一种基因检测知识库构建方法及系统与流程
本发明涉及生物信息数据库领域,具体涉及一种基因检测知识库构建方法及系统。
背景技术:
二代测序技术的发展,使得人类可以越来越方便的获取最基础的基因组序列,了解自身的遗传信息。很多科学研究表明,人类很多疾病、表型和对药物的反应,都源于个体的遗传背景差异,也就是每个人dna序列的差异。从2000年人类基因组计划开始,越来越多的人类基因组被破译,这些基因组构成了人类基因组的参考序列。基因检测的直接目的是,通过对个体的基因组进行测序(或者对部分区域测序),获取其和参考基因组dna水平的序列差异,然后再通过和已有知识库的比对,来预测可能的疾病、表型、药物反应的关联。
dna序列变异和疾病、表型、药物反应关联信息的来源一般有:
gwas(genome-wideassociationstudy):如果对一些有特定疾病、表型和药物使用的人群进行遗传信息挖掘,发现他们和参考序列的差异,那么可以推断,可能是这些dna水平的差异,最终导致了疾病、表型或者是对药物的不同反应。
传统的生物学研究中的生化、遗传学实验:通过传统的生物学研究方法也可以找到大量和疾病、表型或者是对药物反应的基因变异、蛋白质变异、酶变异等,将这些变异反映到dna层面,是dna水平中一类变异的发生。比如,一个疾病是由于一个蛋白质的跨膜区域发生了变异导致离子通道的异常,那么,编码这个蛋白质的dna序列中,只要任何有dna水平的变异影响了蛋白质的跨膜功能域,就可能导致这个疾病的发生。部分实验是在模式生物和细胞系完成。
其他:通过研究疾病样本和对照样本中基因、蛋白质的表达量差异;通过计算生物学的方法进行功能预测等。
以上,证据来源的不同,可信度也会不同,和疾病等关联的元素也不相同。应用到dna水平的序列变异和疾病的关系,会需要不同的记录和后续处理方式,需要设计并搭建好知识库。
知识库搭建是构建基因检测分析流程中的核心环节。但是现在的知识库存在以下问题:
搭建一个知识库需要耗费人力物力,一般需要整合公共数据库,加上专业人员阅读科研文献完成。
公共数据库层次不齐,并且人工收集总会存在一定的偏差。
很多知识库会将dna、rna、蛋白质等不同层面的信息混淆,比如gwas是针对dna层面的结果,而一些疾病相关致病基因的研究是在蛋白质层面,一些和药物代谢相关的研究是在酶和代谢物层面,虽然最终会归到dna层面的变异,但是在数据库搭建的过程中,应该事先进行区分,以便后续算法开发。
基于文本挖掘等一系列的算法,自动搭建知识库,但是完全依靠算法构建的知识库假阳性率和假阴性率都很高,对后续的基因检测疾病风险分析有很大的影响。
很多知识库搭建完成之后,有些已有关联条目也需要根据新科研成果而自动更新,但由于这些数据库固化的结构设计,自动更新很难实现。。
很多知识库都会采用一些公用的数据库管理软件,对生物学背景的研究人员不方便使用。
现有发明专利审核中的生物信息相关数据库,包含了:一种生物信息学数据库系统和数据处理方法(申请号:201410009130.1)和生物信息数据库的构建方法和装置(申请号:201410742604.3)
其中,第一个发明中,公开了一种生物信息数据库系统和数据处理方法,可以实现生物信息学数据的统一管理。该系统中包含了样品、项目和实验模块,主要目的是方便实验的设计和数据处理,提高工作效率;第二个发明中,公开了一种构建生物信息数据库的方法和装置,主要是通过对pubmed摘要的文本挖掘,对疾病相关的文献摘要进行分解、根据突变正则表达式语义库进行基因和突变信息的提取和分类,构建疾病相关的语义库,确定基因突变的得分,最后构建生物信息数据库。
技术实现要素:
针对现有技术中的不足,本发明的目的是提供一种基因检测知识库构建方法及系统,包括:提供基因检测相关知识库搭建方法,以及在数据库搭建完成之后,基于数据库的管理系统。
本发明的目的是采用下述技术方案实现的:
本发明提供一种基因检测知识库构建方法,其改进之处在于,所述方法包括:
构建数据库实体表、公共数据库的关联表和文本挖掘的关联表;
构建关联表打分系统;
构建数据库匹配管理系统,便于专家查找、校验、修改关联表。
进一步地,在所述构建数据库实体表、公共数据库的关联表和文本挖掘的关联表之前,还包括:收集公共数据库并对其整理,并根据公共数据库确定数据库结构。
进一步地,所述构建数据库实体表,包括含疾病、表型和环境因素表格的基因样本信息;
所述环境因素表格为人工录入表格;
在整合不同数据库id之间的匹配信息时,对于生物学元素,采取定位到基因组位置,判定位置的交叉是否大于并集的0.5来判定;
对于疾病的名称,则采用数据库本身录入的匹配表格,如果匹配不上,则放弃录入。
进一步地,所述构建公共数据库的关联表,包括:
将公共数据库分层;
根据分层水平进行打分,打分范围在-1~1,大于0为正相关,小于0为负相关。
进一步地,所述构建文本挖掘的关联表,包括:
收集近25年之内的文献摘要;
将文献摘要进行分类;
对证据级别进行打分,打分包含:动词表达正负关系的打分;不同的文献分值进行叠加,并将绝对值相加,如果直接相加的分值小于阈值,则将证据级的文献摘要提取放入校验表;最终的得分最大值为1分,最小为-1分。
进一步地,根据公共数据库的关联表和文本挖掘的关联表构建关联表打分系统,包括:
整合公共数据库挖掘得到的分值和文献挖掘得到的分值;
最后的关联条目来自于上述两个来源,将所有关联条目根据关联分值进行汇总排序,设置阈值,分别放入核心关联表和候选关联表;
如果公共数据库来源的条目和文献来源的条目分值差异大于阈值,则放入校验表;
对于疾病有确定or值的关联条目,如果不存在相互矛盾或or值倍数差异在0.5~2之内,则直接录入核心关联表;如果存在,则放入校验表。
进一步地,所述数据库匹配管理系统用于数据库查看、数据库搜索、数据库校验、数据库版本管理、log查询页面和操作标准页面。
进一步地,在所述构建数据库匹配管理系统之后,还包括:数据库校验表的人工校验;校验工作将在数据库管理系统中进行条目浏览和手动修改。
总上所述,本发明提供了一种基因检测知识库构建系统,其改进之处在于,所述系统包括:
第一构建模块:用于构建数据库实体表、公共数据库的关联表和文本挖掘的关联表;
第二构建模块:用于构建关联表打分系统;
第三构建模块:用于构建数据库匹配管理系统。
进一步地,所述系统还包括收集公共数据库并对其整理,并根据公共数据库确定数据库结构的收集模块。
与最接近的现有技术相比,本发明提供的技术方案达到的有益效果是:
1.采用半自动化半人工的方式进行知识库搭建,既考虑了人工搭建的偏差和疏漏,又考虑了公共数据库的可信度差异。
2.采用了自动更新的方式对数据库内容进行管理。
3.整合了文本挖掘算法和公共数据库信息采集软件在自动化收集的步骤中。
4.设计了核心库和候选库,并有校验库对分值模糊的条目进行人工校验。
5.设计了匹配的知识库管理系统,便于对数据库进行内容查询、校验、修改和版本管理。
附图说明
为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
图1是一种基因检测知识库构建方法的流程图;
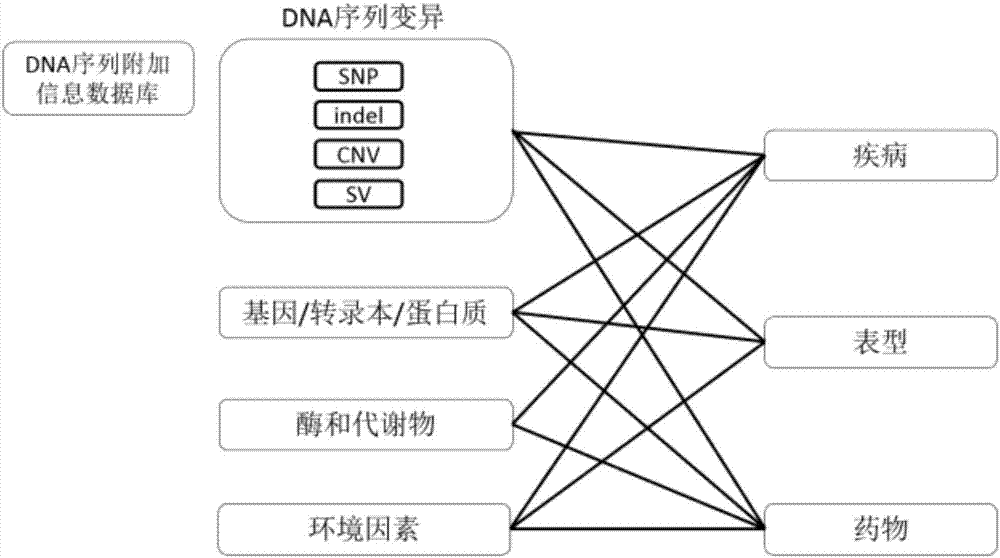
图2是知识数据库结构示意图。
具体实施方式
为使本发明的目的、技术方案和优点更加清楚,下面将对本发明的技术方案进行详细的描述。显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动的前提下所得到的所有其它实施方式,都属于本发明所保护的范围。
第一优选技术方案
知识库的表格一般分为两大类,一类是实体表,一类是关联表。实体表就是对数据库中元素和元素基本信息的记录。一般生物类知识库中的元素有:dna水平的序列差异、基因、转录本、蛋白质、代谢物、环境因素、疾病、表型、药物等。dna水平的序列差异可以根据其复杂程度,分为:
*snp(singlenucleotidepolymorphism):最常见的dna序列差异,为单碱基的突变,占已知dna多态性的90%以上,分布广泛,研究比较多。根据acmg指南,一般根据dbsnp数据库的标准对snp进行规范。snp在人群中的频率,也是在风险计算中很重要的信息,一般采用1000genomes和hapmap的数据。
*indel(insertionanddeletion):小片段的插入缺失,一般在2~16bp的长度范围,不会超过1kb。命名也是根据dbsnp数据库。
*cnv(copynumbervariation):拷贝数变异,最少50bp的长度,大部分在1kb~3mb,可以包含大片段的cnv。它的子类有del(deletion)、dup(duplication)、ins(insertion)和lcv(largescalecnv)。一般根据acmg推荐的dbvar数据库标准,如果不存在条目,则采用dgv数据库。
*sv(structurevariation):大片段的结构变异,广义的定义中sv包含了cnv,为了区别,此处sv仅包含inv(inversion)和translocation,染色体的移位和转置。一般也是根据acmg推荐的dbvar数据库标准,如果不存在条目,则采用dgv数据库。
如果在以上数据库中不存在匹配条目,本发明中会另有标准化输入方式。
基因、转录本、蛋白质参考refseq数据库,如果不匹配,则采用ensembl中的ensg(enst、ensp)命名,并且会记录officialgenesymbol,以便做文本挖掘。
酶和代谢物则根据kegg数据库的注释。
疾病的命名,主要采用acmg推荐的omim数据库,加上icd10、malacard、medgen、orphanet数据库。
表型的命名,主要采用hpo(humanphenotypeontology)的注释,和mesh、umls数据库。
药物的命名,主要采用drugbank数据库。
以上,为知识库的实体表。
关联表是对数据库中,元素之间关联的记录。首先是生物学元素之间的关联:
*dna序列变异中,snp之间的关联为它们之间的连锁不平衡(ld,linkagedisequilibrium)。用于疾病风险计算中的snp独立性分析。根据1000genomesproject数据库的信息。
*基因->转录本->蛋白质(酶)之间,根据转录、翻译的中心法则,元素之间相连。用于预测dna序列变异对所编码蛋白质的影响,再与蛋白质相关的疾病关联。主要根据refseq数据库,关于dna序列对所编码蛋白质的影响,则采用annovar算法。
*基因-基因之间,主要根据geneticinteraction和共进化的信息(biogrid,mint);蛋白质之间,主要根据蛋白质的相互作用网络(biogrid,mint);蛋白质(酶)、代谢物之间,主要根据kegg代谢数据库。这些相关关系主要服务于对突变的解释,以及与疾病(表型、药物反应)之间的关系预测。
*dna序列变异与疾病的关系,主要根据clinvar、hgmd、decipher数据库;基因(蛋白质)与疾病的关系,主要根据omim、uniprot、gad、disgenet等数据库;
*dna序列变异/基因/蛋白质与表型的关系,主要根据hpo数据库;
*dna序列变异/基因/蛋白质与药物的关系,主要根据drugbank、pharmgkb等数据库。
其他,比如环境因素和疾病的关系等,并没有现成的公共数据库,这些都是通过对pubmed进行文本挖掘获得。
采取半自动、半人工的方式搭建知识库
知识库采用模块分层式设计。数据库的实体包括:根据生物学的元素,分为dna、rna、蛋白质、酶和代谢物、环境因素五个层面,根据研究目的,分为疾病、表型、药物等三大模块。每个层面和模块中的元素,都会优先通过收集acmg(theamericancollegeofmedicalgeneticsandgenomics)公认的公共数据库中的信息,构建知识库需要的实体表;另外设计专门的证据库,记录知识库的每个记录的来源信息。
对于实体之间的关联表,将会由专家对所需要挖据的公共数据库进行初筛,并对每个数据库的可信度进行评分。然后,由数据库自动收集软件,将关联信息进行采集,并通过证据评估算法程序,给出每条证据的可信度评分(整合了数据库可信度、数据库条目可信度以及不同数据库记录的交叉程度),将分值归一化到-1~1。
*考虑到公共数据库信息的假阳性和假阴性,本发明包含了针对pubmed文献数据库的文本挖据算法。算法设计的思想是,下载pubmed最新的摘要全库,先根据关键词将每篇文献和三大模块中的内容进行匹配,构建文献和疾病(表型、药物)的关联表,然后将五个层面所有的元素在每个疾病(表型、药物)相关的文献摘要中进行匹配,根据语义对关联进行过滤(不相关)和评分(正相关、负相关),对于复杂疾病如果有确定的or值,记录下来源和or值语句。再将不同文献的评分汇总,并以所有的文献数目进行校正,将分值归一化到-1~1。
最后的关联条目来自于上述两个来源,将所有条目根据关联分值进行汇总排序,设置阈值,分别放入核心关联表和候选关联表;如果公共数据库来源的条目和文献来源的条目有较大出入(分值差异大于0.5,阈值可调整),则放入校验表。其中,对于复杂疾病有确定or值的关联条目,如果不存在相互矛盾或差异较大的关联条目(or值倍数差异在0.5~2之内,阈值可调),则直接录入核心关联表,如果存在,则放入校验表。
将校验表内容交由人工专家进行处理。根据结果将关联条目分别放入核心关联表和候选关联表。
知识库每月可以自动更新(更新周期可调),每次更新将重复以下几个工作:判断公共数据库更新,如果有,重新对条目打分;更新pubmed最新的摘要全库,重复以上算法,对条目打分;根据结果,将关联条目重新放入核心关联表、候选关联表,但是如果一个条目的表归属有变化,将会放入校验表,由专人进行人工校验。
为了使没有数据库专业背景的生物学方向专家对数据库进行人工校验和查询使用,本发明专门开发了配套的数据库管理系统,包含了查询、修改、log记录等功能,可以使生物学方面的专家对数据库内容进行及时校验。另外,数据库会根据是否有修改定期备份,也会根据产品研发进度进行定版,确保在生产环境中使用的数据库都可以溯源。
第二优选技术方案
本发明提供一种基因检测知识库构建方法,其流程图如图1所示,包括下述步骤:
1、第一步为公共数据库的收集和初步整理,现在收集的数据库有:dbsnp、1000genomeprojects、dbvar、dgv、refseq、ensg、clinvar、hgmd、decipher、omim、hpo、mesh、umls、drugbank、biogrid、mint、uniprot、gad、disgenet、pharmgkb、kegg、pubmedabstract。其中除了pubmedabstract,其他数据库的原始数据都处理成tab格式文件,格式如下:
2.第二步为知识数据库结构设计,数据库结构如图2。每个模块中,有专人输入公共数据库的名称和主键,通过自主开发的软件,自动生成数据库的原始整合tab格式文件。在图2中罗列了所有需要准备的文件名称,以及文件的关联来源。此时,也会有辅助文件,来存储不同数据库之间的id匹配等信息。
3.第三步为数据库中实体表的构建,实体表包含了snp(以dbsnp为主),var(除了snp的dna序列变异,以dbvar为主),gene(包含了转录本、蛋白质和酶的信息,以refseq为主),env(环境因素表格),disease(疾病信息,以omim和icd10数据库为主),pheno(表型信息,以hpo为主)和drug(药物,以drugbank为主)。其中,环境因素表格为人工根据经验录入。在整合不同数据库id之间的匹配信息时,对于生物学元素,采取定位到基因组位置,然后判定位置的交叉是否大约并集的0.5来判定;对于疾病的名称,则采用omim等数据库本身录入的匹配表格,除了omim、orphanet、icd10以外其他疾病信息数据库中的疾病名称,如果匹配不上,则放弃录入。
4.第四步为基于公共数据库的关联表构建。由自主开发的软件对关联证据进行打分(范围在-1~1,大于0为正相关,小于0为负相关,大部分数据库记录了正相关信息)。打分的方法为:
*将数据库分类,进行打分,以疾病和基因的关联为例,将数据库分为两个水平,uniprot、omim为第一层,gad、disgenet为第二层,第一层的分值设为0.3分,第二层的分值为0.1分,如果原有数据库的关联已经存在分值,将其归一化到0~1之后,和数据库本身的分值相乘。然后不同数据库的相同记录分值进行叠加,最高分设为1分。
*每个关联表,都会由专家事先将数据库进行分层。
5.第五步为基于文本挖掘的关联表构建。首先采用pubmed官网提供的api接口,下载1990年以来的所有文献的摘要。采用自主开发的软件进行文本挖掘(基于python编程,nltk自然语言处理包)。基本的编程思路如下:
*读入abstract文件(纯文本格式),判定是否有mh(mesh数据库)的注释,如果有,则进行提取,和数据库中所有实体表(和所有alias表)进行比对;如果没有,直接对摘要(ab)进行分词,再将所有分词中,进行词性归类,将所有名词和所有实体表(和所有alias表)进行比对。此时主要和disease_info,disease_alias表格比对(drug、pheno类似)。此步的主要目的是将文献摘要进行分类(允许一篇文献归到多个类别)。
*对于dna序列水平的变异,在snp_alias.txt文件中,snp以rs号为最主要的主键,并收集hgvs名称,根据rsid(e.grs1024323)>碱基变化(e.gnc_000004.11:g.3006043c>t)>转录本变化(e.gnm_001004056.1:c.329c>t)>氨基酸变化(e.gnp_001004057.1:p.ala142val,np_001004057.1:p.a142v)>位置(e.g37:chr4;3006043;c;t),并加上此时基因常用名grk4。如果在语句中直接出现了rs1024323,或者同时出现gkr4和a142v,如下:
e.g:wehypothesizedthat3nonsynonymousgrk4single-nucleotidepolymorphisms,r65l(rs2960306),a142v(rs1024323),anda486v(rs1801058),wouldbeassociatedwithbloodpressureresponsetoatenolol,butnothydrochlorothiazide,andwouldbeassociatedwithlong-termcardiovascularoutcomes(all-causedeath,nonfatalmyocardialinfarction,nonfatalstroke)inparticipantstreatedwithanatenolol-basedversusverapamil-sr-basedantihypertensivestrategy.
e.g:methods:participantsfromtheafricanamericanstudyofkidneydiseaseandhypertension(aask)trialweregenotypedatthreegrk4polymorphisms:r65l,a142v,anda486v.
则将相应的语句提出。
*对于同一类别中的文献,例如此时的atenolol(阿替洛尔),在以上第一个案例中获取了和a142v相关,再在整个摘要中,提取描述相关的语句"a142v(rs1024323),......wouldbeassociatedwithbloodpressureresponsetoatenolol",此时通过分词,获取其中的动词以及褒贬情感分类,记录突变和药物的关系,以及整个语句。
*此时也会对证据级别进行打分,打分包含:动词表达正负关系的打分;比如明确表达相关"associatedwith"给0.3分,如果有"might"等弱化相关关系的词汇,则给0.1分。如果为负向相关,则给-0.3和-0.1分。不同的文献分值进行叠加,并将绝对值相加,如果直接相加的分值小于0.5*绝对值相加的分值(阈值可调),则将该记录提取出,放入校验表。最终的得分最大值为1分,最小为-1分。
6.第六步为关联表打分系统。主要目的是整合公共数据库挖掘得到的分值和文献挖掘得到的分值。最后的关联条目来自于上述两个来源,将所有条目根据关联分值进行汇总排序,设置阈值,分别放入核心关联表和候选关联表;如果公共数据库来源的条目和文献来源的条目有较大出入(分值差异大于0.5,阈值可调整),则放入校验表。其中,对于复杂疾病有确定or值的关联条目,如果不存在相互矛盾或差异较大的关联条目(or值倍数差异在0.5~2之内,阈值可调),则直接录入核心关联表,如果存在,则放入校验表。
7.第七步为匹配的数据库管理系统搭建,该系统的代码事先写好,和最初数据库结构匹配,可以根据数据库真实需求进行二次开发。配套系统需要的硬件配置如下:
(1)数据库部分采用mysql进行开发。
(2)管理网站部分采用django进行开发,用于对数据库数据搜索、修改等,管理所有html页面等。
(3)页面采用html5编写,包含css,javascript代码定义页面样式,编写页面复杂动作等。
(4)采用linuxshell脚本对系统进行备份。
系统搭建在linuxcentos平台,4核cpu,4g内存,1t硬盘。
8.第八步为数据库校验表的人工校验。此步校验工作将在管理系统中,研发专家进行账户登录后,进行条目浏览和手动修改。
9.第九步为匹配管理系统的测试。管理系统包含了:数据库查看、数据库搜索、数据库校验、数据库版本管理、log查询页面和操作标准页面。
本发明提供的构建方法数据库的数据不仅来源于文献摘要,还直接通过公共数据库挖掘,并且挖掘的信息不局限于疾病,还包括了表型、环境因素等,更加全面。另外,本发明中,包含了数据库匹配的管理系统。
第三优选技术方案
基于同样的发明构思,本发明还提供一种基因检测知识库构建系统,包括:
第一构建模块:用于构建数据库实体表、公共数据库的关联表和文本挖掘的关联表;
第二构建模块:用于构建关联表打分系统;
第三构建模块:用于构建数据库匹配管理系统。
所述系统还包括收集公共数据库并对其整理,并根据公共数据库确定数据库结构的收集模块。
以上所述,仅为本发明的具体实施方式,但本发明的保护范围并不局限于此,任何熟悉本技术领域的技术人员在本发明揭露的技术范围内,可轻易想到变化或替换,都应涵盖在本发明的保护范围之内。因此,本发明的保护范围应以所述权利要求的保护范围为准。
- 还没有人留言评论。精彩留言会获得点赞!