测序数据结果分析方法和装置、测序文库构建和测序方法与流程
本发明涉及基因测序领域,具体而言,涉及一种测序数据结果分析方法和装置、测序文库构建和测序方法。
背景技术:
随着基因组学研究的深入,对特定区域序列进行突变鉴定的需求越来越大。序列突变分为两种类型:单碱基替换(singlenucleotidepolymorphism,简写snp,又称单核苷酸多态性)和插入缺失突变,这两种不同的突变类型在检测方法上也有差异。现有的snp突变的鉴定方法主要包括taqman探针法、snapshot法、massarray法、illuminabeadxpress法等,sanger直接测序法、高分辨率溶解曲线分析技术(highresolutionmelting,简写hrm)以及酶切法既可以鉴定snp突变,也可以鉴定插入缺失突变。下面对几种能够对snp突变进行鉴定的方法进行详细的介绍。
taqman探针法如图1所示,taqman探针法是针对染色体上的不同snp位点分别设计pcr引物和taqman探针,进行实时荧光pcr扩增。在探针的5’端和3’端分别标记一个报告荧光基团和一个淬灭荧光基团。当溶液中存在pcr产物时,该探针与模板退火,即产生了适合于核酸外切酶活性的底物,从而将探针5’端连接的荧光分子从探针上切割下来,破坏两荧光分子间的pret,发出荧光。通常用于少量snp位点分析。
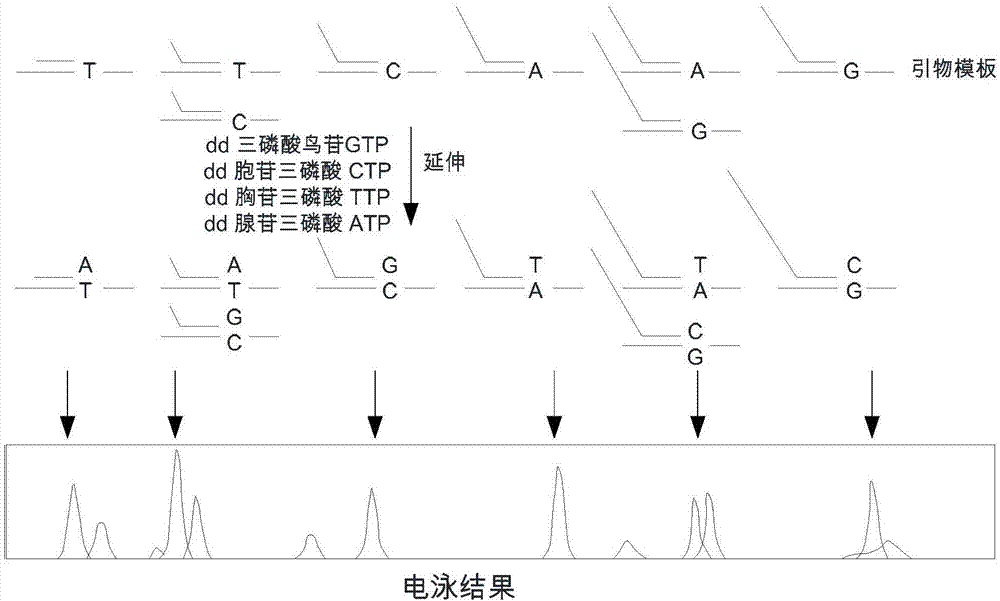
snapshot法如图2所示,snapshot法是基于荧光标记单碱基延伸原理的分型技术,也称小测序,主要针对中等通量的snp分型项目。在一个含有测序酶、四种荧光标记ddntp、紧临多态位点5’-端的不同长度延伸引物和pcr产物模板的反应体系中,引物延伸一个碱基即终止,经abi测序仪检测后,根据峰的移动位置确定该延伸产物对应的snp位点,根据峰的颜色可得知掺入的碱基种类,从而确定该样本的基因型。对于pcr产物模板可通过多重pcr反应体系来获得。通常用于10-30个snp位点分析。
hrm法如图3所示,hrm法是近几年兴起的snp研究工具,它通过实时监测升温过程中双链dna荧光染料与pcr扩增产物的结合情况,来判断是否存在snp,而且不同snp位点、是否是杂合子等都会影响熔解曲线的峰形,因此hrm分析能够有效区分不同snp位点与不同基因型。这种检测方法不受突变碱基位点与类型的局限,无需序列特异性探针,在pcr结束后直接运行高分辨率熔解,即可完成对样品基因型的分析。该方法无需设计探针,操作简便、快速,成本低,结果准确,并且实现了真正的闭管操作。hrm技术是结合饱和荧光染料、未标记探针和实时荧光定量pcr的一种新的检测基因突变与基因分型的分子诊断技术,利用dna双链结构解链一半的温度称为熔解温度(tm),不同序列的dna对应不同的tm值。dna中gc含量越高,tm值越高,gc含量与tm值成正比关系。sybrgreen等花菁类非特异性染料,可以直接插入双链的dna片段当中,并可激发荧光。由此就可以通过在特定温度区间内,通过荧光的强度变化显示出dna的复性及变性的这一过程,这个荧光信号随温度变化而变化形成的曲线就是熔解曲线。任何dna分子在加热变性时都会有自己熔解曲线的形状和位置,主要因为不同核酸分子的片段长短、gc含量、gc分布等是不同的。普通的熔解曲线,以0.5℃/循环,进行缓慢升温,将pcr扩增产物进行变性并实时检测荧光信号,不同的产物会形成不同的熔解曲线的特征峰,普通的realtime-pcr通过特征峰的特异性来判断扩增产物的特异性。
massarray法(也即massarray分子量阵列技术)是一种基因分析工具,通过引物延伸或切割反应与灵敏、可靠的maldi-tof-ms技术相结合,实现基因分型检测。基于massarray平台的iplexgold技术可以设计最高达40重的pcr反应和基因型检测,实验设计灵活,分型结果准确性高。根据应用需要,对数十到数百个snp位点进行数百至数千份样本检测时,massarray具有最佳的性价比,特别适合于对全基因组研究发现的结果进行验证,或者是有限数量的研究位点已经确定的情况。
illuminabeadxpress法是采用beadxpress系统进行批量snp位点检测,可以同时检测1-384个snp位点,往往用于基因组芯片结果确认,适合高通量检测。微珠芯片具有高密度、高重复性、高灵敏度、低上样量、定制灵活等特点,极高的集成密度,从而获得极高的检测筛选速度,在高通量筛选时可显著降低成本。
在上述方法基础上进行的突变鉴定方法,通量较低,有的只能进行单样本进行鉴定分析,成本较高;低频率的突变类型检出效率低;而且操作步骤繁琐,在得到测序下机数据之后还需要生物信息学背景才能分析数据等问题。
针对相关技术中的测序下机结果需要具有技术背景的科技工作人员进行人工辨别样本导致效率较低且成本较高的技术问题,目前尚未提出有效的解决方案。
技术实现要素:
本发明实施例提供了一种测序数据结果分析方法和装置、测序文库构建和测序方法,以至少解决相关技术中的测序下机结果需要具有技术背景的科技工作人员进行人工辨别样本导致效率较低且成本较高的技术问题。
根据本发明实施例的一个方面,提供了一种测序数据结果分析方法,包括:获取测序文库的测序数据结果,其中,测序文库包括混合的多个样本,每个样本对应一个标签序列组合,且不同样本对应的标签序列组合不同,其中,标签序列组合包括多个标签序列,测序数据结果为对混合的多个样本进行测序得到的测序片段集,测序片段集包括无序的多个测序片段;确定每个测序片段的标签序列组合;根据每个测序片段的标签序列组合确定其对应的样本。
进一步地,多个测序片段包括第一测序片段,确定第一测序片段的标签序列组合包括:在第一测序片段中提取所有的标签序列;将在第一测序片段中提取出的每个标签序列与多个编号已知的参考标签序列分别进行比对,以确定第一测序片段中每个标签序列对应的编号;将第一测序片段中所有标签序列的编号的组合确定为第一测序片段的标签序列组合的编号。
进一步地,在将在第一测序片段中提取出的每个标签序列与多个编号已知的参考标签序列分别进行比对之前,该方法还包括:获取预先存储的多个编号已知的参考标签序列。
进一步地,在测序数据结果为通过pariend测序方法获取的情况下,每个测序片段包括正读forwardread序列和反读reverseread序列,在第一测序片段中提取所有的标签序列包括:分别在第一测序片段的正读序列和反读序列中提取标签序列,其中,第一测序片段的标签序列组合包括从正读序列中提取的标签序列和从反读序列中提取的标签序列。
进一步地,在根据每个测序片段的标签序列组合确定其对应的样本之后,该方法还包括:获取每个样本的参考序列;在每个测序片段中提取样本的序列;将提取出的每个样本序列与对应样本的参考序列进行比对,以确定每个样本的突变信息。
进一步地,获取每个样本的参考序列包括:接收客户终端通过控件上传的每个样本的参考序列;在确定每个样本的突变信息之后,该方法还包括:将每个样本的突变信息反馈至客户终端。
进一步地,获取测序文库的测序数据结果包括:接收客户终端通过控件上传的测序数据结果;在根据每个测序片段的标签序列组合确定其对应的样本之后,该方法还包括:将多个测序片段与多个样本的对应关系反馈至客户终端。
根据本发明实施例的一个方面,提供了一种测序文库的构建和测序方法,包括:利用第一对引物对目标基因片段进行第一轮pcr反应,得到第一轮pcr产物;利用第二对引物对第一轮pcr产物进行第二轮pcr反应,得到样本,其中,第二对引物包括多个标签序列;对不同的目标基因片段分别执行上述的两轮pcr反应,得到多个样本,其中,不同的目标基因片段对应的标签序列组合不同,标签序列组合为第二对引物中包括的多个标签序列的组合;对测序文库执行测序,得到测序数据结果,其中,测序文库为混合的多个样本,测序数据结果为无序的多个测序片段;对测序数据结果执行本发明的测序数据结果分析方法,得到分析结果。
进一步地,测序文库中包括的多个样本是等量混合的。
进一步地,在执行第二轮pcr反应时采用的pcr板上具有多个孔,每个孔对应放置一个样本,每个孔的编号为放置的样本采用的标签序列组合的编号。
根据本发明实施例的一个方面,提供了一种试剂盒,该试剂盒包括:多个试剂孔,其中,每个试剂孔设置有对应的标签,每个试剂孔的标签用于指示向对应的试剂孔中放置的试剂中添加的标签序列。
进一步地,试剂盒包括一个标签板,标签板用于设置多个标签,标签板上的多个标签与多个试剂孔的位置一一对应。
根据本发明实施例的一个方面,提供了一种测序数据结果分析装置,包括:获取单元,用于获取测序文库的测序数据结果,其中,测序文库包括混合的多个样本,每个样本对应一个标签序列组合,且不同样本对应的标签序列组合不同,其中,标签序列组合包括多个标签序列,测序数据结果为对混合的多个样本进行测序得到的测序片段集,测序片段集包括无序的多个测序片段;第一确定单元,用于确定每个测序片段的标签序列组合;第二确定单元,用于根据每个测序片段的标签序列组合确定其对应的样本。
根据本发明实施例的另一方面,还提供了一种存储介质,该存储介质包括存储的程序,其中,在程序运行时控制存储介质所在设备执行本发明的测序数据结果分析方法。
根据本发明实施例的另一方面,还提供了一种处理器,该处理器用于运行程序,其中,程序运行时执行本发明的测序数据结果分析方法。
在本发明实施例中,通过获取测序文库的测序数据结果,其中,测序文库包括混合的多个样本,每个样本对应一个标签序列组合,且不同样本对应的标签序列组合不同,其中,标签序列组合包括多个标签序列,测序数据结果为对混合的多个样本进行测序得到的测序片段集,测序片段集包括无序的多个测序片段;确定每个测序片段的标签序列组合;根据每个测序片段的标签序列组合确定其对应的样本,解决了相关技术中的测序下机结果需要具有技术背景的科技工作人员进行人工辨别样本导致效率较低且成本较高的技术问题,进而实现了能够直接确定包括多个混合样本进行测序的下机数据中每个数据对应的样本的技术效果。
附图说明
此处所说明的附图用来提供对本发明的进一步理解,构成本申请的一部分,本发明的示意性实施例及其说明用于解释本发明,并不构成对本发明的不当限定。在附图中:
图1是现有技术的一种taqman探针法检测snp的原理示意图;
图2是现有技术的一种snapshot方法检测snp的原理示意图;
图3是现有技术的一种hrm技术检测snp的原理示意图;
图4是根据本发明实施例的一种可选的测序数据结果分析方法的流程图;
图5是根据本发明实施例的一种可选的barcode板的示意图;
图6是根据本发明实施例的一种可选的测序文库的构建和测序方法的流程图;
图7a是根据本发明实施例的一种可选的第一轮扩增原理的示意图;
图7b是根据本发明实施例的一种可选的第一轮扩增产物的示意图;
图8是根据本发明实施例的一种可选的测序数据结果分析装置的示意图。
具体实施方式
为了使本技术领域的人员更好地理解本发明方案,下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分的实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都应当属于本发明保护的范围。
需要说明的是,本发明的说明书和权利要求书及上述附图中的术语“第一”、“第二”等是用于区别类似的对象,而不必用于描述特定的顺序或先后次序。应该理解这样使用的数据在适当情况下可以互换,以便这里描述的本发明的实施例能够以除了在这里图示或描述的那些以外的顺序实施。此外,术语“包括”和“具有”以及他们的任何变形,意图在于覆盖不排他的包含,例如,包含了一系列步骤或单元的过程、方法、系统、产品或设备不必限于清楚地列出的那些步骤或单元,而是可包括没有清楚地列出的或对于这些过程、方法、产品或设备固有的其它步骤或单元。
本申请提供了一种测序数据结果分析方法的实施例。
图4是根据本发明实施例的一种可选的测序数据结果分析方法的流程图,如图4所示,该方法包括如下步骤:
步骤s101,获取测序文库的测序数据结果;
步骤s102,确定每个测序片段的标签序列组合;
步骤s103,根据每个测序片段的标签序列组合确定其对应的样本。
测序文库是预先构建的基因文库,测序文库中包括混合的多个样本,每个样本可以是通过对目标基因片段进行处理之后得到的,其中,目标基因片段是指需要进行试验(例如,突变鉴定试验)的基因片段,但是由于在测序时需要对多段基因的混合物进行测序,在得到的测序结果中无法区分出各个目标基因片段,因此,需要对每个目标基因片段进行处理,在目标基因片段的基础上,至少加入用于标记的标签序列得到样本,以使得每个样本能够与其它的样本区分开,因此,每个样本对应一个标签序列组合,且不同样本对应的标签序列组合不同,其中,标签序列组合包括多个标签序列,测序数据结果为对混合的多个样本进行测序得到的测序片段集,测序片段集包括无序的多个测序片段。
具体而言,测序文库的构建方式可以是采用通用的文库构建方法,例如,通过分别对多个目标基因片段(目标基因片段是指基因中的一个片段,多个目标基因片段可以是不同样本对象的基因中的同一段基因片段)进行两轮pcr扩增得到多个样本,其中,在进行pcr扩增时的引物中包括多个标签序列,标签序列是用于作为对目标基因片段的标记,测序文库中的每个样本包括多个标签序列,且每个样本的多个标签序列的组合都是不同的。
例如,多个标签序列包括r1、r2和f1、f2等等,每个目标基因片段通过两个标签序列进行标记,第一个目标基因片段通过r1、f1进行标记,第二个目标基因片段通过r1、f2进行标记,第三个目标基因片段通过r2、f1进行标记,第四个目标基因片段通过r2、f2进行标记,也即,每个目标基因片段的标签序列组合是不同的,上述对不同目标基因片段进行标记的标签序列组合进行举例仅作示例性的说明,不构成对本申请技术方案的限制。
在得到测序文库之后,可以将测序文库通过测序平台进行测序,得到测序下机数据,也即,测序数据结果,由于在测序时测序文库中的多个样本是混合的,因此,测序数据结果中的测序片段是无序的,每个测序片段对应于一个样本,但是具体的每个测序片段对应于哪一个样本则是未知的。因此,在获取测序文库的测序数据结果之后,确定每个测序片段的标签序列组合,根据每个测序片段的标签序列组合将多个测序片段与多个样本一一对应起来。
需要说明的是,该实施例提供的数据处理方法是通过软件执行的,具体而言,可以是通过终端设备上安装的程序或应用执行的。可选的,该实施例可以通过服务器执行,在获取测序文库的测序数据结果时,服务器可以通过接收客户终端通过控件(例如,网页上的输入框)上传的测序数据结果,服务器在根据每个测序片段的标签序列组合将多个测序片段与多个样本一一对应起来之后,将多个测序片段与多个样本的对应关系反馈至客户终端。
例如,该实施例提供的数据处理方法可以是通过服务器执行的,步骤s101获取测序数据结果的方式可以是服务器接收请求端(请求对测序数据结果进行数据处理的其它终端)通过网络发送的测序数据结果,在步骤s103服务器得到测序文库中每个样本与测序数据结果中每个测序片段之间的对应关系之后,服务器可以通过网络将对应关系发送至请求端。进一步地,服务器还可以通过网络获取请求端上传的每个目标基因片段(样本序列)的参考序列,在将每个测序片段中的样本序列与对应的参考序列进行比对之后将突变鉴定结果反馈至请求端。
上述服务器可以是通过网页接收请求端发送的数据,服务器可以是采用linux系统,并使用apeche软件,数据库可以采用mysql(例如,mariadb)数据库系统,网页可以采用perl、php或python等语言脚本搭建而成。例如,在服务器中的程序可以由perl脚本结合shell执行脚本组成,网站分析界面由php语言结合javascript语言搭建而成。
在一个可选的实施方式中,以测序数据结果中包括的多个测序片段之一(第一测序片段)为例,确定第一测序片段的标签序列组合的步骤包括:在第一测序片段中提取所有的标签序列;将在第一测序片段中提取出的每个标签序列与多个编号已知的参考标签序列分别进行比对,以确定第一测序片段中每个标签序列对应的编号;将第一测序片段中所有标签序列的编号的组合确定为第一测序片段的标签序列组合的编号。其中,每个测序片段中至少包括多个标签序列和样本序列(目标基因片段的测序结果)。
可选的,测序数据结果是以数据压缩包的格式存在的,在获取测序文库的测序数据结果时,对数据压缩包进行解压缩,可以得到多个测序片段。可选的,每个测序片段可以是以一个数据包的格式存在的,每个数据包中包括多段测序数据,每段测序数据可以是样本序列、一个标签序列或样本中包括的其它序列之一,在第一测序片段的数据包中提取出标签序列,并与多个编号已知的参考标签序列进行比对,其中,多个编号已知的参考标签序列是根据测序文库的构建方法确定的,将数据包中提取出的标签序列与在构建测序文库时采用的标签序列的库进行比对,确定数据包中提取出的标签序列的编号。
在将在第一测序片段中提取出的每个标签序列与多个编号已知的参考标签序列分别进行比对之前,需要获取预先存储的多个编号已知的参考标签序列。可选的,多个编号已知的参考标签序列可以是客户端上传的数据,也可以是服务器本地预先存储的数据。
可选的,在测序数据结果为通过pariend测序方法获取的情况下,每个测序片段包括正读forwardread序列和反读reverseread序列,在第一测序片段中提取所有的标签序列包括:分别在第一测序片段的正读序列和反读序列中提取标签序列,其中,第一测序片段的标签序列组合包括从正读序列中提取的标签序列和从反读序列中提取的标签序列。
根据每个测序片段的标签序列组合将多个测序片段与多个样本一一对应起来可以包括:根据第一测序片段中每个标签序列对应的编号确定第一测序片段对应的样本,对每个测序片段采用与第一测序片段相同的处理方式以确定每个测序片段对应的样本。
在根据每个测序片段的标签序列组合确定其对应的样本之后,该方法还可以包括:获取每个样本的参考序列;在每个测序片段中提取样本的序列;将提取出的每个样本序列与对应样本的参考序列进行比对,以确定每个样本的突变信息。
进一步地,获取每个样本的参考序列的步骤可以包括:接收客户终端通过控件上传的每个样本的参考序列;在确定每个样本的突变信息之后,该方法还包括:将每个样本的突变信息反馈至客户终端。
相似的,获取测序文库的测序数据结果的步骤可以包括:接收客户终端通过控件上传的测序数据结果;在根据每个测序片段的标签序列组合确定其对应的样本之后,该方法还包括:将多个测序片段与多个样本的对应关系反馈至客户终端。
该实施例通过获取测序文库的测序数据结果,其中,测序文库包括混合的多个样本,每个样本对应一个标签序列组合,且不同样本对应的标签序列组合不同,其中,标签序列组合包括多个标签序列,测序数据结果为对混合的多个样本进行测序得到的测序片段集,测序片段集包括无序的多个测序片段;确定每个测序片段的标签序列组合;根据每个测序片段的标签序列组合确定其对应的样本,解决了相关技术中的测序下机结果需要具有技术背景的科技工作人员进行人工辨别样本导致效率较低且成本较高的技术问题,进而实现了能够直接确定包括多个混合样本进行测序的下机数据中每个数据对应的样本的技术效果。
该实施例提供的数据处理方法能够提高突变的鉴定效率,无需生物信息背景即可获得高通量的分析结果,并且进一步地,在识别出每个样本对应的数据的基础上,将每个样本的测序结果与参考基因进行对比,得到突变鉴定结果,是一种新的高通量的突变鉴定方法,能够简化实验步骤。
作为一种可选的实施方式,描述该实施例提供的数据处理方法在一种可选的应用场景下的过程:
第一步,请求端将测序结果数据(压缩包的格式)上传(通过php)上传至服务器;
第二步,服务器调用服务器本地的解压软件(例如gunzip)对上传的数据进行解压缩;
第三步,服务器(利用perl脚本)提取每组pairend序列(双端测序序列)的barcode(标签序列)组合;
第四步,服务器(利用perl脚本)结合barcode组合确定测序文库中每个样品的编号:
具体的,测序文库可以通过如图5所示的试验孔板(或称barcode板,标签序列板)放置,在该试验孔板上每个孔对应一个标签序列组合,每个标签序列组合是不同的,如图5所示,标签序列包括f1~f12、r1~r8共20种标签,构成12×8=96个标签序列组合(f1r1、f2r1等等),每个标签序列组合对应孔板上的一个孔,每个孔中放置一个样品,在测序时,可以将试验孔板直接放置到测序的仪器中进行测序;
因此,服务器如果获取到所有的标签序列的数据库,已知上述f1~f12、r1~r8的20种标签,就可以将测序数据结果中的每个测序片段中的标签序列与已知的标签序列进行对比,确定出每个测序片段的标签序列组合(例如,f1r1、f2r1等等),在确定出每个测序片段的标签序列组合之后,可以确定出每个样本的编号(例如,样本编号也可以通过标签序列组合进行标示),并且也可以确定出测序数据结果中每个测序片段与试验孔板上每个孔的对应关系,可选的,所有的标签序列的数据库可以是请求端上传的,也可以是在服务器的数据库中预先存储的通用标签序列数据库;
第五步,服务器(利用本地的短序比对数据工具如bwa软件等)将每个样本序列与请求端上传的参考基因组序列进行比对:
具体的,在该步骤之前,服务器需要获取请求端上传的参考基因组序列,获取请求端上传的参考基因组序列的步骤在该步骤之前执行即可,也即,在服务器接收到参考基因组序列之后才可以执行该步骤;
第六步,服务器(利用perl脚本)分析、整理、统计每个样本的突变信息;
第七步,请求端(通过php)下载服务器分析的结果。
需要说明的是,在附图的流程图虽然示出了逻辑顺序,但是在某些情况下,可以以不同于此处的顺序执行所示出或描述的步骤。
本申请还提供了一种存储介质的实施例,该实施例的存储介质包括存储的程序,其中,在程序运行时控制存储介质所在设备执行本发明实施例的测序数据结果分析方法。
本申请还提供了一种处理器的实施例,该实施例的处理器用于运行程序,其中,程序运行时执行本发明实施例的测序数据结果分析方法。
本申请还提供了一种测序文库的构建和测序方法的实施例。
图6是根据本发明实施例的一种可选的测序文库的构建和测序方法的流程图,如图6所示,该方法包括如下步骤:
步骤s201,利用第一对引物对目标基因片段进行第一轮pcr反应,得到第一轮pcr产物:
可选的,可以通过将搭桥序列和目标片段扩增特异引物序列这两种序列整合在上、下游扩增的第一对引物上,通过第一对引物将不同的样本材料的目标区域片段(目标基因片段)扩增和富集。需要说明的是,在该步骤之前,还包括提取待测样品基因组dna(基因)的步骤,任何方法提取的dna样品均可,对样品量及浓度也无特殊要求。
如图7a所示,第一对引物包括第一上游引物序列与第一下游引物序列,将第一上游引物序列与第一下游引物序列和单个样品的dna序列的溶液混合,并进行pcr反应,获得第一pcr产物。具体的,第一上游引物序列或第一下游引物序列可以从5’端到3’端依次包含搭桥序列以及第一上游目标片段扩增特异引物序列或第一下游目标片段扩增特异引物序列,搭桥序列是与第二轮pcr的引物序列互补配对的序列,第一上游目标片段扩增特异引物序列与第一下游目标片段扩增特异引物序列分别与dna序列两条单链的3’末端互补,其中,目标片段扩增特异引物序列的长度可以取15~25bp,搭桥序列的长度可以取15~30bp。第一上游引物序列和第一下游引物序列中也可以加入标签序列,其加入的标签序列的长度可以随机,可以是1~50bp,再通过随机组合,可以一次性的区分大量的样本。
步骤s202,利用第二对引物对第一轮pcr产物进行第二轮pcr反应,得到样本:
第二对引物中包括多个标签序列,不同的目标基因片段对应的标签序列组合不同,标签序列组合为第二对引物中包括的多个标签序列的组合。第二轮pcr产物如图7b所示,每个样本的两端分别有一个标签序列。
可选的,可以将接头序列、测序引物、标签序列以及搭桥序列整合在通用的第二对引物上,第二轮引物为通用引物组合,已排列成固定的96孔板(如图5所示)或者384孔板的组合,做成混合的试剂盒。第二对引物包括第二上游引物序列和第二下游引物序列,将步骤s201中所得到pcr产物分别与第二上游引物序列和第二下游引物序列不同组合的溶液混合,或者,也可以直接使用配置好的混合试剂盒(例如上述的96孔或者384孔的第二轮引物混合试剂盒),进行pcr反应,获得第二pcr产物。具体的,第二上游引物序列5’端到3’端可以依次包含接头序列、测序引物序列、标签序列和第一上游引物序列中的搭桥序列,第二下游引物序列5’端到3’端可以依次包含接头序列、测序引物序列、标签序列和第一下游引物序列中的搭桥序列;每对笫二上游引物序列与第二下游引物序列的标签序列使得每个dna序列经过pcr反应后具有不同于其它dna序列的标签序列,标签序列的长度可以取1~20bp,利用pairend测序(双端测序),结合两侧的标签序列,可以同时区分1~+∞个样品的混合。
可选的,测序引物序列与标签序列之间可以引入4~10个碱基,提高测序所得标签序列的准确度。
步骤s203,对不同的目标基因片段分别执行上述的两轮pcr反应,得到多个样本:
需要说明的是,不同的目标基因片段对应的标签序列组合不同,标签序列组合为第二对引物中包括的多个标签序列的组合。
可选的,测序文库中包括的多个样本是等量混合的。对不同的目标样本进行两轮pcr反应,将等量混合的第二轮pcr产物进行纯化,可以得到测序文库,测序文库中包括混合的多个样本。
通过上述两轮扩增的方法可以节约合成引物的费用,同时利用该方法只需通过两步扩增即可完成测序文库的构建,不仅提高测序文库质量及建库效率,而且构建所得的测序文库因具有常规测序平台上所使用的接头序列以及测序引物序列,使得该测序文库能够利用常规上机测序所用的试剂进行高通量测序,而无需额外提供测序引物以及对所混入的phix文库的测序引物进行变更。
步骤s204,对测序文库执行测序,得到测序数据结果:
具体而言,测序文库为混合的多个样本,可以使用高通量测序平台对测序文库进行二代测序,得到测序下机数据,也即测序数据结果,测序数据结果为无序的多个测序片段。可选的,在得到所述测序文库后,并在进行高通量测序之前,还包括对所述测序文库进行质检的步骤。
步骤s205,对测序数据结果执行本发明的测序数据结果分析方法,得到分析结果。
应用该实施例提供的测序方法,通过两轮pcr,结合二代测序,将测序下机数据直接提供数据处理方法进行处理,即可以实现对混合多样本的测序数据结果进行自动识别的效果。需要说明的是,可以通过调节第二轮引物对组合的数目,来调节混合样本的数目。可选的,在对测序数据结果进行识别之后,还可以自动对每个样本的目标基因片段的突变进行鉴定分析。
可选的,优先使用步骤s202采用的引物序列中的标签序列区分样本,如果区分失败可以利用测序公司提供的标识序列区别来自不同样本的多个dna序列。
可选的,在执行第二轮pcr反应时采用的pcr板上具有多个孔,每个孔对应放置一个样本,每个孔的编号为放置的样本采用的标签序列组合的编号。
该实施例提供的测序方法通过一种高通量扩增子突变鉴定的建库方法及对应的分析软件鉴定突变信息,相比于其他类的突变鉴定方法,在建库和分析方法上有了新的改进,并且配套有自动解码及鉴定突变的软件,且成本更加便宜,建库所需时间更短,操作更加简单,单次能够区分混合样本的数目不受限制。该方法能自动对混合样本的测序的结果进行拆分,自动鉴定出单个材料的突变类型。该操作方法比较简单,无需任何生物信息学背景即可完成大量样本的鉴定工作。
本申请还提供了一种试剂盒的实施例,该试剂盒包括多个试剂孔,其中,每个试剂孔设置有对应的标签,每个试剂孔的标签用于指示向对应的试剂孔中放置的试剂中添加的标签序列。可选的,标签可以是布置在一个标签板上的,具体的,试剂盒可以包括一个标签板,标签板上可以通过粘贴、印刷等方式设置多个标签,多个标签与多个试剂孔的位置一一对应。
例如,该实施例提供的试剂盒可以包括如图5所示的barcode(标签)板,该barcode板上的每个标签与一个试剂孔对应,每个试剂孔的标签指示一个试剂孔的编号,每个试剂孔对应的试剂能够添加入两种标签序列,每个标签的编号包括所添加的两个标签序列的编号。通过如图5所示的20个编号不同的标签序列,可以标记最多96个试剂。
本申请还提供了一种测序数据结果分析装置的实施例。
图8是根据本发明实施例的一种可选的测序数据结果分析装置的示意图,如图8所示,该装置包括获取单元10,第一确定单元20和第二确定单元30,其中,获取单元,用于获取测序文库的测序数据结果,其中,测序文库包括混合的多个样本,每个样本对应一个标签序列组合,且不同样本对应的标签序列组合不同,其中,标签序列组合包括多个标签序列,测序数据结果为对混合的多个样本进行测序得到的测序片段集,测序片段集包括无序的多个测序片段;第一确定单元,用于确定每个测序片段的标签序列组合;第二确定单元,用于根据每个测序片段的标签序列组合确定其对应的样本。
该实施例通过获取单元,用于获取测序文库的测序数据结果,其中,测序文库包括混合的多个样本,每个样本对应一个标签序列组合,且不同样本对应的标签序列组合不同,其中,标签序列组合包括多个标签序列,测序数据结果为对混合的多个样本进行测序得到的测序片段集,测序片段集包括无序的多个测序片段;第一确定单元,用于确定每个测序片段的标签序列组合;第二确定单元,用于根据每个测序片段的标签序列组合确定其对应的样本,解决了相关技术中的测序下机结果需要具有技术背景的科技工作人员进行人工辨别样本导致效率较低且成本较高的技术问题,进而实现了能够直接确定包括多个混合样本进行测序的下机数据中每个数据对应的样本的技术效果。
作为上述实施例的一个可选实施方式,多个测序片段包括第一测序片段,第一确定单元包括:提取模块,用于在第一测序片段中提取所有的标签序列;比对模块,用于将提取出的多个标签序列分别与多个编号已知的参考标签序列进行比对,以确定第一测序片段中每个标签序列对应的编号。
上述的装置可以包括处理器和存储器,上述单元均可以作为程序单元存储在存储器中,由处理器执行存储在存储器中的上述程序单元来实现相应的功能。
存储器可能包括计算机可读介质中的非永久性存储器,随机存取存储器(ram)和/或非易失性内存等形式,如只读存储器(rom)或闪存(flashram),存储器包括至少一个存储芯片。
上述本申请实施例的顺序不代表实施例的优劣。
在本申请的上述实施例中,对各个实施例的描述都各有侧重,某个实施例中没有详述的部分,可以参见其他实施例的相关描述。在本申请所提供的几个实施例中,应该理解到,所揭露的技术内容,可通过其它的方式实现。
其中,以上所描述的装置实施例仅仅是示意性的,例如所述单元的划分,可以为一种逻辑功能划分,实际实现时可以有另外的划分方式,例如多个单元或组件可以结合或者可以集成到另一个系统,或一些特征可以忽略,或不执行。另一点,所显示或讨论的相互之间的耦合或直接耦合或通信连接可以是通过一些接口,单元或模块的间接耦合或通信连接,可以是电性或其它的形式。
另外,在本申请各个实施例中的各功能单元可以集成在一个处理单元中,也可以是各个单元单独物理存在,也可以两个或两个以上单元集成在一个单元中。上述集成的单元既可以采用硬件的形式实现,也可以采用软件功能单元的形式实现。
所述集成的单元如果以软件功能单元的形式实现并作为独立的产品销售或使用时,可以存储在一个计算机可读取存储介质中。基于这样的理解,本申请的技术方案本质上或者说对现有技术做出贡献的部分或者该技术方案的全部或部分可以以软件产品的形式体现出来,该计算机软件产品存储在一个存储介质中,包括若干指令用以使得一台计算机设备(可为个人计算机、服务器或者网络设备等)执行本申请各个实施例所述方法的全部或部分步骤。而前述的存储介质包括:u盘、只读存储器(rom,read-onlymemory)、随机存取存储器(ram,randomaccessmemory)、移动硬盘、磁碟或者光盘等各种可以存储程序代码的介质。
以上所述仅是本申请的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本申请原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本申请的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!