一种宏基因组数据分类方法和装置与流程
本发明属于基因数据处理领域,尤其涉及一种宏基因组数据分类方法和装置。
背景技术:
基于DNA的宏基因组学理论上覆盖了环境样品中的全部微生物,因此可以更加全面真实地反映微生物群落组成,同时大大拓展了筛选新的基因或生物活性物质的来源。根据所用策略不同,宏基因组学研究可分为序列驱动的(sequence-driven)和功能驱动的(function-driven),其中,序列驱动是指通过测序分析微生物群落的结构和功能,功能驱动是指基于构建宏基因组文库筛选新基因或新物质的宏基因组学研究。
宏基因组研究的目标是研究微生物群里的结构组成,例如,对海洋样本的测序科研揭示起环境的多样性,同样,对人类样本的研究可以人类微生物和人类健康之间的关系。一旦一个宏基因组的样本被测序,第一项任务就是要找到存在其中的各种微生物物种。基于比对和序列组成,将宏基因组的读长(read)归类到已有的生物物种,现在有许多工具可以用。
基于序列结构组成的宏基因组分类方法,是利用序列本身的构成特征进行分类的方法。普遍的过程是用统计学的方法对样本数据进行抽样,利用筛选出来的特征表达,将序列数据抽象为生物意义上的特征向量,然后将这些特征向量组成特征矩阵,选择合适的分类器模型,对生物序列进行分类分析。Karlin研究了多种微生物的基因组序列,发现同一物种的基因序列的碱基构成具有相似性(例如GC的含量),而不同物种的碱基使用偏向性差异很大。基于这一理论基础,Teelin等人开发了TERTRA工具,Chan等人开发了基于自组织生长算法的工具。在特征的使用方面,微生物的物种丰度、基因功能、代谢通路、系统发育关系等可作为该群落或样本的特征用来进行样本分类。David等人使用微生物的全基因组序列的表型特点;G、C含量、基因组大小、微生物能量来源、生存湿度W及耗氧量等作为样本特征,利用R-SVM分类器对宏基因组序列进行了分类。
常用的分类器有朴素贝叶斯分类模型、期望最大化模型、最大似然估计模型、马尔可夫模型等。目前,一种宏基因组的分类器是监督分类,起使用结构组成的相关的序列特征,应用在已知类别标签的序列中,提取特征信息,输入分类器,训练分类模型,最后对未知标签的序列进行分类。CARMA就是一种基于监督的宏基因组分类工具,它根据隐马尔科夫模型,对长度80bps(Base pairs)的较短序列的分类效果很好。TACOA用了基于核函数的kNN算法能够对读长大于800bps的序列进行预测,该软件可以保持参考基因组数据库的实时更新,并且可以使用IMMs(Interpolated Markov Models)来建模,对长度大于100bps的序列的分类准确度很高。NBC将朴素贝叶斯分类算法应用到宏基因组分类上,而且实现了网络在线服务,使得宏基因组分类的结果可以得到方便快捷的在网页上展示。张学工等人提出了一种不需要参考序列,使用R-SVM算法的基于监督的宏基因组分类算法,利用特征选择算法筛选出序列结构信息中的有用特征来提高分类准确率。
然而,上述现有的监督分类算法,由于特征提取方法和分类器模型性能的缘故,在针对低分类层次、多物种分类的大规模宏基因组数据分类问题时分类精度比较低,且运行时间开销太大。
技术实现要素:
本发明的目的在于提供一种宏基因组数据分类方法和装置,以较小的时间开销提高基因组的分类精度。
本发明第一方面提供一种宏基因组数据分类方法,所述方法包括:
计算待测序序列的特征向量;
对所述特征向量进行聚类得到M组包含读长的簇G1至GM,所述M为不小于1的整数;
获取所述簇G1至GM中每个簇的中心集合Ki;
通过将所述每个簇的中心集合Ki的每一读长与参考基因序列对比,判断所述每个簇的基因组类别。
本发明第二方面提供一种宏基因组数据分类装置,所述装置包括:
计算模块,用于计算待测序序列的特征向量;
聚类模块,用于对所述特征向量进行聚类得到M组包含读长的簇G1至GM,所述M为不小于1的整数;
获取模块,用于获取所述簇G1至GM中每个簇的中心集合Ki;
类别判断模块,用于通过将所述每个簇的中心集合Ki的每一读长与参考基因序列对比,判断所述每个簇的基因组类别。
从上述本发明技术方案可知,通过对待测序序列的特征向量进行聚类得到若干组包含读长的簇,并由此获取所述簇的中心集合,由于只是将所述每个簇的中心集合的每一读长与参考基因序列对比,判断每个簇的基因组类别,因此,与现有技术相比,本发明提供的技术方案既降低了分类所用的时间开销即提高了运算速度,又显著提高了对测序序列所属基因组类别的分类精度。
附图说明
图1是本发明实施例一提供的宏基因组数据分类方法的实现流程示意图;
图2是本发明实施例二提供的宏基因组数据分类装置的结构示意图;
图3是本发明实施例三提供的宏基因组数据分类装置的结构示意图;
图4是本发明实施例四提供的宏基因组数据分类装置的结构示意图;
图5-a是本发明实施例五提供的宏基因组数据分类装置的结构示意图;
图5-b是本发明实施例六提供的宏基因组数据分类装置的结构示意图;
图5-c是本发明实施例七提供的宏基因组数据分类装置的结构示意图;
图6-a是本发明实施例八提供的宏基因组数据分类装置的结构示意图;
图6-b是本发明实施例九提供的宏基因组数据分类装置的结构示意图;
图6-c是本发明实施例十提供的宏基因组数据分类装置的结构示意图。
具体实施方式
为了使本发明的目的、技术方案及有益效果更加清楚明白,以下结合附图及实施例,对本发明进行进一步详细说明。应当理解,此处所描述的具体实施例仅仅用以解释本发明,并不用于限定本发明。
本发明实施例提供一种宏基因组数据分类方法,所述方法包括:计算待测序序列的特征向量;对所述特征向量进行聚类得到M组包含读长的簇G1至GM,所述M为不小于1的整数;获取所述簇G1至GM中每个簇的中心集合Ki;通过将所述每个簇的中心集合Ki的每一读长与参考基因序列对比,判断所述每个簇的基因组类别。本发明实施例还提供相应的宏基因组数据分类装置。以下分别进行详细说明。
请参阅附图1,是本发明实施例一提供的宏基因组数据分类方法的实现流程示意图,主要包括以下步骤S101至步骤S104,详细说明如下:
S101,计算待测序序列的特征向量。
作为本发明一个实施例,计算待测序序列的特征向量可通过如下步骤S1011和S1012实现:
S1011,将待测序序列分割成L-k+1个长度为k的k-mer,其中,L为待测序序列的长度。
在基因学领域,k-mer是指一个长度为k的子串,一般是从序列的某一位置开始的k个连续组成碱基。假设测序序列长度为L,在本发明实施例中,可以将待测序序列依次按长度为k=3、4、6截取片段,每个片段就是一个k-mer,如此,一个长度为L的待测序序列总共可分割为L-k+1个长度为k的k-mer。
S1012,统计经步骤S1011分割所得的L-k+1个k-mer中每个k-mer的出现频率,将L-k+1个k-mer中k-mer的出现频率组成维度为4k的向量作为待测序序列的特征向量。
具体地,针对被分割为L-k+1个长度为k的k-mer的待测序序列,统计这些k-mer中不同k-mer的出现频率,然后,对这些k-mer进行编码,分别将A(腺嘌呤)、T(鸟嘌呤)、C(胞嘧啶)、G(胸腺嘧啶)采用0、1、2、3这些数字表示,再进行四进制编码,将每个k-mer的数字表示作为向量的维度索引,该k-mer的出现频率作为向量值,从而组成一个维度为4k的向量,而该向量就是被分割为L-k+1个长度为k的k-mer的待测序序列的特征向量。
需要说明的是,为了降低后续处理时的计算量和/或复杂度,从而减小运行时的时间开销,在本发明实施例中,可以对待测序序列的特征向量进行降维处理,具体可以使用基于互信息选择对待测序序列的特征向量进行降维处理。
S102,对经步骤S101计算所得待测序序列的特征向量进行聚类得到M组包含读长的簇G1至GM,此处,M为不小于1的整数。
具体地,可以使用聚类工具箱vlfeat中的kmeans算法将经步骤S101计算所得待测序序列的特征向量进行聚类,从而得到M组包含读长的簇(即cluster),此处编号为G1、G2、…、Gi…、GM-1、GM。
S103,获取簇G1至GM中每个簇的中心集合Ki。
经步骤S102聚类所得的簇中,每个簇中有很多读长可能是有重叠的碱基的读长,在本发明实施例中,具体可以是将每个簇里的所有读长构成一个图,而每个读长是图的一个顶点,然后计算图的最大独立集,将这个最大独立集包含的那些读长构成每个簇的中心集合Ki。
S104,通过将每个簇的中心集合Ki的每一读长与参考基因序列对比,判断每个簇的基因组类别。
作为本发明一个实施例,通过将每个簇的中心集合Ki的每一读长与参考基因序列对比,判断每个簇的基因组类别可通过如下步骤S1041和S1042实现:
S1041,通过将每个簇的中心集合Ki的每一读长与参考基因序列对比,统计每个簇的中心集合Ki的每一读长的基因组类别。
具体可以将每个簇的中心集合Ki的每一读长与参考基因序列对比,使用工具BLAST,统计出每个簇的中心集合Ki的每一读长的基因组类别。需要说明的是,本发明的技术方案并不是将每个簇的所有读长与参考基因序列对比,而是只选择每个簇的中心集合Ki的每一读长与参考基因序列对比,如此,减小了每个簇的类别的搜索范围,减小了对比量,从而减小了时间上的开销。
S1042,若中心集合Ki中任一读长Ri的基因组类别Ci的出现频率不小于预设阈值,则将读长Ri的基因组类别Ci确认为读长Ri所属簇的基因组类别。
在将每个簇的中心集合Ki的每一读长与参考基因序列对比过程中,统计的结果可能是同一读长却属于不同的基因组类别,此时,可以以该读长的基因组类别的出现频率来确定其基因组类别。例如,假设预设阈值是70%,若对比和统计的结果显示读长Ri的基因组类别属于C’i的出现频率是30%,属于C”i的出现频率是43%,属于Ci的出现频率是75%,则将读长Ri的基因组类别确定为Ci,并且将读长Ri的基因组类别Ci确认为读长Ri所属中心集合Ki的基因组类别或所属簇的基因组类别。
为了将经步骤S104错分或误分的序列剔除,提高宏基因组数据分类整体的分类准确率,在本发明实施例中,可在步骤S104后,进一步采用多核学习训练分类器对所述已确认基因组类别的簇再次进行分类。具体可以是从已确认基因组类别的簇的中心集合Ki中随机选取一定比例,例如60%的读长作为训练集,用多核学习工具shogun训练分类模型,将余下比例,例如40%的读长作为测试集,采用多核学习训练分类器对其进行分类,滤除每个中心集合Ki中由于上一步聚类错误判别的读长。
从上述附图1示例的宏基因组数据分类方法可知,通过对待测序序列的特征向量进行聚类得到若干组包含读长的簇,并由此获取所述簇的中心集合,由于只是将所述每个簇的中心集合的每一读长与参考基因序列对比,判断每个簇的基因组类别,因此,与现有技术相比,本发明提供的技术方案既降低了分类所用的时间开销即提高了运算速度,又显著提高了对测序序列所属基因组类别的分类精度。
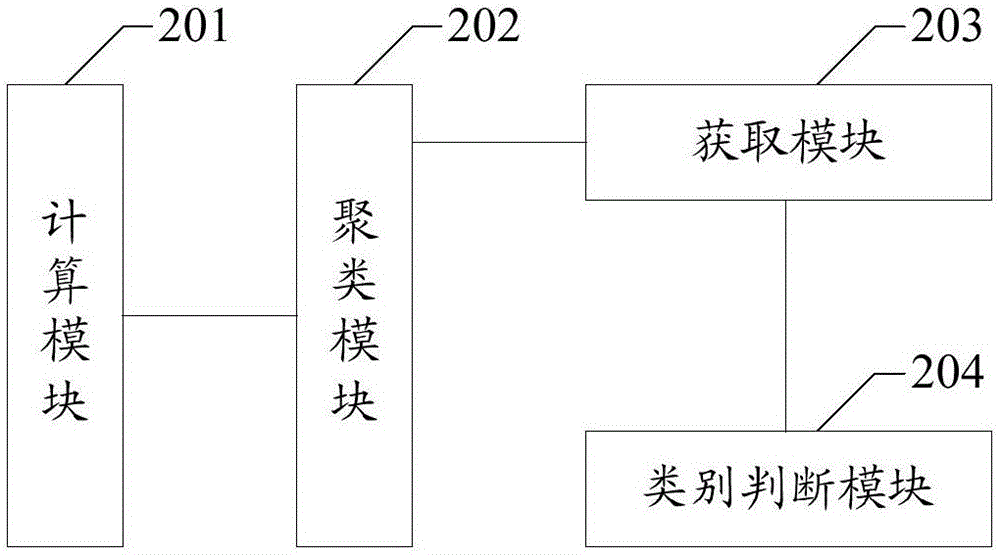
请参阅附图2,是本发明实施例二提供的宏基因组数据分类装置的结构示意图。为了便于说明,附图2仅示出了与本发明实施例相关的部分。附图2示例的宏基因组数据分类装置可以是附图1示例的宏基因组数据分类方法的执行主体。附图2示例的宏基因组数据分类装置主要包括计算模块201、聚类模块202、获取模块203和类别判断模块204,其中:
计算模块201,用于计算待测序序列的特征向量。
聚类模块202,用于对计算模块201计算所得待测序序列的特征向量进行聚类得到M组包含读长的簇G1至GM,其中,M为不小于1的整数。
具体地,聚类模块202可以使用聚类工具箱vlfeat中的kmeans算法将经计算模块201计算所得待测序序列的特征向量进行聚类,从而得到M组包含读长的簇(即cluster),此处编号为G1、G2、…、Gi…、GM-1、GM。
获取模块203,用于获取簇G1至GM中每个簇的中心集合Ki。
经聚类模块202聚类所得的簇中,每个簇中有很多读长可能是有重叠的碱基的读长,在本发明实施例中,获取模块203具体可以将每个簇里的所有读长构成一个图,而每个读长是图的一个顶点,然后计算图的最大独立集,将这个最大独立集包含的那些读长构成每个簇的中心集合Ki。
类别判断模块204,用于通过将每个簇的中心集合Ki的每一读长与参考基因序列对比,判断每个簇的基因组类别。
需要说明的是,以上附图2示例的宏基因组数据分类装置的实施方式中,各功能模块的划分仅是举例说明,实际应用中可以根据需要,例如相应硬件的配置要求或者软件的实现的便利考虑,而将上述功能分配由不同的功能模块完成,即将所述宏基因组数据分类装置的内部结构划分成不同的功能模块,以完成以上描述的全部或者部分功能。而且,实际应用中,本实施例中的相应的功能模块可以是由相应的硬件实现,也可以由相应的硬件执行相应的软件完成,例如,前述的聚类模块,可以是具有执行前述对计算模块(或计算器)计算所得待测序序列的特征向量进行聚类得到M组包含读长的簇G1至GM的硬件,例如聚类器,也可以是能够执行相应计算机程序从而完成前述功能的一般处理器或者其他硬件设备;再如前述的类别判断模块,可以是执行通过将每个簇的中心集合Ki的每一读长与参考基因序列对比,判断每个簇的基因组类别的硬件,例如类别判断器,也可以是能够执行相应计算机程序从而完成前述功能的一般处理器或者其他硬件设备(本说明书提供的各个实施例都可应用上述描述原则)。
附图2示例的计算模块201可以包括分割单元301和统计单元302,如附图3所示本发明实施例三提供的宏基因组数据分类装置,其中:
分割单元301,用于将待测序序列分割成L-k+1个长度为k的k-mer,其中,L为待测序序列的长度。
在基因学领域,k-mer是指一个长度为k的子串,一般是从序列的某一位置开始的k个连续组成碱基。假设测序序列长度为L,在本发明实施例中,分割单元301可以将待测序序列依次按长度为k=3、4、6截取片段,每个片段就是一个k-mer,如此,一个长度为L的待测序序列总共可分割为L-k+1个长度为k的k-mer。
统计单元302,用于统计L-k+1个k-mer中每个k-mer的出现频率,将L-k+1个k-mer中k-mer的出现频率组成维度为4k的向量确认为待测序序列的特征向量。
具体地,针对被分割为L-k+1个长度为k的k-mer的待测序序列,统计单元302统计这些k-mer中不同k-mer的出现频率,然后,对这些k-mer进行编码,分别将A(腺嘌呤)、T(鸟嘌呤)、C(胞嘧啶)、G(胸腺嘧啶)采用0、1、2、3这些数字表示,再进行四进制编码,将每个k-mer的数字表示作为向量的维度索引,该k-mer的出现频率作为向量值,从而组成一个维度为4k的向量,而该向量就是被分割为L-k+1个长度为k的k-mer的待测序序列的特征向量。
附图2示例的类别判断模块204可以包括对比单元401和确定单元402,如附图4所示本发明实施例四提供的宏基因组数据分类装置,其中:
对比单元401,用于通过将每个簇的中心集合Ki的每一读长与参考基因序列对比,统计每个簇的中心集合Ki的每一读长的基因组类别。
具体地,对比单元401可以将每个簇的中心集合Ki的每一读长与参考基因序列对比,使用工具BLAST,统计出每个簇的中心集合Ki的每一读长的基因组类别。需要说明的是,本发明的技术方案并不是将每个簇的所有读长与参考基因序列对比,而是只选择每个簇的中心集合Ki的每一读长与参考基因序列对比,如此,减小了每个簇的类别的搜索范围,减小了对比量,从而减小了时间上的开销。
确定单元402,用于若中心集合Ki中任一读长Ri的基因组类别Ci的出现频率不小于预设阈值,则将读长Ri的基因组类别Ci作为读长Ri所属簇的基因组类别。
在对比单元401将每个簇的中心集合Ki的每一读长与参考基因序列对比过程中,统计的结果可能是同一读长却属于不同的基因组类别,此时,可以以该读长的基因组类别的出现频率来确定其基因组类别。例如,假设预设阈值是70%,若对比和统计的结果显示读长Ri的基因组类别属于C’i的出现频率是30%,属于C”i的出现频率是43%,属于Ci的出现频率是75%,则确定单元402将读长Ri的基因组类别确定为Ci,并且将读长Ri的基因组类别Ci确认为读长Ri所属中心集合Ki的基因组类别或所属簇的基因组类别。
附图2至4任一示例的宏基因组数据分类装置还可以包括降维模块501,如附图5-a至5-c所示本发明实施例五至七提供的宏基因组数据分类装置。降维模块501用于计算模块201计算待测序序列的特征向量之后,聚类模块202对特征向量进行聚类得到M组包含读长的簇G1至GM之前,对待测序序列的特征向量进行降维处理,具体可以使用基于互信息选择对待测序序列的特征向量进行降维处理。经过降维模块501的降维处理后,可以降低后续处理时的计算量和/或复杂度,从而减小运行时的时间开销。
附图2至4任一示例的宏基因组数据分类装置还可以包括再分类模块601,如附图6-a至6-c所示本发明实施例八至十提供的宏基因组数据分类装置。再分类模块601用于类别判断模块204通过将每个簇的中心集合Ki的每一读长与参考基因序列对比,判断每个簇的基因组类别之后,采用多核学习训练分类器对已确认基因组类别的簇再次进行分类。
为了将经类别判断模块204错分或误分的序列剔除,提高宏基因组数据分类整体的分类准确率,在本发明实施例中,可在类别判断模块204通过将每个簇的中心集合Ki的每一读长与参考基因序列对比,判断每个簇的基因组类别后,再分类模块601进一步采用多核学习训练分类器对已确认基因组类别的簇再次进行分类。具体可以是再分类模块601从已确认基因组类别的簇的中心集合Ki中随机选取一定比例,例如60%的读长作为训练集,用多核学习工具shogun训练分类模型,将余下比例,例如40%的读长作为测试集,采用多核学习训练分类器对其进行分类,滤除每个中心集合Ki中由于上一步聚类错误判别的读长。
需要说明的是,上述装置各模块/单元之间的信息交互、执行过程等内容,由于与本发明方法实施例基于同一构思,其带来的技术效果与本发明方法实施例相同,具体内容可参见本发明方法实施例中的叙述,此处不再赘述。
本领域普通技术人员可以理解上述实施例的各种方法中的全部或部分步骤是可以通过程序来指令相关的硬件来完成,该程序可以存储于一计算机可读存储介质中,存储介质可以包括:只读存储器(ROM,Read Only Memory)、随机存取存储器(RAM,Random Access Memory)、磁盘或光盘等。
以上对本发明实施例所提供的宏基因组数据分类方法和装置进行了详细介绍,本文中应用了具体个例对本发明的原理及实施方式进行了阐述,以上实施例的说明只是用于帮助理解本发明的方法及其核心思想;同时,对于本领域的一般技术人员,依据本发明的思想,在具体实施方式及应用范围上均会有改变之处,综上所述,本说明书内容不应理解为对本发明的限制。
- 还没有人留言评论。精彩留言会获得点赞!