评估和校验三代测序的序列组装结果的方法与装置与流程
本发明属于基因组测序领域,涉及一种评估和校验三代测序的序列组装结果的方法与装置。
背景技术:
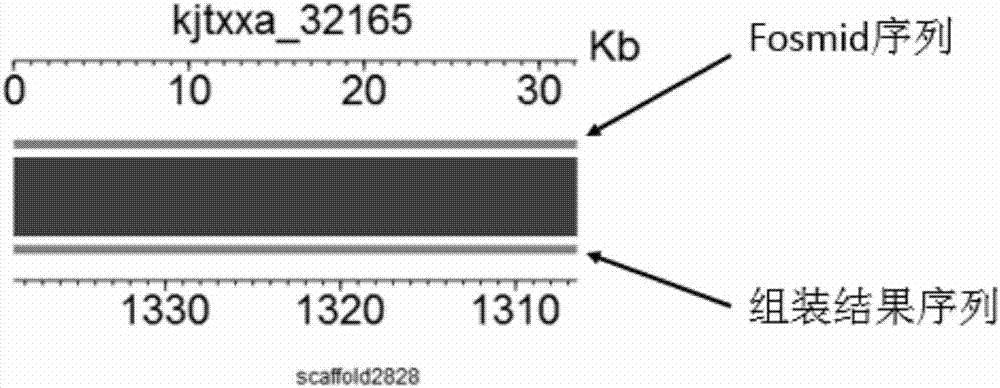
:重叠群(contig)是由序列(reads)通过对重叠(overlap)区域拼接组装成没有间隙(gap)的序列段;骨架序列(scaffold)通过双末端位置信息确定出的重叠群(contig)排列,中间有gap。把组装出的contigs或scaffolds从大到小排列,当其累计长度刚刚超过全部组装序列总长度50%时,最后一个contig或scaffold的大小即为n50的大小,n50对评价组装序列的连续性、完整性有重要意义;n70和n90的计算方法与n50类似,只是百分数变为70%或90%。二代测序由于读长的限制(一般为50bp-300bp),采用拼接的两种算法olc和dbg都无法跨过比较长的重复区域,在组装时遇到这些重复区域都会被断开。虽然可以采用不同梯度大片段(比如2k,5k,10k,20k,40k等)数据利用双末端位置的比对关系把两个重叠群连接起来拼接成骨架序列(scaffold),但contign50长度还是不长(一般为1k-70k)。三代测序—pacbio单分子实时测序(smrt)技术由于具有超长读长(平均读长一般在8k-13k)的特点,能对高重复序列、转座子区域与高度变异区域等基因组复杂区域进行高水平组装,使得重叠群(contig)n50和骨架序列(scaffold)n50长度更长,组装结果更完整准确,随着三代测序成本越来越低,三代组装基因组项目也越来越多。目前三代组装软件主要有pbcr、falcon、mecat、canu、hgap等,这些软件都包含自纠错和纠错后序列自组装的功能。由于三代序列平均错误率高达15%,故这些软件都需要先进行自纠错,再利用纠错后序列进行组装,最后得到组装结果,由于组装结果可能存在一定的单碱基错误或结构变异,所以后续需要用三代原始序列进行polish纠错,及用二代序列进行pilon纠错,得到最终的三代组装结果,三代组装的主要过程如图1所示。在得到组装结果后,我们会通过不同的方法对组装结果的质量进行评价。比如:(一)利用同一个个体的bac/fosmid序列(或者同种物种的bac/fosmid序列),通过与基因组序列比对,检验基因组常染色质覆盖度,如图2所示,上面是一段fosmid序列,下面是我们的组装结果序列,它们比对的效果非常好,证明这段fosmid序列已经被组装出来且效果非常好。(二)利用已有的est序列,通过与基因组序列比对,检验基因区的覆盖度。(三)单碱基覆盖深度评估,二代序列比对到三代组装结果并统计三代组装结果每个碱基的覆盖深度。如图3所示,二代序列的平均覆盖深度80x,x轴代表不同区间的覆盖深度,y轴代表不同区间覆盖深度对应的比例,从此图来看,小于10x覆盖深度的比例越低,反应组装结果单碱基的组装质量越高(四)gc含量分布分析。如图4所示,横坐标是gc含量,纵坐标是平均深度。二代序列比对到三代组装结果并统计三代组装结果每个碱基的覆盖深度,以10kb为窗口无重复进行计算。根据这个图我们可以分析这个物种的gc含量,可以对该样品是否有外源dna污染进行判断。另外也可以看出我们部分区域的组装质量效果。图4(b)结果显示组装结果gc含量深度分布正常,但图4(a)所示,有部分低深度覆盖区域,造成此现象的可能有两个原因,一是三代序列在这些区域覆盖深度较低,导致组装结果存在一定的碱基错误或缺失,虽然经过三代polish纠错和二代pilon纠错,但并没有纠正过来;二是这部分区域组装是准确的,三代在这部分区域覆盖深度很高,但二代在这部分区域覆盖深度较低,可能由于测序错误导致比对不到这部分区域,或者这部分区域没有测到或测到的部分较低。技术实现要素:为了有效解决三代组装结果的部分区域在二代序列中覆盖深度较低是何种原因导致的,本发明提供了一种评估和校验三代测序的序列组装结果的方法与装置。本发明所提供的评估三代测序的序列组装结果的方法,大致包括如下步骤:(1)将同一样本的二代测序序列与三代测序的序列组装结果进行对比。(2)根据步骤(1)的比对结果,从所述三代测序的序列组装结果中挑选出在所述二代测序序列中平均覆盖深度低的区域,然后将所选的每一个区域均在所述三代测序的序列组装结果中进行延伸,从而获取若干个延伸后序列。(3)将三代测序序列与步骤(2)获得的每一个延伸后序列进行单独比对。(4)根据步骤(3)的比对结果,统计步骤(2)中所选的每一个区域(即每一个二代低覆盖深度区域)在所述三代测序序列中的平均覆盖深度。(5)根据步骤(4)的统计结果,确定步骤(2)中所选的每一个区域的组装质量的高低,进而实现对所述三代测序的序列组装结果的评估。具体的,所述方法包括如下步骤:(1)将同一样本的二代测序序列与三代测序的序列组装结果进行对比(可以使用比对软件bwa或soapaligner等软件完成),统计所述三代测序的序列组装结果中每个碱基在所述二代测序序列中的覆盖深度(可以使用soapcoverage软件进行),进而以1-5kb(具体如1kb)为窗口,计算得到所述三代测序的序列组装结果中每个窗口区域在所述二代测序序列中的平均覆盖深度。(2)根据步骤(1)的结果,从所述三代测序的序列组装结果中挑选出在所述二代测序序列中平均覆盖深度低的全部窗口区域,然后将所选的每一个窗口区域均在所述三代测序的序列组装结果中向前后各延伸10-40kb(具体如30kb),从而获取若干个延伸后序列。(3)将三代测序序列与步骤(2)获得的每一个延伸后序列进行单独比对(比对软件可以用bwa)。(4)根据步骤(3)的比对结果,统计步骤(2)中所选的每一个窗口区域(即原1-5kb二代低覆盖深度区域)在所述三代测序序列中的平均覆盖深度。(5)根据步骤(4)的统计结果,按照如下对步骤(2)中所选的每一个窗口区域(即原1-5kb二代低覆盖深度区域)进行组装质量高低的标记,进而评估所述三代测序的序列组装结果的整体组装质量:如果步骤(2)中所选的某一个窗口区域a在所述三代测序序列中的平均覆盖深度小于等于5x,则将所述窗口区域a标记为“组装质量相对较低的区域”;如果步骤(2)中所选的某一个窗口区域b在所述三代测序序列中的平均覆盖深度大于5x,则将所述窗口区域b标记为“组装质量相对较高的区域”。在所述方法的步骤(5)中,具体是按照如下评估所述三代测序的序列组装结果的整体组装质量的:所标记的所述“组装质量相对较高的区域”的数量与所述“组装质量相对较低的区域”和所述“组装质量相对较高的区域”两者总数量的比值越大,则所述三代测序的序列组装结果的整体组装质量越高。其中,步骤(2)中所选的窗口区域中如果连续2个或多个同时被标记为“组装质量相对较低的区域”,则将它们合并记成一个“组装质量相对较低的区域”;如果连续2个或多个同时被标记为“组装质量相对较高的区域”,则将它们合并记成一个“组装质量相对较高的区域”。在所述方法的步骤(1)中,所述二代测序序列为二代高通量测序所得的原始数据经过过滤处理后的序列(去除了接头以及低质量碱基)。在本发明的一个实施例中,所述样本为玉米基因组,所述二代测序序列具体为玉米基因组的hiseq2500平台250pe测序所得原始序列过滤掉接头以及低质量碱基后的部分。进一步地,本发明中由原始数据到有效数据过滤需要经过三步处理:1)过滤接头:测序read匹配上adapter序列的50%或者以上则删除整条reads;2)过滤低质量数据:如果测序read中质量值低于20的碱基占整条read的10%-50%(具体如20%)或者以上则删除整条read;3)去n:如果测序reads中n含量占整条read的1%-10%(具体如2%)或者以上,则删除整条read。其中,n表示测序未测出的碱基。在所述方法的步骤(3)中,所述三代测序序列为未纠错序列或者已经自纠错后的序列。其中,三代未纠错序列有两种,一种是pacbiorsii机器测序得到的subreads序列,另一种是sequal机器测序得到的bam格式数据转换成fasta格式的序列;自纠错后的序列指的是三代原始数据自纠错后得到的序列。在本发明的一个实施例中,所述样本为玉米基因组,所述三代测序序列具体为玉米基因组的pacbio单分子实时测序(smrt)所得原始序列经过自纠错后的序列。在所述方法的步骤(2)中,所述平均覆盖深度低指的是平均覆盖深度低于“低深度定义阀值”,为如下任一:(a1)当所述二代测序的平均覆盖深度为30x时,所述“低深度定义阀值”为3x。(a2)当所述二代测序的平均覆盖深度大于30x且小于等于50x时,所述“低深度定义阀值”为4-5x。(a3)当所述二代测序的平均覆盖深度大于50x且小于等于70x时,所述“低深度定义阀值”为6-8x。(a4)当所述二代测序的平均覆盖深度大于70x时,所述“低深度定义阀值”为9-10x。在本发明的一个实施例中,所述样本为玉米基因组,所述二代测序的平均覆盖深度为60x,所述“低深度定义阀值”为6x。在所述方法的步骤(2)中,所述“将所选的每一个窗口区域(即原1kb二代低覆盖深度区域)均在所述三代测序的序列组装结果中向前后各延伸30kb”,若所选窗口区域在其所在的骨架序列(scaffold)中,其前和/或后不足30kb,则延伸至存在的部分结束即可。即:如果向前后各延伸30kb,获取延伸后的序列,每个延伸后区域长度共计61kb;scaffold前后如果不足30k的,取有的部分,比如窗口是5-6k区域,往前只取0-5k,往后去6-36k,即1-36kb;scaffold后面不足的,从窗口末尾取到最后一个碱基。由于三代序列的长度范围为几百bp到几十kb,而这些低深度覆盖区域可能低至几十bp,所以本发明对低深度覆盖区域前后各延伸了30kb,以保证三代序列能和这些区域进行比对。在所述方法的步骤(1)中,所述二代测序的平均数据量达到基因组大小的30x以上,最好为50x以上。在本发明的一个实施例中,所述样本为玉米基因组,所述二代测序的平均数据量具体为玉米基因组大小的60x。在所述方法的步骤(1)中,进行所述比对时允许的错配碱基数最好小于等于2。在所述方法中,所述三代测序为pacbio单分子实时测序(smrt);所述三代测序的平均数据量最好在基因组大小的50x以上。在本发明的一个实施例中,所述样本为玉米基因组,所述三代测序的平均数据量具体为玉米基因组大小的80x。本发明所提供的三代测序的序列组装结果的评估系统(装置),包括数据处理装置a、数据处理装置b、数据处理装置c、数据处理装置d和数据处理装置e。所述数据处理装置a内设模块a1、模块a2、模块a3;所述模块a1能够对二代测序序列与三代测序的序列组装结果进行对比;所述模块a2能够根据所述模块a1比对的结果,统计所述三代测序的序列组装结果中每个碱基在所述二代测序序列中的覆盖深度;所述模块a3能够根据所述模块a2的统计结果,以1-5kb(具体如1kb)为窗口,计算得到所述三代测序的序列组装结果中每个窗口区域在所述二代测序序列中的平均覆盖深度。在本发明中,所述模块a1具体为bwa软件或soapaligner软件;所述模块a2具体为soapcoverage软件。所述数据处理装置b内设模块b1和模块b2;所述模块b1能够根据所述数据处理装置a获得的结果,从所述三代测序的序列组装结果中挑选出在所述二代测序序列中平均覆盖深度低的全部窗口区域;所述模块b2能够将所述模块b1所选的每一个窗口区域均在所述三代测序的序列组装结果中向前后各延伸10-40kb(具体如30kb),从而获取若干个延伸后序列。所述数据处理装置c内设模块c1;所述模块c1能够将三代测序序列与所述数据处理装置b获得的每一个延伸后序列进行单独比对。在本发明中,所述模块c1具体为bwa软件。所述数据处理装置d内设模块d1;所述模块d1能够根据所述数据处理装置c获得的比对结果,统计所述数据处理装置b中所述模块b1所选的每一个窗口区域(即原1kb二代低覆盖深度区域)在所述三代测序序列中的平均覆盖深度。所述数据处理装置e内设模块e1和模块e2;所述模块e1能够根据所述数据处理装置d获得的统计结果,按照如下对所述数据处理装置b中所述模块b1所选的每一个窗口区域(即原1kb二代低覆盖深度区域)进行组装质量高低的标记:如果某一个窗口区域a在所述三代测序序列中的平均覆盖深度小于等于5x,则将所述窗口区域a标记为“组装质量相对较低的区域”;如果某一个窗口区域b在所述三代测序序列中的平均覆盖深度大于5x,则将所述窗口区域b标记为“组装质量相对较高的区域”;所述模块e2能够根据所述模块e1的标记结果统计计算所述数据处理装置b中所述模块b1所标记的所述“组装质量相对较高的区域”的数量与所述“组装质量相对较低的区域”和所述“组装质量相对较高的区域”两者总数量的比值;其中,所述模块b1所选窗口区域中如果连续2个或多个同时被标记为“组装质量相对较低的区域”,则将它们合并记成一个“组装质量相对较低的区域”;如果连续2个或多个同时被标记为“组装质量相对较高的区域”,则将它们合并记成一个“组装质量相对较高的区域”。所述方法或所述系统在如下任一中的应用也属于本发明的保护范围:(a)评估三代测序的序列组装结果;(b)筛选并标注三代测序的序列组装结果中质量差的区域。实验证明,利用本发明所提供的评估和校验三代测序的序列组装结果的方法,可以成功筛选出三代组装结果中质量不是太高的区域,并将其标注出来。这样在后续的物种研究中,如果需要使用到这些质量不高的区域有提醒的功能,及为后续的改进提供快速的筛选手段。同时也能证明三代组装结果的准确性和质量,能提高组装结果的准确性。附图说明图1为三代序列组装,及利用三代序列和三代序列分别对三代组装结果纠错的流程图。图2为用fosmid序列和组装结果进行比对验证的图;图3为组装结果的二代序列覆盖深度分布的图。图4为组装结果的gc含量二代覆盖深度分布的图。图5为本发明利用三代序列覆盖深度去评估和校验三代组装结果中二代低覆盖深度区域的流程图。具体实施方式下述实施例中所使用的实验方法如无特殊说明,均为常规方法。下述实施例中所用的材料、试剂等,如无特殊说明,均可从商业途径得到。图5示出本发明利用三代序列覆盖深度去评估和校验三代组装结果中二代低覆盖深度区域的一个实施例的流程图。如图5所示,步骤202是二代序列与三代组装结果进行比对,二代序列比对到三代组装结果并统计三代组装结果每个碱基的覆盖深度。一般建议二代的平均数据量能达到基因组的50x以上,最低30x。这个阶段的比对可以使用比对软件bwa或soapaligner等软件完成。允许的错配碱基数一般小于等于2。并统计三代组装结果每个位点覆盖深度。统计是可以使用soapcoverage软件进行。步骤204,低覆盖深度区域延伸与选取:以1kb为窗口,挑选平均覆盖深度低(低深度的阀值可参考表1的建议)的区域,并向前后各延伸30kb,获取延伸后的序列,由于三代序列的长度范围为几百bp到几十kb,而这些低覆盖区域可能低至几十bp,所以我们对低深度覆盖区域前后各延伸了30kb,以保证三代序列能和这些区域进行比对。scaffold前后如果不足30k的,取有的部分,比如窗口是5-6k区域,往前只取0-5k,往后去6-36k,即1-36kb;scaffold后面不足的,从窗口末尾取到最后一个碱基。表1低深度阀值推荐表二代测序深度低深度定义阀值30x3x大于30x且小于等于50x4-5x大于50x且小于等于70x6-8x大于70x9-10x步骤206,三代序列与延伸序列比对:用三代序列与延伸后的序列做比对,这里三代序列可以是未纠错序列或者已经自纠错后的序列。比对软件可以用bwa。三代序列的数据量建议在基因组大小的50x以上。步骤208,碱基覆盖深度统计:获得原1kb二代低覆盖深度区域的三代平均碱基覆盖深度。步骤210,组装结果标记,对二代覆盖深度低或无覆盖的区域,三代平均覆盖深度小于等于5x的标记为组装质量低的区域,三代平均覆盖深度大于5x标记为组装质量高的区域。如果连续2个或多个1kb低深度区域三代都不够5x的,将它们合并成一个区域。如果连续2个或多个同时被标记为“组装质量相对较高的区域”,则将它们合并记成一个“组装质量相对较高的区域”。被标记为“组装质量高的区域”在全部所选窗口区域中所占比例越高,则所述三代测序的序列组装结果的整体组装质量越高。实施例1、本发明方法的玉米基因组具体应用实例(一)三代序列组装使用三代组装软件falcon对80x的三代玉米基因组数据(pacbio单分子实时测序(smrt)结果)进行组装,并用三代原始数据对组装结果做polish纠错,再用60x二代数据对polish后的组装结果进一步纠错,得到玉米基因组的最终的组装结果。(二)二代序列与三代组装结果进行比对使用soapaligner软件将60xpe250的二代序列(过滤后的二代序列,去了接头和低质量碱基后的序列。由原始数据到有效数据过滤经三步处理:1)过滤接头:测序read匹配上adapter序列的50%或者以上则删除整条reads;2)过滤低质量数据:如果测序read中质量值低于20的碱基占整条read的20%或者以上则删除整条read;3)去n:如果测序reads中n含量占整条read的2%或者以上,则删除整条read)比对到三代组装结果,每个read允许最大2bp的错配,用soapcoverage软件统计组装结果每个位点在二代序列中的覆盖深度。(三)低覆盖深度区域延伸与选取以1kb为窗口,挑选三代组装结果中每个1kb窗口区域在二代序列中平均覆盖深度小于等于6x的区域,并向前后各延伸30kb。如表2所示,这样的区域总是有88个。scaffold前后如果不足30k的,取有的部分,比如窗口是5-6k区域,往前只取0-5k,往后去6-36k,即1-36kb;scaffold后面不足的,从窗口末尾取到最后一个碱基。(四)三代序列与延伸序列比对用bwa软件将80x经过自纠错的三代序列与延伸后的序列做比对。(五)碱基覆盖深度统计获得原1kb二代低覆盖深度区域的三代平均碱基覆盖深度。(六)组装结果标记如表2所示,三代平均覆盖深度小于等于5x的标记为组装质量较低的区域,共计35个;三代平均覆盖深度大于5x标记为组装质量高的区域,共计53个。通过本发明能很好的标记出三代组装结果中二代覆盖深度低的区域哪些是高组装质量区域,哪些是较差的区域。表2原1kb二代序列低深度区域中三代的覆盖深度情况小于6x区域数量三代覆盖大于5x的区域三代覆盖小于等于5x的区域885335综上所述,通过本发明可以筛选出三代组装结果中质量不是太高的区域,并将其标注出来。在后续的物种研究中,如果需要使用到这些质量不高的区域有提醒的功能,及为后续的改进提供快速的筛选手段。同时也能证明三代组装结果的准确性和质量,能提高组装结果的准确性。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1