一种基于局部图的三代测序序列校正方法与流程
本发明涉及三代测序(pacbiosmrt和oxfordnanopore测序)测序序列错误矫正方法,特别涉及一种基于局部图的三代测序序列校正方法。
背景技术:
目前三代测序技术主要包含pacbio公司的单分子实时测序(singlemolecule,real-time,smrt)测序技术和oxfordnanopore公司的纳米孔(nanopore)测序技术。与二代测序技术相比,三代测序数据具有读长(或测序序列)很长(longread,平均10-15kb左右)和测序序列无gc偏好性等特点,这些数据特征可以有力弥补了一代和二代测序技术很多缺陷,从而使其具有广泛应用市场:在基因组测序方面,研究者利用三代测序的测序序列完成了大基因组组装、基因组复杂区深度解析、人类基因组150个gap区域和结构变异的解析;在转录组测序方面,研究者利用测序序列已包含完整cdna信息深入分析全转录组可变剪接和亚型;在dna修饰测序方面,研究者利用模板修饰碱基降低聚合酶合成速率来有效检测dna未知的修饰(例如dna甲基化)。目前,三代测序技术将成为二代测序技术的有力补充或替代,近两年广泛应用于基因组组装、长片段indel检测和矫正以及甲基化修饰的检测等研究中。
三代测序高测序错误率(15%,其主要是插入或缺失)给三代测序数据处理带来了巨大的挑战,与此同时,高错误率使二代测序软件很难用于三代测序数据分析。为了解决高测序错误率,研究者采用多重序列比对获取高一致性公共序列的方法校正三代测序序列,校正后序列正确率可以达到97-99%。目前组装流程中的序列校正软件主要有三个:dagcon,fc_consensus,和falconsense;他们通过两步完成序列校正过程:1)将模板序列与候选序列进行两两比对获得模板的多重比对结果;2)通过多重序列比对推断止确的序列。dagcon将多重序列比对结果转化成有向校正图,通过寻找有向图的最优路径从而获取校正序列;fc_consensus和falconsense通过对每个碱基位各种操作计数从而获得校正序列(图x)。dagcon的校正精度可以达到99.9%,但其速度很慢。虽然fc_consensus和falconsense的校正速度较快,然而校正精度只有96-98%,并且fc_consensus和falconsense输入是来源于daligner和mhap的两两比对结果,mhap和daligner的两两比对结果是我们mecat2pw软件的2-3倍,其大量多于的两两比对结果增加了多重序列比对的时间,增大了计算量。三代测序校正过程非常耗时,目前主流组装流程canu(用falconsense校正)和falcon(用fc_consensus软件)完成54x人三代数据的校正需要30-50万核时。
因此,创建一种高效三代测序测序序列校正方法可以大幅降低目前已有校正方法的计算资源消耗,具有良好商业价值。
技术实现要素:
本专利针对目前三代测序序列校正方法速度慢和校正正确率不高问题,设计了基于局部图的三代测序序列校正系统和方法。
具体的,本发明提供了一种基于局部图的三代测序序列校正系统,其特征在于该系统包括两两比对模块、多重序列比对模块、校正操作比对模块、校正操作分类模块、一致区域的碱基位校正和复杂区域局部图碱基序列校正模块及模板序列校正分割和去融合处理模块,两两比对模块分别与单分子实时测序数据库和纳米孔测序数据连接,单分子实时测序数据库和纳米孔测序数据库分别输入到两两比对模块。
单分子实时测序数据库包含pacbio数据,纳米孔测序数据库包含nanopore数据。
上述系统中,优选的,两两比对模块、多重序列比对模块、校正操作比对模块、校正操作分类模块、一致区域的碱基位校正和复杂区域局部图碱基序列校正模块及模板序列校正分割和去融合处理模块按顺序依次连接,前一模块数据依次输入后一连接模块中进行处理。
上述系统中,优选的,两两比对模块中嵌合两两比对结果优选规则信息和过滤规则信息。尤其优选的,两两比对模块数据中包括两条测序序列的核心种子位置对信息,核心种子位置对信息的格式为九列格式信息,九列格式信息为:
第一列是测序序列a编号,
第二列是测序序列b编号,
第三列是a序列的正负链信息,正为0,负为1,
第四列是b序列的正负链信息,
第五列是核心位置对在a序列上的位置pa,
第六列是核心位置对在b序列上的位置pb,
第七列是核心位置对的全局投票得分,
第八列是a序列的长度la,
第九列是b序列的长度lb
过滤规则信息为在校正模板序列t时,提取所有与t相关的两两比对结果记录:即第一列或第二列含有t编号的记录。
优选的,对t相关两两比对结果过滤规则如下:
(1)过滤掉两条序列重叠长度小于90%的两条序列较小长度的记录。
过滤规则为:通过两个序列核心位置对信息,计算每条记录两个测序序列重叠长度,如果pa>=pb,左边的a和b的重叠长度ol=pb,否则ol=pa;如果la-pa>=lb-pb,右边的a和b的重叠长度or=lb-pb,否则or=la-pa;a和b序列重叠长度长度为o=ol+or,当o长度大于a的序列长度la的90%或者大于b的序列长度la的90%,该两两比对记录将被保留,否则该记录将被过滤掉或忽略。
按照上述重叠长度过滤原则过滤掉分析所有t序列相关的两两比对记录,获得t序列的过滤后记录,另外其中一条测序序列的长度小于5000,该记录将被过滤。
(2)优选200候选两两比对记录进入候选校正步骤。
在重叠度过滤后,将t序列过滤后所有记录按照全局投票打分进行降序排序,取最高200个全局投票得分记录进入候选多重序列比对和t序列校正过程。
优选的,多重序列比对模块运行方法为:根据过滤后每条两两比对的核心位置对信息,从核心位置从右到左取出模板序列和输入序列的左边部分的两条序列进行局部两两比对。
优选的,局部两两比对方法为:针对于pacbio数据,采用diff进行局部序列分析;针对nanopore数据,采用smith-waterman方法进行局部序列比对。
优选的,局部两两比对方法为:对取出两条左边序列进行按照500bp动态分段进行比对,先取出第一段500bp的两个序列,进行局部两两比对,比对完后从尾部向头回溯,寻找到有连续5个碱基匹配的的位置,为了保证500bp左边的序列起始正确性,从这个连续5bp匹配位置开始,再取500bp两个完成下一个段局部序列比对,重复上述过程,完成左边两条序列的局部比对过程。
类似左边两条序列局部比对过程,从核心位置从左到右取出模板序列和输入序列的右边部分的两条序列进行局部两两比对,其分段比对过程与左右相似。
优选的,局部两两比对的终止条件:1)完成两条序列局部比对;2)在每个500bp的分段序列中,错配,插入和删除碱基数小于500*0.2=100,如果遇到一段500bp中错误大于100bp,终止模板和输入序列的局部序列比对,过滤掉该两两比对记录。将所有200条模板序列相关的两两比对记录完成两两局部比对产生模板序列的多重序列比对结果。
上述系统中,优选的校正操作比对模块执行基于多重序列比对的一致性校正操作统计分析。具体的,统计模板序列每个位置匹配(mat)、删除(del)、插入(ins)、替代(mismat)四类校正操作的个数,获取模板序列每个位置一致性校正操作统计表。
当模板每个位置中mat+del<6时,该位点的mat值和del值设为0,当模板每个位置ins<6,ins值设为0,因为覆盖度小于6,该位点将不被校正。
优选的,校正操作分类模块执行基于校正操作统计表对校正操作分类规则。
优选的,规则如下:
根据多重序列比对的一致性校正操作统计表,对每个模板碱基位进行分类,其分成三类:1)如果
优选的,一致区域的碱基位校正和复杂区域局部图碱基序列校正模块中嵌合如下规则:
碱基为属于fmp碱基位,将保持该碱基序列不变,其中85%以上属于这样碱基校正位,对于剩下的碱基位,从该碱基为向左和向右寻找fmp碱基为,找到左边最近的fmp碱基位和右边最近fmp的碱基位,将两个碱基位之间的所有多重比对序列取出,建立局部图采用dagcon方法进行这部分复杂区域的校正:每个碱基是一个节点,每个边记录多重序列比对中走这个节点的次数,通过做经过次数做多的边,获取最终复杂区域的校正。
优选的,模板序列校正分割和去融合处理模块嵌合如下规则:
保证校正后序列正确率大于97%,当模板序列没有6x的覆盖度时,输出校正后序列时将剪切该段序列,当剪切序列在中间时,将造成一条测序序列多段校正输出的现象,在校正模板序列时,当校正某个位置,其输入序列没有超过1000bp的序列经过该位点左边和右边,该位置认为是融合位置,融合位置被剪断。
优选的,本发明还提供了一种基于局部图的三代测序粗劣校正方法,该方法包括如下步骤:
步骤1:两两比对结果优选和过滤,
步骤2:过滤后两两比对结果的多重序列比对,
步骤3:基于多重序列比对的一致性校正操作统计分析,
步骤4:基于校正操作统计表对校正操作分类,
步骤5:高度一致区域的碱基位校正和复杂区域局部图碱基序列校正,
步骤6:模板序列校正分割和去融合处理,
其中,步骤1中,基于两两比对方法(mecat2pw)的输出结果,每条mecat2pw的输出结果记录了两条测序序列的核心种子位置对信息,其记录是九列格式信息:第一列是测序序列a编号,第二列是测序序列b编号,第三列是a序列的正负链信息(正为0,负为1),第四列是b序列的正负链信息,第五列是核心位置对在a序列上的位置(pa),第六列是核心位置对在b序列上的位置(pb),第七列是核心位置对的全局投票得分,第八列是a序列的长度(la),第九列是b序列的长度(lb),在校正模板序列t时,提取所有与t相关的两两比对结果记录:即第一列或第二列含有t编号的记录。
对t相关两两比对结果过滤步骤如下:
步骤1-1:过滤掉两条序列重叠长度小于90%的两条序列较小长度的记录:通过两个序列核心位置对信息,计算每条记录两个测序序列重叠长度,如果pa>=pb,左边的a和b的重叠长度ol=pb,否则ol=pa;如果la-pa>=lb-pb,右边的a和b的重叠长度or=lb-pb,否则or=la-pa;a和b序列重叠长度长度为0=ol+or,当o长度大于a的序列长度la的90%或者大于b的序列长度la的90%,该两两比对记录将被保留,否则该记录将被过滤掉或忽略
按照上述重叠长度过滤原则过滤掉分析所有t序列相关的两两比对记录,获得t序列的过滤后记录,另外其中一条测序序列的长度小于5000,该记录被过滤。
步骤1-2:优选200候选两两比对记录进入候选校正步骤:在重叠度过滤后,将t序列过滤后所有记录按照全局投票打分进行降序排序,取最高200个全局投票得分记录进入候选多重序列比对和t序列校正过程,即输入序列(inputs)和模板序列(template)。
步骤2中,根据过滤后每条两两比对的核心位置对信息,从核心位置从右到左取出模板序列和输入序列的左边部分的两条序列进行局部两两比对,比对步骤如下:
步骤2-1:分段完成两两局部序列比对:对取出两条左边序列进行按照500bp动态分段进行比对,先取出第一段500bp的两个序列,进行局部两两比对,比对完后从尾部向头回溯,寻找到有连续5个碱基匹配的的位置,为了保证500bp左边的序列起始正确性,从这个连续5bp匹配位置开始,再取500bp两个完成下一个段局部序列比对,重复上述过程,完成左边两条序列的局部比对过程,类似左边两条序列局部比对过程,从核心位置从左到右取出模板序列和输入序列的右边部分的两条序列进行局部两两比对,其分段比对过程与左右相似。
步骤2-2:局部比对的终止条件:1)完成两条序列局部比对;2)在每个500bp的分段序列中,错配,插入和删除碱基数小于500*0.2=100,如果遇到一段500bp中错误大于100bp,终止模板和输入序列的局部序列比对,过滤掉该两两比对记录。将所有200条模板序列相关的两两比对记录用2-1和2-2完成他们的两两局部比对过程,产生模板序列的多重序列比对结果。
步骤3中,基于多重序列比对的一致性校正操作统计分析的方法如下:
统计模板序列每个位置匹配(mat)、删除(del)、插入(ins)、替代(mismat)等四类校正操作的个数,获取模板序列每个位置一致性校正操作统计表。
对于pacbio数据,由于diff比对中没有碱基替代比对,所以统计表中碱基校正操作只有三个类型:匹配、删除和插入。由于覆盖度小于6的位置,该位点保持原有模板序列比校正更可靠,所以当模板每个位置中mat+del<6时,该位点的mat值和del值设为0,当模板每个位置ins<6,ins值设为0,因为覆盖度小于6,该位点将不被校正。
步骤4中,基于校正操作统计表对校正操作分类的方法如下:
根据多重序列比对的一致性校正操作统计表,对每个模板碱基位进行分类,其分成三类:1)如果
步骤5中,高度一致区域的碱基位校正和复杂区域局部图碱基序列校正方法如下:
碱基为属于fmp碱基位,将保持该碱基序列不变,其中85%以上属于这样碱基校正位,对于剩下的碱基位,从该碱基为向左和向右寻找fmp碱基为,找到左边最近的fmp碱基位和右边最近fmp的碱基位,将两个碱基位之间的所有多重比对序列取出,建立局部图采用dagcon方法进行这部分复杂区域的校正:每个碱基是一个节点,每个边记录多重序列比对中走这个节点的次数,通过做经过次数做多的边,获取最终复杂区域的校正。
步骤6中,模板序列校正分割和去融合处理方法如下:
保证校正后序列正确率大于97%,当模板序列没有6x的覆盖度时,输出校正后序列时剪切该段序列,当剪切序列在中间时,将造成一条测序序列多段校正输出的现象。由于pacbio测序序列存在1%的融合现象(不相关两短序列测序成一条测序序列),在校正模板序列时,当校正某个位置,其输入序列没有超过1000bp的序列经过该位点左边和右边,该位置认为是融合位置,融合位置将被剪断。
有益效果
本发明针对目前三代测序序列校正方法速度慢和校正正确率不高问题,设计了基于局部图的三代测序序列校正方法和系统,在高度一致碱基位采用碱基计数方法,在复杂区域建立局部图方法。基于局部图三代测序序列校正方法完成了三代测序校正软件的开发,精度可以达到99%,速度是目前常用软件,例如软件fc_consensus和falconsense等的7-10倍。
附图说明
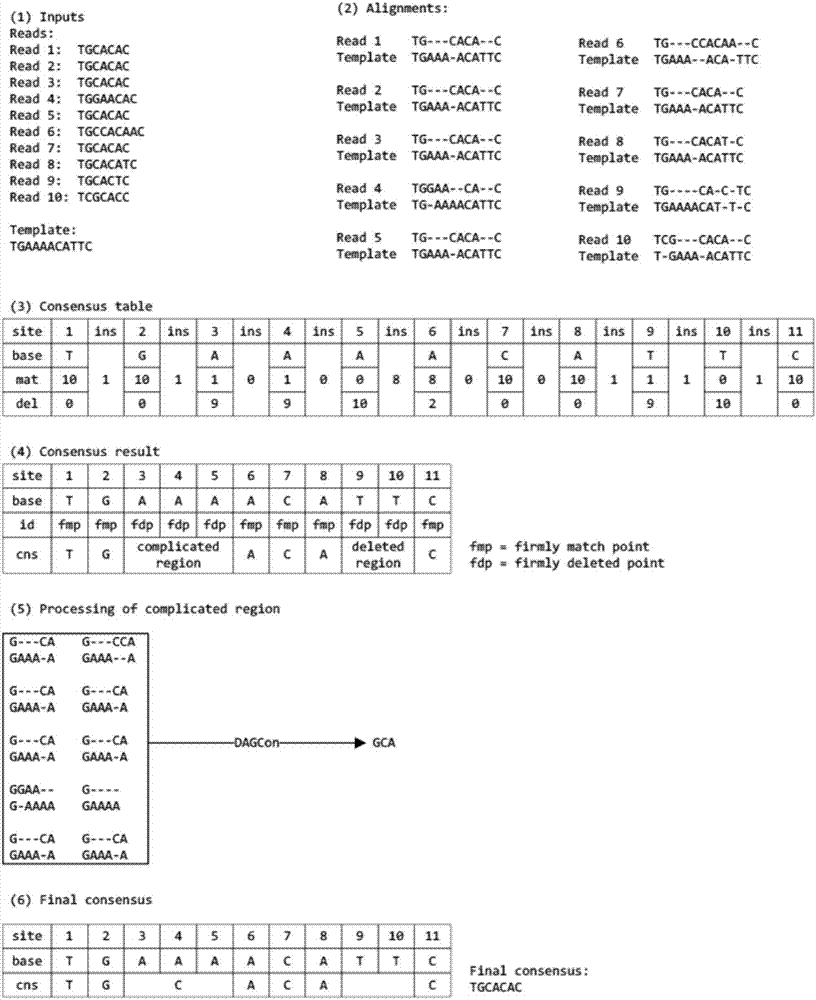
图1基于局部图的三代测序序列校正示意图
具体实施方式
实施例1
基于两两比对结果,基于局部图的三代测序序列校正方法一下实施方式完成大量三代测序序列校正,其详细设计过程如下:
两两比对结果过滤:按照1-1的规则过滤所有两两比对所有结果,过滤记录可以消除重复子序列和错误的read信息对校正结果的影响。过滤剩余两两比对结果进行分卷,每卷包含200000条测序序列的两两比对结果记录,并卷内的比对信息按照测序序列的编号进行排序,以方便每条测序序列记录集中,方便后续校正处理。具体方法如下:
基于两两比对方法(mecat2pw)的输出结果,每条mecat2pw的输出结果记录了两条测序序列的核心种子位置对信息,其记录是九列格式信息:第一列是测序序列a编号,第二列是测序序列b编号,第三列是a序列的正负链信息(正为0,负为1),第四列是b序列的正负链信息,第五列是核心位置对在a序列上的位置(pa),第六列是核心位置对在b序列上的位置(pb),第七列是核心位置对的全局投票得分,第八列是a序列的长度(la),第九列是b序列的长度(lb)。在校正模板序列t时,提取所有与t相关的两两比对结果记录:即第一列或第二列含有t编号的记录。对t相关两两比对结果过滤规则如下:
1-1过滤掉两条序列重叠长度小于90%的两条序列较小长度的记录:通过两个序列核心位置对信息,计算每条记录两个测序序列重叠长度,如果pa>=pb,左边的a和b的重叠长度ol=pb,否则ol=pa;如果la-pa>=lb-pb,右边的a和b的重叠长度or=lb-pb,否则or=la-pa;a和b序列重叠长度长度为o=ol+or,当o长度大于a的序列长度la的90%或者大于b的序列长度la的90%,该两两比对记录将被保留,否则该记录将被过滤掉(忽略)。按照上述重叠长度过滤原则过滤掉分析所有t序列相关的两两比对记录,获得t序列的过滤后记录,另外其中一条测序序列的长度小于5000,该记录将被过滤。
1-2优选200候选两两比对记录进入候选校正步骤:在重叠度过滤后,将t序列过滤后所有记录按照全局投票打分进行降序排序,取最高200个全局投票得分记录进入候选多重序列比对和t序列校正过程,即图1中输入序列(inputs)和模板序列(template)。
测序序列的压缩内存存储:通常一条测序序列与多条测序序列有重叠关系,需要从测序序列库中调出多条相关的序列。如果将所有测序序列存在磁盘中,读取大量零散相关的测序序列过程将导致cpu长期处于等待i/o状态而使cpu利用率低下。为了加速测序序列的读取速度和提高cpu利用率,所有测序序列进行压缩编码(2个bit编码一个核酸)加载到内存,并对每条lr存储位置索引,方便读取。按照上述方案,约160g的三代测序测序序列大概占用40g的内存,即测序碱基1/4的内存。
每卷测序序列的校正:每个含200000测序序列的分卷取出一条目标校正测序序列序列和它的所有两两关系记录,根据过滤规则1-2获取最多200条候选记录,根据两两比对记录,从内存中取出相关的所有测序序列,按照如下方法做多重序列比对和目标序列校正:
过滤后两两比对结果的多重序列比对
根据过滤后每条两两比对的核心位置对信息,从核心位置从右到左取出模板序列和输入序列的左边部分的两条序列进行局部两两比对:a.针对于pacbio数据,由于其主要错误是删除和插入,采用diff进行局部序列分析;b.针对nanopore数据,由于主要错误是错配,采用smith-waterman方法进行局部序列比对。详细比对过程如图1。
2-1分段完成两两局部序列比对:对取出两条左边序列进行按照500bp动态分段进行比对,先取出第一段500bp的两个序列,进行局部两两比对,比对完后从尾部向头回溯,寻找到有连续5个碱基匹配的的位置,为了保证500bp左边的序列起始正确性,从这个连续5bp匹配位置开始,再取500bp两个完成下一个段局部序列比对,重复上述过程,完成左边两条序列的局部比对过程。类似左边两条序列局部比对过程,从核心位置从左到右取出模板序列和输入序列的右边部分的两条序列进行局部两两比对,其分段比对过程与左右相似。
2-2局部比对的终止条件:1)完成两条序列局部比对;2)在每个500bp的分段序列中,错配,插入和删除碱基数小于500*0.2=100,如果遇到一段500bp中错误大于100bp,终止模板和输入序列的局部序列比对,过滤掉该两两比对记录。将所有200条模板序列相关的两两比对记录用2-1和2-2完成他们的两两局部比对过程,产生模板序列的多重序列比对结果(图1的第2步)。
基于多重序列比对的一致性校正操作统计分析
统计模板序列每个位置匹配(mat)、删除(del)、插入(ins)、替代(mismat)等四类校正操作的个数,获取模板序列每个位置一致性校正操作统计表:对于pacbio数据,由于diff比对中没有碱基替代比对,所以统计表中碱基校正操作只有三个类型:匹配、删除和插入(图1的第3步)。由于覆盖度小于6的位置,该位点保持原有模板序列比校正更可靠,所以当模板每个位置中mat+del<6时,该位点的mat值和del值设为0,当模板每个位置ins<6,ins值设为0,因为覆盖度小于6,该位点将不被校正。
基于校正操作统计表对校正操作分类
根据多重序列比对的一致性校正操作统计表,对每个模板碱基位进行分类,其分成三类:1)如果
高度一致区域的碱基位校正和复杂区域局部图碱基序列校正
如果该碱基为属于fmp碱基位,将保持该碱基序列不变,其中85%以上属于这样碱基校正位,对于剩下的碱基位,从该碱基为向左和向右寻找fmp碱基为,找到左边最近的fmp碱基位和右边最近fmp的碱基位,将两个碱基位之间的所有多重比对序列取出,建立局部图采用dagcon方法进行这部分复杂区域的校正:每个碱基是一个节点,每个边记录多重序列比对中走这个节点的次数,通过做经过次数做多的边,获取最终复杂区域的校正。
模板序列校正分割和去融合处理
为了保证校正后序列正确率大于97%,当模板序列没有6x的覆盖度时,输出校正后序列时将剪切该段序列,当剪切序列在中间时,将造成一条测序序列多段校正输出的现象。由于pacbio测序序列存在1%的融合现象(不相关两短序列测序成一条测序序列),在校正模板序列时,当校正某个位置,其输入序列没有超过1000bp的序列经过该位点左边和右边,该位置认为是融合位置,融合位置将被剪断。
重复上述过程,完成该分卷测序序列所有序列校正,其中很多覆盖多小于6x的测序序列,将被丢弃,不能完成其序列的校正。每卷校序列校正完毕后输出一个校正该卷序列文件。
并行运算和校正序列合并:采用共享内存方式完成每卷测序序列校正,所有测序序列的压缩内存将被共享,每10条序列分一个线程进行校正,动态分配线程完成每卷序列的校正。逐一完成所有分卷序列的校正,将校正后每卷序列,合并成一个文件,完成三代测序大数据的校正过程。
上述局部图的三代策略序列校正方法实现了计算资源消耗降低和准确度增加,具体的测量实例数据参加如下表1和表2。
数据中mecat方法即为上述具体实施方式部分的实施例1的方法。
表1:计算资源消耗
上述测试在相同计算节点(2.0ghzcpu和512gb内存)进行。数据大小指校正之后数据量,数据量越大效率越高。相对于其他方法,本发明的方法mecat数据校正利用率最高(最高可达3倍以上),处理效率最高(最高可达21倍以上)。
表2:校正序列准确度
随机挑选了每个数据集100m原始数据进行校正,对校正前后的数据准确度进行比较(序列通过dnadiff软件比对到参考基因组以评估准确度)。可以看出本发明的方法mecat准确度可达99%以上,比原始数据准确度要高并且优于同类其他软件。
- 还没有人留言评论。精彩留言会获得点赞!