一种低复杂度的天然无序蛋白质的预测方法与流程
本发明属于生物信息学领域,涉及一种高效、低计算复杂度的天然无序蛋白质的预测方案。
背景技术:
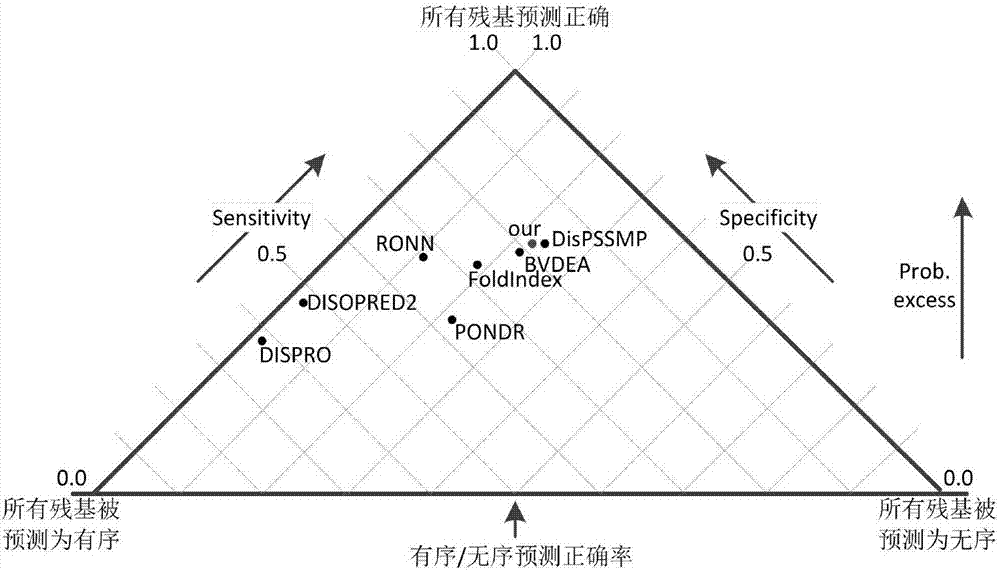
:天然无序蛋白质是指一个蛋白质至少有一个缺少唯一的三维结构且具有动态构象的区域,在药物设计、蛋白质表达和功能注释等方面都有重要的作用。因为研究发现一些天然无序蛋白质参与细胞中的重要调节功能,对阿尔茨海默病、帕金森病与某些癌症等疾病有重要影响。由于无序蛋白质区域提纯和结晶困难,通过实验来测定不但费用高昂且耗时很长。因此,通过计算的方法由蛋白质序列来测定无序区域的研究是十分重要的。在过去的十数年间,提出了许多无序蛋白质预测方案,大致可分为两类:第一类利用无序蛋白质序列的氨基酸倾向性,第二类利用机器学习的方法。其中,第一类方法十分简单但是准确度不高。第二类方法主要基于人工神经网络和支持向量机,可以得到较高的预测准确度,但是要求计算一系列特征计算复杂度很高。技术实现要素:本发明的目的是克服现有技术存在的上述不足,设计一种低复杂度的天然无序蛋白质的预测方法,可以使用少量的特征和计算,得到较高的预测准确度、较快的运算速度和鲁棒性。本发明提供的低复杂度的天然无序蛋白质的预测方法的具体步骤如下:(1)针对学习样本dis数据集,令w表示其中一条蛋白质序列,用长度为n的滑动窗口截取n长的连续残基片段进行计算。此时假设w的长度即为n。(2)计算w的香农熵,公式为:其中,fk代表第k种氨基酸在w中出现的频率。(3)计算拓扑熵:将w映射为0-1序列,其中疏水性氨基酸包括异亮氨酸、亮氨酸和缬氨酸,芳香族氨基酸包括苯丙氨酸、色氨酸和酪氨酸被映射为1,其余为0。计算w的拓扑熵:其中pw(n)代表w中长度为n的不同子字的个数,n满足:代表的从l开始的长度为2n+n-1的连续符号。(4)对于长度为n的序列w,计算其remark465,deleage/roux以及bfactor(2std)三种倾向性的加权平均值:其中代表序列w到第p种的倾向性的值。(5)对于一条长度为l>n的序列w,将每个滑动窗口计算得到的五个特征值作为一个矢量分配给窗口的每个残基;针对每个残基,累加得到的矢量并除以累加次数,得到最终的特征矢量;截取n长片段wj=w(j)…w(j+n-1),1≤j≤l-n+1,计算其香农熵、拓扑熵和三种倾向性的加权平均值这五种特征,得到一个5×1矢量vj:vj=[hs(wj)htop(wj)m1(wj)m2(wj)m3(wj)]t(5)之后计算序列w的特征矩阵f=[x1x2…xl…xl],其中(6)利用5-fold交叉验证,训练分类器。将学习样本中的无序残基和有序残基的特征矢量输入分类器进行学习,得到分类器的参数:投影方向w和分类阈值。计算训练集的特征矩阵:其中ns代表训练集中蛋白质序列的个数,fi代表长度为li的第i条蛋白质序列的特征矩阵,1≤i≤ns。最佳投影方向为:其中ndis和nord分别代表训练集中无序残基和有序残基的总个数,xdis和xord分别代表所有无序残基和有序残基的特征矩阵,如公式(7)所定义,和分别代表xdis和xord中的第j个列向量。在w上的投影为y=wtx。通过线性搜索,可以得到在y上的分类阈值。本发明的优点和积极效果:1、本发明仅使用了5种特征和线性分类器,就使天然无序蛋白质的预测方法具有较高的运算速度和鲁棒性。2、仿真结果表明,在相似的预测准确度下,本发明设计的天然无序蛋白质的预测方法与现有的同类型预测方法相比,大大减少了特征个数和计算复杂度。附图说明图1:实现本发明预测天然无序蛋白质方法的流程图。图2:针对pu159数据集,本发明设计的天然无序蛋白质的预测方法与现有的同类型预测方法的预测准确度比较。图3:针对r80数据集,本发明设计的天然无序蛋白质的预测方法与现有的同类型预测方法的预测准确度比较。具体实施方式实施例1:本发明提供的天然无序蛋白质的预测方法具体步骤如下:针对一条未判定无序区域的蛋白质序列w(以r80数据集中一条标号为1g4m的蛋白质序列为例),利用本发明提供的无序蛋白质预测方案进行预测的具体步骤如下:步骤一:该序列长度为393,用n=35的滑动窗口对序列进行截取。针对每个窗口区间计算五种特征的值。序列w=mgdkgtrvfkkaspngkltvylgkrdfvdhidlvepv…针对第一个长度为n的窗口,按照公式(1)(3)(4),计算窗口所截取的序列片段的五种特征的值,并将这五个值分别赋给片段中的每个残基;之后,滑动窗口,计算从第二个残基开始的长度为n的序列片段的五种特征的值并累加给片段中每个残基;重复上述过程,直至窗口覆盖到最后一个残基。统计序列中每个残基的累加次数,用残基的各个累加的特征的值除以累加次数,得到其最终的特征矢量。计算得到的序列w的特征矩阵如下,其中每一列为对应该位置残基的特征矢量:步骤二:利用学习样本计算得到的投影方向和阈值,对x投影和判定,其中35个无序残基有29个被正确判定为无序,358个有序残基有314个被正确判定为有序。为了验证该预测方法的有效性,利用r80数据集和pu159数据集对该方法进行了天然无序蛋白质的预测。其中,r80数据集中包含80条蛋白质序列,每条蛋白质序列都含有至少一个无序区域;pu159数据集中包含79条完全无序序列和80条完全有序序列。表1中列出了针对pu159数据集,本发明设计的天然无序蛋白质的预测方法与现有的同类型预测方法的预测准确度比较。表2列出了针对r80数据集,本发明设计的天然无序蛋白质的预测方法与现有的同类型预测方法的预测准确度比较。表3列出了各个预测准确度参数的定义,其中tp表示预测正确的无序残基个数,tn表示预测正确的有序残基个数,fn表示原本是无序残基被错判为有序残基的个数,fp表示原本是有序残基被错判为无序残基的个数。表1methodssens.spec.prob.ex.mccourmethod0.8120.7830.5960.594dispssmp0.8250.7650.5900.589bvdea0.7960.7850.5810.586ronn0.6750.8880.5630.580foldindex0.7220.8150.5360.540disopred20.4690.9810.4490.543pondr0.6320.7820.4140.420dispro0.3830.9820.3650.467prelink0.3190.9910.3100.430表2methodssens.spec.prob.ex.mccourmethod0.7270.8970.6240.515dispssmp0.7670.8480.6150.463bvdea0.8170.7280.5450.451ronn0.6030.8780.4810.395foldindex0.4880.8110.2990.224disopred20.4050.9720.3770.470pondr0.5570.8160.3730.278dispro0.4180.9930.4110.578prelink0.2370.9470.1830.219表3参考文献1.jingy,marcinjm,paullf,vladimirnu,lukaszk,rapid:fastandaccuratesequence-basedpredictionofintrinsicdisordercontentonproteomicscale,biochimicaetbiophysicaacta,1671-1680,2013.2.vnuversky,themysteriousunfoldome:structureless,underappreciated,yetvitalpartofanygivenproteome,j.biomed.biotechnol,2010.3.wrightp,dysonh,intrinsicallyunstructuredproteins:re-assessingtheproteinstructure-functionparadigm,j.mol.biol.,293:321-331,1999.4.iremek,turgayi,okanke,predictionofdisorderwithnewcomputationaltool:bvdea.expertsystemswithapplications,38:14451-14459,2011.5.oldfieldcj,ulrichel,chengy,dunkerak,markleyjl,addressingtheintrinsicdisorderbottleneckinstructuralproteomics,proteins,59:444-453,2005.6.jaimep,cliffordef,tzviyazbm,edwinhr,ornam,jacquessb,israelsjls,foldindex:asimpletooltopredictwhetheragivenproteinsequenceisintrinsicallyunfolded,bioinformatics,21(16):3435-3438,2005.7.rlinding,rbrussell,vneduva,tjgibson,globplot:exploringproteinsequencesforglobularityanddisorder.nucleicacidsresearch,31(13):3701-3708,2003.8.ferenco,judito,proteinswithout3dstructure:definition,detectionandbeyond,bioinformatics,27(11):1449-1454,2011.9.kpeng,svucetic,pradivojac,cjbrown,akdunker,zobradovic,optimizinglongintrinsicdisorderpredictorswithproteinevolutionaryinformation,journalofbioinformaticsandcomputationalbiology,3(1):35-60,2005.10.yangzr,thomsonr,mcneilp,esnoufrm,ronn:thebio-basisfunctionneuralnetworktechniqueappliedtothedetectionofnativelydisorderedregionsinproteins.bioinformaticsadvanceaccesspublished9,2005.11.jjward,jssodhi,ljmcguffin,bfbuxton,dtjones,predictionandfunctionalanalysisofnativedisorderinproteinsfromthethreekingdomsoflife.j.mol.biol.,337:635-645,2004.12.suc,chenc,ouy,proteindisorderpredictionbycondensedpssmconsideringpropensityfororderordisorder,bmcbioinformatics,307-319,2006.13.ishidat,kinoshitak,predictionofdisorderedregionsinproteinsbasedonthemetaapproach,bioinformatics24:1344-1348,2008.14.schlessingera,improveddisorderpredictionbycombinationoforthogonalapproaches,plosone,4:4433,2009.15.chengj,sweredoskimj,baldip,accuratepredictionofproteindisorderedregionsbyminingproteinstructuredata,dataminingandknowledgediscovery,11:213-222,2005.16.weathersea,paulaitisme,woolftb,hohjh,reducedaminoacidalphabetissufficienttoaccuratelyrecognizeintrinsicallydisorderedprotein,febsletters,576:348-352,2004.17.davidk,topologicalentropyofdnasequences.bioinformatics,27(8):1061-1067,2011.18.mikas,ratschg,westonj,scholkophb,mullerskr,fisherdiscriminantanalysiswithkernels,neuralnetworksforsignalprocessing,1999.19.kohavi,ron,astudyofcross-validationandbootstrapforaccuracyestimationandmodelselection.proceedingsofthefourteenthinternationaljointconferenceonartificialintelligence,sanmateo,ca:morgankaufmann,2(12):1137-1143,1995.20.uverskyvn,gillespiejr,finkal,whyare"nativelyunfolded"proteinsunstructuredunderphysiologicconditions,proteins41:415-427,2000。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1