一种精准分析DMD基因结构变异断点的方法及系统与流程
本发明属于生物信息学领域,更具体地涉及一种精准分析dmd基因结构变异断点的方法及系统。
背景技术:
假肥大型肌营养不良症(duchennemusculardystrophy,dmd)是一种以进行性四肢近端骨骼肌萎缩无力、小腿腓肠肌假性肥大为特征,同时累及心肌和呼吸肌,部分患者伴有智力障碍的致死性x连锁隐性遗传病,其发病率为活产男婴的1/3500。患者从3~5岁起病,12岁左右失去行走能力,20多岁死亡。becker肌营养不良症(beckermusculardystrophy,bmd)与dmd互为等位基因异质性疾病,其发病率为1/30000,发病年龄比dmd晚,进展速度慢。目前国内外尚缺乏对本病特效的治疗方法,故通过对先证者的确诊、携带者的产前诊断和提供正确的遗传咨询以杜绝假肥大型肌营养不良症患儿的出生是降低本病发病率的关键措施。
dmd/bmd是由定位于xp21.2-21.3的编码抗肌萎缩蛋白(dystrophin)的dmd基因突变所致,大约有60~65%的dmd患儿是由于dmd基因外显子缺失所致,5~10%是由于dmd基因外显子重复所致,其余约30%是由于dmd基因微小突变导致。dmd基因外显子拷贝数变异发生呈现出显著的热点效应,44~55号,及3~22号外显子为最常发生缺失区域,3~11号,21~37号为最常发生重复区段。因此,探究dmd基因发生拷贝数变异的分子基础,我们需要对基因拷贝数变异(copynumbervariations,cnv)断点进行精确检测。
目前,现有技术对基因拷贝数变异的检测方法有:
1、细胞遗传学方法:如荧光原位杂交技术(fish)及dna纤维荧光原位杂交技术(stretched-fiberfish),该方法分辨率较低、且费时费力;
2、基于pcr技术的靶向分析方法:如多重扩增探针杂交技术(maph)、依赖于连接的多重探针扩增技术(mlpa)等,该方法分辨率较高、成本低廉,但普遍通量低;
3、基于芯片和测序的高通量分析方法:如基于芯片的比较基因组杂交技术(array-cgh)、基于芯片的探针技术等,可实现基因拷贝数差异检测,该方法通量大、分辨率高,但难以精准分析基因拷贝数变异的断点位置;
为解决现有技术的不足,开发一种精准分析dmd基因拷贝数变异断点的方法及系统,为探索疾病发生的dna遗传分子机制提供了有力的技术支持,是本领域急需解决的问题。
技术实现要素:
本发明的目的在于提供一种精准分析dmd基因拷贝数变异断点的方法及系统。
本发明所采取的技术方案是:
一种精准分析dmd基因拷贝数变异断点的方法,包括如下步骤:
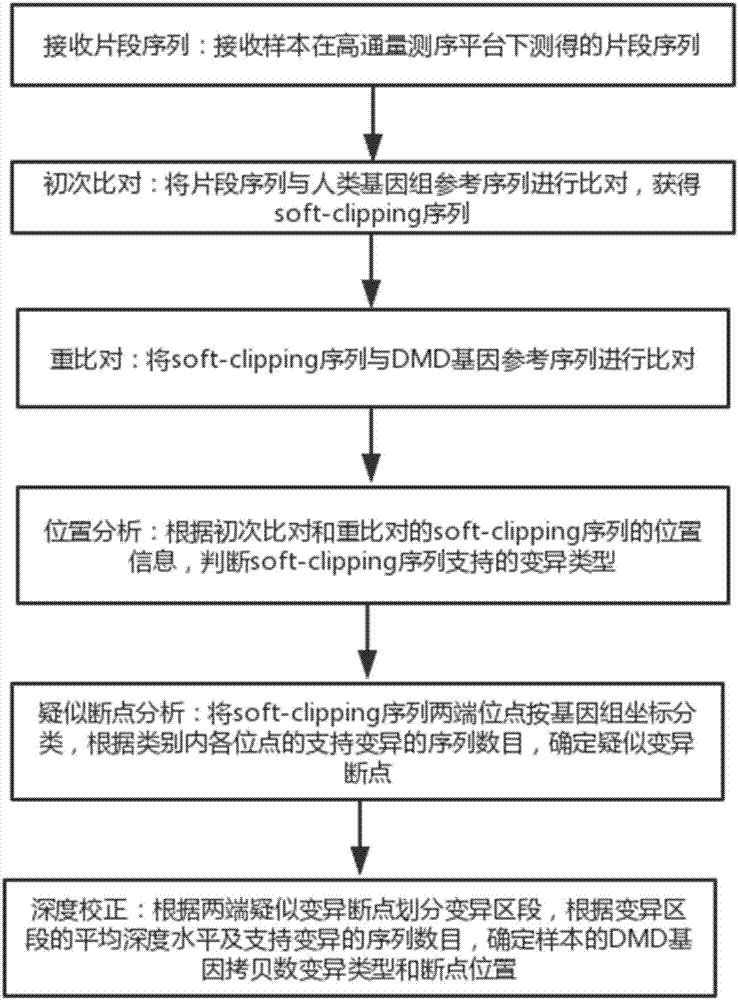
接收片段序列:接收样本在高通量测序平台下测得的片段序列;
初次比对:将片段序列与人类基因组参考序列进行比对,获得soft-clipping序列;
重比对:将soft-clipping序列与dmd基因参考序列进行比对;
位置分析:根据初次比对和重比对的soft-clipping序列的位置信息,判断soft-clipping序列支持的变异类型;
疑似断点分析:将soft-clipping序列两端位点按基因组坐标分类,根据类别内各位点的支持变异的序列数目,确定疑似变异断点;
深度校正:根据两端疑似变异断点划分变异区段,根据变异区段的平均深度水平及支持变异的序列数目,确定样本的dmd基因拷贝数变异类型和断点位置。
进一步的,所述方法还包括步骤:
样本质控:根据初次比对的结果,设定样本质控信息的阈值,筛选质控合格的样本。
进一步的,所述方法还包括步骤:
序列质控:重比对前,去除soft-clipping序列长度小于20bp的junctionreads。
进一步的,高通量测序平台为半导体测序平台。
进一步的,步骤”位置分析”中,判断soft-clipping序列支持的变异类型的方法具体如下:
对于3’端soft-clipping序列,如果重比对位置位于初次比对上的终止位置3’端,则该soft-clipping序列支持dmd片段缺失;如果重比对位置位于初次比对上的终止位置5’端,则该soft-clipping序列支持dmd片段重复;
对于5’端soft-clipping序列,如果重比对位置位于初次比对上的终止位置5’端,则该soft-clipping序列支持dmd片段缺失;如果重比对位置位于初次比对上的终止位置3’端,则该soft-clipping序列支持dmd片段重复。
进一步的,步骤”疑似断点分析”具体包括:
将soft-clipping序列两端位点的基因组坐标进行取模、排序和聚类;
在类别内统计各位点的junctionreads数目作为支持变异的序列数目;
支持变异的序列数目最多的位点为疑似变异断点。
进一步的,步骤”深度校正”具体包括:
根据两端疑似变异断点划分疑似变异区段;
统计疑似变异区段的平均深度水平及支持变异的序列数目,所述平均深度水平为疑似变异区段与疑似变异区段外的平均深度的比值;所述支持变异的序列数目为疑似变异区段的juntionreads数目;
确定样本的dmd基因拷贝数变异类型和断点位置,方法具体如下:
疑似变异区段的平均深度水平≤5%,判定为基因拷贝数纯合缺失;
疑似变异区段的平均深度水平>5%且≤50%,判定为基因拷贝数杂合缺失;
疑似变异区段的平均深度水平≥140%,判定为基因拷贝数重复;
上述判定的疑似变异区段存在交集情况下,纯合缺失的优先级高于杂合缺失;
确定变异类型后,支持变异的序列数目最多的疑似变异区段两端的位点则判定为样本的dmd基因拷贝数变异断点。
一种精准分析dmd基因拷贝数变异断点的系统,包括:
接收片段序列模块:用于接收样本在高通量测序平台下测得的片段序列;
初次比对模块:用于将片段序列与人类基因组参考序列进行比对,获得soft-clipping序列;
重比对模块:用于将soft-clipping序列与dmd基因参考序列进行比对;
位置分析模块:用于根据初次比对和重比对的soft-clipping序列的位置信息,判断soft-clipping序列支持的变异类型;
疑似断点分析模块:用于将soft-clipping序列两端位点按基因组坐标分类,根据类别内各位点的支持变异的序列数目,确定疑似变异断点;
深度校正模块:用于根据两端疑似变异断点划分变异区段,根据变异区段的平均深度水平及支持变异的序列数目,确定样本的dmd基因拷贝数变异类型和断点位置。
进一步的,所述系统还包括:
样本质控模块:用于根据初次比对的结果,设定样本质控信息的阈值,筛选质控合格的样本。
进一步的,所述系统还包括:
序列质控模块:用于重比对前,去除soft-clipping序列长度小于20bp的junctionreads。
本发明的有益效果是:
本发明开发了一种精准分析dmd基因拷贝数变异断点的方法和系统,包括接收片段序列、初次比对、重比对、位置分析、疑似断点分析、深度校正这些流程,能够精准分析出dmd基因拷贝数变异的类型和断点位置,检测误差率平均小于4bp,精准度高,稳定性好;此外,本发明可同时涵盖外显子和内含子的变异检测,解决了现有技术无法精准分析基因拷贝数变异断点的弊端,为实现快速并行的dmd断点检测服务和dmd结构变异分子机制研究提供的技术支持。
附图说明
图1:基因发生结构缺失、重复时出现junctionreads的情况示意图;
图2:本发明的精准分析dmd基因拷贝数变异断点的方法的流程图;
图3:本发明采用位置分析基因缺失变异的判定原理,图中,蓝色粗实心箭头为初次比对序列匹配部分;倾斜的灰色细实心箭头表示初次比对未匹配的soft-clipping序列,水平的灰色细实心箭头表示重比对的位置;
图4:本发明采用位置分析基因重复变异的判定原理;图中,蓝色粗实心箭头为初次比对序列匹配部分;倾斜的灰色细实心箭头表示初次比对未匹配的soft-clipping序列,水平的灰色细实心箭头表示重比对的位置;
图5:本发明的精准分析dmd基因拷贝数变异断点的系统的流程图;
图6:本发明实施例1的步骤流程图。
具体实施方式
本发明涉及较多的生物信息学领域术语,未进一步解释的术语以本领域的常规解释为准,不作赘述。
本发明术语“soft-clipping序列”是生物信息学领域的常规术语,是指片段序列与参考序列在比对过程中出现终止,未比对上目标区域而被剪切掉的片段,称为soft-clipping序列。
本发明术语“junctionreads”是生物信息学领域的常规术语,是指在比对时横跨两个区段的一段测序序列;对于基因组发生片段缺失或重复的情况下,junctionreads覆盖了缺失或重复变异断点,其示意图如图1所示。
参照图2,一种精准分析dmd基因拷贝数变异断点的方法,包括如下步骤:
接收片段序列:接收样本在高通量测序平台下测得的片段序列;
初次比对:将片段序列与人类基因组参考序列进行比对,获得soft-clipping序列;
重比对:将soft-clipping序列与dmd基因参考序列进行比对;
位置分析:根据初次比对和重比对的soft-clipping序列的位置信息,判断soft-clipping序列支持的变异类型;
疑似断点分析:将soft-clipping序列两端位点按基因组坐标分类,根据类别内各位点的支持变异的序列数目,确定疑似变异断点;
深度校正:根据两端疑似变异断点划分变异区段,根据变异区段的平均深度水平及支持变异的序列数目,确定样本的dmd基因拷贝数变异类型和断点位置。
其中,dmd基因参考序列优选为dmd基因上下游3~10kb参考基因组序列,进一步优选为dmd基因上下游5kb参考基因组序列。
进一步作为优选的实施方式,所述方法还包括步骤:
样本质控:根据初次比对的结果,设定样本质控信息的阈值,筛选质控合格的样本。
其中,质控合格的样本优选为满足以下条件:q20碱基比例≥75%,比对到目标区域的reads比例≥80%,50x覆盖的目标区域比例≥90%,目标区域的平均深度≥300x。
进一步作为优选的实施方式,所述方法还包括步骤:
序列质控:重比对前,去除soft-clipping序列长度小于20bp的junctionreads。
作为优选的实施方式,高通量测序平台为半导体测序平台。
作为优选的实施方式,步骤”位置分析”中,判断soft-clipping序列支持的变异类型的方法具体如下:
对于3’端soft-clipping序列,如果重比对位置位于初次比对上的终止位置3’端,则该soft-clipping序列支持dmd片段缺失;如果重比对位置位于初次比对上的终止位置5’端,则该soft-clipping序列支持dmd片段重复;
对于5’端soft-clipping序列,如果重比对位置位于初次比对上的终止位置5’端,则该soft-clipping序列支持dmd片段缺失;如果重比对位置位于初次比对上的终止位置3’端,则该soft-clipping序列支持dmd片段重复。
其中,以上判断方法原理示意图如图3和图4所示。
作为优选的实施方式,步骤”疑似断点分析”具体包括:
将soft-clipping序列两端位点的基因组坐标进行取模、排序和聚类;
在类别内统计各位点的junctionreads数目作为支持变异的序列数目;
支持变异的序列数目最多的位点为疑似变异断点。
其中,支持变异的序列数目也可以是soft-clipping序列数目,因为soft-clipping序列来自于junctionreads,此外,类别内支持变异的序列数目越多的位点,表示测序数据对该位点的可信度越高。
作为优选的实施方式,步骤”深度校正”具体包括:
根据两端疑似变异断点划分疑似变异区段;
统计疑似变异区段的平均深度水平及支持变异的序列数目,所述平均深度水平为疑似变异区段与疑似变异区段外的平均深度的比值;所述支持变异的序列数目为疑似变异区段的juntionreads数目;
确定样本的dmd基因拷贝数变异类型和断点位置,方法具体如下:
疑似变异区段的平均深度水平≤5%,判定为基因拷贝数纯合缺失;
疑似变异区段的平均深度水平>5%且≤50%,判定为基因拷贝数杂合缺失;
疑似变异区段的平均深度水平≥140%,判定为基因拷贝数重复;
上述判定的疑似变异区段存在交集情况下,纯合缺失的优先级高于杂合缺失;
确定变异类型后,支持变异的序列数目最多的疑似变异区段两端的位点则判定为样本的dmd基因拷贝数变异断点。
参照图5,一种精准分析dmd基因拷贝数变异断点的系统,包括:
接收片段序列模块:用于接收样本在高通量测序平台下测得的片段序列;
初次比对模块:用于将片段序列与人类基因组参考序列进行比对,获得soft-clipping序列;
重比对模块:用于将soft-clipping序列与dmd基因参考序列进行比对;
位置分析模块:用于根据初次比对和重比对的soft-clipping序列的位置信息,判断soft-clipping序列支持的变异类型;
疑似断点分析模块:用于将soft-clipping序列两端位点按基因组坐标分类,根据类别内各位点的支持变异的序列数目,确定疑似变异断点;
深度校正模块:用于根据两端疑似变异断点划分变异区段,根据变异区段的平均深度水平及支持变异的序列数目,确定样本的dmd基因拷贝数变异类型和断点位置。
作为优选的实施方式,所述系统还包括:
样本质控模块:用于根据初次比对的结果,设定样本质控信息的阈值,筛选质控合格的样本。
作为优选的实施方式,所述系统还包括:
序列质控模块:用于重比对前,去除soft-clipping序列长度小于20bp的junctionreads。
以下结合附图和具体实施例对本发明方案作进一步详细描述,以下实施例用于说明本发明,但不用来限制本发明的范围。
本发明实施例涉及的样本均取自中山大学附属第一医院神经科实验室,dmd患者/携带者样本已通过常规分子诊断确诊。
实施例1
图6为本发明的一个具体实施方式流程图,更具体地,包括如下步骤:
(1)接收片段序列
接收样本在半导体测序平台下测得的片段序列。
(2)初次比对
采用lifetechnologies公司提供的tmap套件,将片段序列与人类基因组参考序列ncbibuild37/hg19(获自http://www.ncbi.nlm.nih.gov/)进行比对,获得soft-clipping序列。
(3)样本质控
利用samtools工具(http://www.htslib.org/),将初次比对结果进行排序和索引建立,提取质控信息,根据每个样本的质控信息文件进行筛选,筛选质控合格的样本,其中,质控合格的样本满足以下条件:q20碱基比例≥75%,比对到目标区域的reads比例≥80%,50x覆盖的目标区域比例≥90%,目标区域的平均深度≥300x。
(4)序列质控
重比对前,去除soft-clipping序列长度小于20bp的junctionreads。
(5)重比对
采用tmap套件,将质控合格的soft-clipping序列与dmd基因上下游5kb参考基因组序列进行比对。
(6)位置分析
整合初次比对和重比对的soft-clipping序列两端位点的位置信息,所述位置信息包括基因组坐标和比对方向,按照下述流程判断每条soft-clipping序列支持的变异类型:
对于3’端soft-clipping序列,如果重比对位置位于初次比对上的终止位置3’端,则该soft-clipping序列支持dmd片段缺失;如果重比对位置位于初次比对上的终止位置5’端,则该soft-clipping序列支持dmd片段重复;
对于5’端soft-clipping序列,如果重比对位置位于初次比对上的终止位置5’端,则该soft-clipping序列支持dmd片段缺失;如果重比对位置位于初次比对上的终止位置3’端,则该soft-clipping序列支持dmd片段重复。
(7)疑似断点分析
将所有soft-clipping序列两端位点的基因组坐标汇总,对100取模,随后进行排序和聚类,在类别内统计各位点的junctionreads数目,类别内junctionreads数目最多的位点为疑似变异断点。
(8)深度校正
根据两端疑似变异断点划分疑似变异区段,统计疑似变异区段的平均深度水平及支持变异的序列数目,所述平均深度水平为疑似变异区段与疑似变异区段外的平均深度的比值;所述支持变异的序列数目为疑似变异区段的junctionreads数目;以此确定样本的dmd基因拷贝数变异类型和断点位置,具体判断方法如下:疑似变异区段的平均深度水平≤5%,判定为基因拷贝数纯合缺失;疑似变异区段的平均深度水平>5%且≤50%,判定为基因拷贝数杂合缺失;疑似变异区段的平均深度水平≥140%,判定为基因拷贝数重复;上述判定的疑似变异区段存在交集情况下,纯合缺失的优先级高于杂合缺失;确定变异类型后,支持变异的序列数目最多的疑似变异区段两端的位点则判定为样本的dmd基因拷贝数变异断点。
实际样本检测结果
针对7例样本的dmd基因区段,采用本领域常规的dna探针进行杂交捕获,采用ionpitmhi-qtmsequencing200kit(lifetechnologies,a26772)进行半导体测序,获得相应的片段序列,应用本发明提供的精准分析dmd基因拷贝数变异断点的方法和系统,并且采用sanger测序进行验证评价,每个样本重复分析3次,结果一致,如表1所示。
表1、7例样本的dmd基因拷贝数变异断点分析结果
如表1所示,对7例检测结果进行了sanger验证,结果显示通本发明分析的断点位置与sanger验证结果平均误差<4bp,其中9个断点(共14个断点)位置完全相同,说明本发明分析方法和系统的检测误差率小,精准度高,稳定性好。
深度校正结果
表2示出本发明提供的精准分析dmd基因拷贝数变异断点的方法和系统在处理10例样本采用深度校正前后的情况。
表2、10例样本的深度校正情况
注:1、平均深度水平为疑似变异区段与疑似变异区段外的平均深度的比值;
2、下划线标记的位点为样本的疑似变异断点位置。
如表2所示,以上10例样本经过深度校正,精准确定dmd基因变异类型和断点位置,排除疑似位点中的假阳性结果,不采用深度校正,无法准确获取样本的变异断点信息。其中,样本13根据判断标准,既存在cnv纯合缺失,又存在cnv杂合缺失,由于疑似变异区段存在交集,纯合缺失的优先级高于杂合缺失,因此判定为cnv纯合缺失。
- 还没有人留言评论。精彩留言会获得点赞!