequencingnucleotides(厂家:lifetechnologies,货号:a26432)的试剂盒操作说明书进行上机测序。二、iontorrent平台开发的配套的生物信息学算法如下:1、数据质控(1)去除低质量的reads:包括大于q15的碱基比例不小于80%、reads长度不小于50bp;(2)去重:由于建库过程中扩增步骤而导致的同一条reads被多次复制的当作一条reads;(3)比对reads唯一性:对于多次比对到基因组不同部位的reads,将其从结果中去除;2、数据校正(1)基因组的端粒、着丝粒、卫星、微卫星区域进行掩盖;(2)将基因组切割成50kb一个的窗口,统计落在每一个窗口中的reads个数;(3)窗口内如果参考基因组的n碱基的比例大于10%,直接去除该窗口,否者对reads进行中位数校正;(4)对窗口内的reads根据每个窗口的gc百分数进行loess校正;(5)对窗口内的reads根据每个窗口的实际比对率进行lowess校正;(6)根据女性或者男性样本的基线数据分别根据性别进行校正;3、确定拷贝数变异(1)校正后的数据根据circularbinarysegmentation(cbs)算法确定拷贝数变异数值;(2)同样根据hiddenmarkovmodel(hmm)算法确定拷贝数变异数值;(3)根据z-score对区间内拷贝数变异显著性进行进一步统计,并综合以上的结果给出明确的判定;4、结果展示(1)所有染色体拷贝数变异的核型图;(2)每条染色体具体区间拷贝数发生变异的详细图;(3)变异结果的图标展示;(4)变异结果的遗传学解读;5、参考范围的确定:通过对88例明确拷贝数位点区域的测序结果评估,计算假阳性、假阴性并给出特异性和敏感性的roc曲线后可以确定r值的参考范围在[-0.2,0.2]之间,高于或者低于以上范围则是拷贝数变异的位点。同时结合z值对拷贝数进一步确定,z值的参考范围是[-3,3]。对于r值大于0.2(或小于-0.2),z值大于(或小于)3的区间,则提示该染色体区域存在拷贝数重复(或缺失)的情况,需要进一步的临床诊断确认,进一步确诊方法主要是对该区域进行核型分析或fish等其他方法进行验证。6、判断嵌合体的存在和比例:上机后的数据按照前面步骤1、2的数据质控和数据校正后,采用人工模拟染色体嵌合体配比样本log2ratio值和嵌合体比例之间的关系,具体如下:设立了7个梯度的模拟嵌合体比例,分别为12.5%,35%,47.5%,50%,62.5%,75%,87.5%。经过测序数据分析得出不同比例的嵌合体log2ratio值。然后将log2ratio值转化为标准化读长频率。做标准曲线(见图5)。根据标准曲线从标准化读长频率可以推测出样本是否存在嵌合体以及嵌合体比例。通过对88例样本中的116个染色体缺失或重复片段检测和9例核型分析验证过的嵌合体样本中嵌合比例的测序结果评估,结果显示大于1mb的染色体缺失重复可以100%检测,并且和微阵列芯片验证结果一致。9例嵌合体样本嵌合体全部检出。表1为88例样本中116个拷贝数变异大小和检出情况以及与微阵列芯片验证的符合率表2为ngs检测嵌合体与核型分析比较a指ngs检测嵌合体与核型分析检测嵌合体两者结果的变异系数。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1