数据处理方法、装置、存储介质及处理器与流程
本发明涉及生物领域,具体而言,涉及一种数据处理方法、装置、存储介质及处理器。
背景技术:
染色体异常是导致出生缺陷的重要原因,在众多染色体异常疾病中,胎儿染色体非整倍体病变是胎儿最常见的染色体畸形。因此,产前诊断可以在孕早期或中期就对胎儿做出诊断,以便进行提前干预或治疗,是降低出生缺陷、提高出生人口素质的重要手段。
目前,对于染色体异常的产前诊断通常分为非侵入性和侵入性两种手段。侵入性手段包括:绒毛活检术,羊膜穿刺术和经腹脐静脉穿刺术等等。虽然采用侵入性手段的诊断结果较准确,但是具有相当高的风险性,容易造成孕妇流产或宫内感染。产前筛查手段(外周血生化筛查和超声颈部透明带)虽然不使用侵入式方法,但是检测率和假阳率并不能达到期望的水平。
NIPT,简称无创产前检测,是应用于孕期产检的一项技术,这项技术是基于孕妇外周血血浆中存在游离胎儿DNA,具有很高的检测准确性,同时也避免了侵入性检测所带来的流产和宫内感染风险。
NIPT检测方法是利用假设检验Z-test来检测染色体异常。首先需要抽取孕妇外周血,分离出血浆,提取出血浆游离DNA,构建二代测序文库,利用二代测序仪得到孕妇血浆游离DNA的序列信息。让后将得到的测序数据通过基本的质控、和人类参考基因组HG19比对、GC校正、计算Z score等步骤来得到完成染色体异常诊断。
然而,由于参考基因组HG19是针对欧洲人的基因组参考序列,而对于中国孕妇人群会存在参考偏差(reference bias),从而导致染色体异常的产前诊断不够准确。
另外,母体自身存在的基因拷贝数变异CNV也会对胎儿的检测带来很大干扰,也会影响诊断的准确性。
由于现有技术无法确定染色体异常诊断中干扰因素,进而无法取出干扰因素,从而导致染色体异常诊断的不够准确。
针对现有技术无法确定染色体异常诊断中干扰因素的技术问题,目前尚未提出有效的解决方案。
技术实现要素:
本发明实施例提供了一种数据处理方法、装置、存储介质及处理器,以至少解决现有技术无法确定染色体异常诊断中干扰因素的技术问题。
根据本发明实施例的一个方面,提供了一种数据处理方法,该方法包括:获取样本染色体上的基因序列;比对所述基因序列与人类基因组参考序列,确定所述样本染色体中与所述人类基因组参考序列唯一匹配的碱基序列reads;统计每个观测区域bin中所述碱基序列reads的数量,其中,所述观测区域bin为所述样本染色体按照预定分窗条件进行分窗后得到的多个区域bin;根据多个观测区域bin中所述碱基序列reads的数量确定隐马尔科夫模型;根据所述隐马尔科夫模型确定基因拷贝数变异CNV,其中,所述拷贝数变异CNV包括:至少一个观测区域bin。
进一步地,所述隐马尔科夫模型的五元素包括:观测序列、隐状态的序列、隐状态的初始概率、隐状态的转移概率、隐状态的发射概率,根据多个观测区域bin中所述碱基序列reads的数量确定隐马尔科夫模型包括:确定所述观测序列{Xm}为:多个所述观测区域bin中所述碱基序列reads的数量的序列,m为所述观测区域bin序号,Xm表示序号为m的所述观测区域bin中所述碱基序列reads的数量;确定所述隐状态的序列{Ym}为:产生不同数目的所述碱基序列reads所对应的所述观测区域bin的隐藏状态的序列,其中,在碱基序列reads的数目高于预定阈值的情况下,确定所述观测区域bin的状态为拷贝数增加,用k=1表示;在碱基序列reads的数目等于预定阈值的情况下,确定所述观测区域bin的状态为拷贝数中性,用k=0表示;在碱基序列reads的数目低于预定阈值的情况下,确定所述观测区域bin的状态为拷贝数缺失,用k=-1表示;Ym=k,Ym表示序号为m的所述观测区域bin的状态;确定所述隐状态的初始概率为:P(Y1=k)=αk,其中,在k=-1的情况下,αk为所述观测区域bin的状态为拷贝数缺失的概率;在k=0的情况下,αk为所述观测区域bin的状态为拷贝数中性的概率;在k=1时的情况下,αk为所述观测区域bin的状态为拷贝数增加的概率;确定所述隐状态的转移概率为:P(Ym+1=km+1|Ym=km)=pjαkm+(1-pj)I(Km+1=km),表示在序号为km状态,为Ym的情况下,序号为km+1,状态为Ym+1的概率,其中,I为指示函数,在km+1=km的情况下,I=1,在km+1≠km的情况下,I=1,pj为预设概率值,αkm为序号为m的所述观测区域bin在状态为k的情况下的概率;确定所述隐状态的发射概率为:
表示在序号为m,隐状态为Ym=k的情况下,所述观测区域bin中所述碱基序列reads的数量为Xm的概率,σk为预定标准差。
进一步地,根据隐马尔科夫模型确定基因拷贝数变异CNV包括:确定所述隐马尔科夫模型的前向概率,其中,所述前向概率通过如下公式表示:
其中,F(1,Y1)=P(X1|Y1);确定所述隐马尔科夫模型的后向概率,其中,所述后向概率通过如下公式表示:
其中,B(L,ZL)=1,L为观测序列的长度;确定所述隐马尔科夫模型中每个隐状态的后验概率,其中,所述后验概率通过如下公式表示:
Ymk=P(Ym=k|X1,...,XL)∞F(m,Ym=k)B(m,Ym=k);
根据所述前向概率、所述后向概率、以及所述后验概率确定基因拷贝数变异CNV。
进一步地,在根据隐马尔科夫模型确定基因拷贝数变异CNV之后,所述方法还包括:在多个所述观测区域bin中删除所述基因拷贝数变异CNV所包含的观测区域bin。
进一步地,在多个所述观测区域bin中删除所述基因拷贝数变异CNV所包含的观测区域bin之后,所述方法还包括:通过平滑样条法对校正区域中所述碱基序列reads的数量进行GC校正,其中,所述校正区域为删除所述基因拷贝数变异CNV后的观测区域bin。
进一步地,在通过平滑样条法对校正区域中所述碱基序列reads的数量进行GC校正之后,所述方法还包括:计算经过GC校正后多个所述校正区域中所述碱基序列reads的数量的校正均值和校正标准差;根据所述校正均值和所述校正标准差构建加权线性回归模型;根据所述加权线性回归模型校正所述样本染色体的基线带来的偏差;确定所述样本染色体校正后的所述碱基序列reads的数量。
进一步地,在确定校正后的所述样本染色体所包含的所述碱基序列reads的数量之后,所述方法还包括:根据校正后的所述样本染色体所包含的所述碱基序列reads的数量利用假设检验Z-test计算所述样本染色体的Z值。
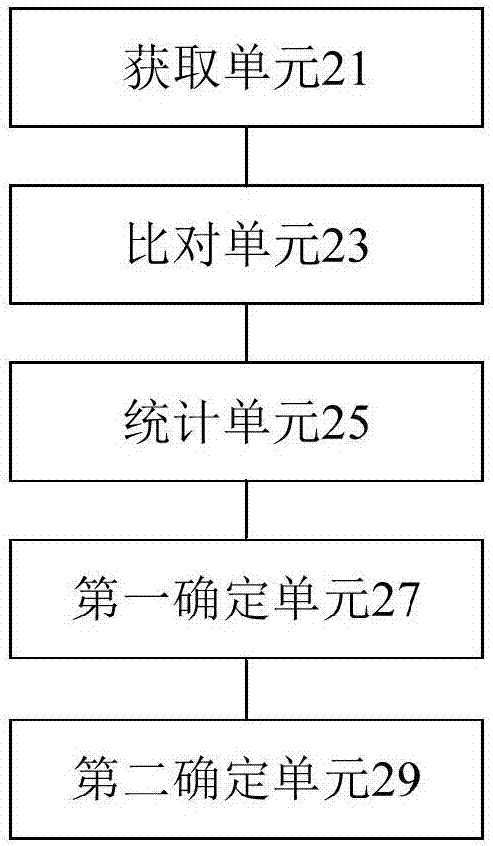
根据本发明实施例的另一方面,还提供了一种数据处理装置,该装置包括:获取单元,用于获取样本染色体上的基因序列;比对单元,用于比对所述基因序列与人类基因组参考序列,确定所述样本染色体中与所述人类基因组参考序列唯一匹配的碱基序列reads;统计单元,用于统计每个观测区域bin中所述碱基序列reads的数量,其中,所述观测区域bin为所述样本染色体按照预定分窗条件进行分窗后得到的多个区域bin;第一确定单元,用于根据多个观测区域bin中所述碱基序列reads的数量确定隐马尔科夫模型;第二确定单元,用于根据所述隐马尔科夫模型确定基因拷贝数变异CNV,其中,所述拷贝数变异CNV包括:至少一个观测区域bin。
根据本发明实施例的另一方面,还提供了一种存储介质,该存储介质包括存储的程序,其中,在程序运行时控制存储介质所在设备执行本发明的数据处理方法。
根据本发明实施例的另一方面,还提供了一种处理器,该处理器用于运行程序,其中,程序运行时执行本发明的数据处理方法。
在本发明实施例中,在获取样本染色体上的基因序列后,将获取的基因序列与预定的人类基因参考序列进行对比,得到样本染色体中与人类基因组参考序列唯一匹配的碱基序列reads,然后按照预定分窗条件对样本染色体分窗,得到多个观测区域bin,并统计每个观测区域bin中碱基序列reads的数量,再根据多个观测区域bin中碱基序列reads的数量确定隐马尔科夫模型,可以根据隐马尔科夫模型确定基因拷贝数变异CNV,从而确定通过隐马尔科夫模型确定染色体异常诊断中由于基因拷贝数变异CNV所产生的干扰因素,解决了现有技术无法确定染色体异常诊断中干扰因素的技术问题,进而在基因序列或人类基因参考序列将确定的基因拷贝数变异CNV去除后,便可准确诊断染色体是否异常,降低假阳性,达到了提高染色体异常检测的准确性的技术效果。
附图说明
此处所说明的附图用来提供对本发明的进一步理解,构成本申请的一部分,本发明的示意性实施例及其说明用于解释本发明,并不构成对本发明的不当限定。在附图中:
图1是根据本发明实施例的一种可选的数据处理方法的流程图;
图2是根据本发明实施例的一种可选的数据处理装置的示意图。
具体实施方式
为了使本技术领域的人员更好地理解本发明方案,下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分的实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都应当属于本发明保护的范围。
需要说明的是,本发明的说明书和权利要求书及上述附图中的术语“第一”、“第二”等是用于区别类似的对象,而不必用于描述特定的顺序或先后次序。应该理解这样使用的数据在适当情况下可以互换,以便这里描述的本发明的实施例能够以除了在这里图示或描述的那些以外的顺序实施。此外,术语“包括”和“具有”以及他们的任何变形,意图在于覆盖不排他的包含,例如,包含了一系列步骤或单元的过程、方法、系统、产品或设备不必限于清楚地列出的那些步骤或单元,而是可包括没有清楚地列出的或对于这些过程、方法、产品或设备固有的其它步骤或单元。
为了便于描述,下面对本发明实施例中的部分名词或术语进行详细说明:
基因拷贝数变异(copy number variations,简称CNV):用来表示染色体缺失或扩增。
reads:测序得到的基因序列(碱基序列),是高通量测序仪产生的测序数据。
GC校正:在基因序列(碱基序列)中,鸟嘌呤G和胞嘧啶C所占比率为GC含量,对基因序列中GC含量校正即为GC校正。
隐马尔科夫模型:英文名称为:Hidden Markov Model,HMM,是一种统计分析模型,用来描述隐含位置参数的马尔科夫过程,可以从可观察的参数中确定该过程的隐含参数,然后理由隐含参数做进一步分析。
本申请提供了一种数据处理方法的实施例。
图1是根据本发明实施例的一种可选的数据处理方法的流程图,如图1所示,该方法包括如下步骤:
步骤S102,获取样本染色体上的基因序列;
步骤S104,比对基因序列与人类基因组参考序列,确定样本染色体中与人类基因组参考序列唯一匹配的碱基序列reads;
步骤S106,统计每个观测区域bin中碱基序列reads的数量,其中,观测区域bin为样本染色体按照预定分窗条件进行分窗后得到的多个区域bin;
步骤S108,根据多个观测区域bin中碱基序列reads的数量确定隐马尔科夫模型;
步骤S110,根据隐马尔科夫模型确定基因拷贝数变异CNV,其中,拷贝数变异CNV包括:至少一个观测区域bin。
通过上述步骤,在获取样本染色体上的基因序列后,将获取的基因序列与预定的人类基因参考序列进行对比,得到样本染色体中与人类基因组参考序列唯一匹配的碱基序列reads,然后按照预定分窗条件对样本染色体分窗,得到多个观测区域bin,并统计每个观测区域bin中碱基序列reads的数量,再根据多个观测区域bin中碱基序列reads的数量确定隐马尔科夫模型,可以根据隐马尔科夫模型确定基因拷贝数变异CNV,从而确定通过隐马尔科夫模型确定染色体异常诊断中由于基因拷贝数变异CNV所产生的干扰因素,解决了现有技术无法确定染色体异常诊断中干扰因素的技术问题,进而在基因序列或人类基因参考序列将确定的基因拷贝数变异CNV去除后,便可准确诊断染色体是否异常,降低假阳性,达到了提高染色体异常检测的准确性的技术效果。
需要说明的是,在本申请中,基因序列,即DNA序列,又可以被称为碱基序列。
在步骤S102提供的方案中,获取基因序列可以待检测生物样本的基因序列。
可选地,本发明提供的技术方案可以应用在无创产前检测中,将待检测孕妇的基因信息作为样本基因信息。
例如,可以一定数量的孕周大于等于12周,且核型分析无染色体异常的孕妇样本构建参考数据库,其中,孕妇样本中胎儿的男、女比例无统计学差异。
可选地,在抽取孕妇外周血,分离出血浆,提取出血浆游离DNA,构建二代测序文库,利用Illumina平台测序等高通量测序仪得到孕妇血浆游离DNA的序列信息。
可选地,获取基因序列的过程中,可以先对获取的基因序列进行预处理来剔除基因序列中的干扰信息,增强检测的准确性。
可选地,为了确保测序数据的质量,避免接头序列污染,可以对原始数据进行去除接头及低质量的reads,其中,原始数据可以是直接通过测序仪得到的测序数据或序列信息。
在步骤S104提供的方案中,人类基因组参考序列可以是HG19序列,通过将基因序列与比对到人类基因组参考HG19序列上,然后在根据比对结果来统计基因序列在每条染色体上的分布情况,进而统计每条染色体上的唯一匹配上的碱基序列(Unique Reads)的条数。
需要说明的是,HG19是国际生物信息学数据库收集存储单位发布的基因组信息,但是HG19是基于欧洲人建立的基因组信息,而对于中国孕妇人群会存在参考偏差(reference bias)。
在步骤S104提供的方案中,可以通过设置分窗的高度和宽度来确定预定分窗条件。
作为一种可选的示例,在进行无创产前检测中,由于无创产前检测(NIPT)样本测序覆盖度很低,因此,在进行后续分析之前,需要将整个染色体的碱基序列reads分窗统计,利用1Kbp的bin作为构建块来确定最优的观测区域bin的宽度,通过设置不同宽度的窗2K,5K,10K,20K,50K,100K,200K,500K,1000K,及不同长度的重叠区域45K,50K,100K,通过对邻近的1Kbp构建块的观测区域bin的含量取均值得到不同窗宽的观测区域bin的含量,并计算变异系数(标准差与均值之比),以期在bin的宽度和bin的数目之间保持平衡,确保染色体含量的稳定性。
可选地,在在本发明中可以选择观测区域bin为100kbp,重叠区域为50kbp。
需要说明的是,观测区域bin的含量可以是观测区域bin中碱基序列reads的数量。
为了避免一些重复序列的干扰,在对样本染色体分窗后,可以去除包含基因组串联重复及散在重复序列的观测区域bin。
可选地,基因拷贝数变异CNV包括母体基因拷贝数变异CNV、以及种群基因拷贝数变异CNV。
需要说明的是,种群基因拷贝数变异CNV是指基于种群水平的基因拷贝数变异CNV,例如,针对HG19,由于HG19是基于欧洲人建立的基因组信息,HG19中可能携带有欧洲白种人共有的变异基因,即种群基因拷贝数变异CNV。
作为一种可选的实施例,在样本是中国孕妇人群(即基因序列为中国人的基因序列),而参考基因组HG19是基于欧洲人建立的基因组,因此在检测过程中会存在一些种群水平上的假定基因拷贝数变异CNV。
此外,在进行无创产前检测的情况下,母体自身存在的CNV也会对胎儿三体的检测带来干扰。
为了排除母体基因拷贝数变异CNV,以及种群基因拷贝数变异CNV的干扰,可以通过隐马尔科夫模型来确定多个观测区域bin中存在母体基因拷贝数变异CNV和/或种群基因拷贝数变异CNV的异常观测区域。
作为一种可选的实施例,隐马尔科夫模型的五元素包括:观测序列、隐状态的序列、隐状态的初始概率、隐状态的转移概率、隐状态的发射概率,根据多个观测区域bin中碱基序列reads的数量确定隐马尔科夫模型中的五种元素。
采用本发明上述实施例,通过根据多个观测区域bin中碱基序列reads的数量确定隐马尔模型的五元素中观测序列、隐状态的序列、隐状态的初始概率、隐状态的转移概率、隐状态的发射概率,可以准确建立隐马尔模型,从而根据隐马尔模型可以确定检测出基因序列和人类基因参考序列中的基因拷贝数变异CNV。
可选地,可以确定观测序列{Xm}为:多个观测区域bin中碱基序列reads的数量的序列,m为观测区域bin序号,Xm表示序号为m的观测区域bin中碱基序列reads的数量。
可选地,可以确定隐状态的序列{Ym}为:产生不同数目的碱基序列reads所对应的观测区域bin的状态的序列,其中,在碱基序列reads的数目高于预定阈值的情况下,确定观测区域bin的状态为拷贝数增加,用k=1表示;在碱基序列reads的数目等于预定阈值的情况下,确定观测区域bin的状态为拷贝数中性,用k=0表示;在碱基序列reads的数目低于预定阈值的情况下,确定观测区域bin的状态为拷贝数缺失,用k=-1表示;Ym=k,Ym表示序号为m的观测区域bin的状态。
需要说明的是,如果一个观测区域bin内含有母体基因拷贝数变异CNV,那么该观测区域bin与不含母体基因拷贝数变异CNV的观测区域bin相比,碱基序列reads的数量的变化大约是50%左右。
例如,在包含母体基因拷贝数变异CNV的观测区域bin中碱基序列reads的数量,可以比不含母体基因拷贝数变异CNV的观测区域bin中碱基序列reads的数量多50%,或少50%。
可选地,可以确定隐状态的初始概率为:P(Y1=k)=αk,其中,在k=-1的情况下,αk为观测区域bin的状态为拷贝数缺失的概率;在k=0的情况下,αk为观测区域bin的状态为拷贝数中性的概率;在k=1时的情况下,αk为观测区域bin的状态为拷贝数增加的概率。
可选地,可以通过更新公式来更新αk,更新公式为:
可选地,可以确定隐状态的转移概率为:P(Ym+1=km+1|Ym=km)=pjαkm+(1-pj)I(km+1=km),表示在序号为km状态,为Ym的情况下,序号为km+1,状态为Ym+1的概率,其中,I为指示函数,在km+1=km的情况下,I=1,在km+1≠km的情况下,I=1,pj为预设概率值,αkm为序号为m的观测区域bin在状态为k的情况下的概率。
可选地,pj是一个概率,如1/pj是母体基因拷贝数变异CNV的平均宽度(单位为观测区域bin的数目)。
可选地,预设概率值pj=0.001。
可选地,可以确定隐状态的发射概率为:
表示在序号为m,隐状态为Ym=k的情况下,观测区域bin中碱基序列reads的数量为Xm的概率,σk为预定标准差。
可选地,为了更新σk,可以对每个k计算XmYmk,Ymk为序号为m,状态为k的隐状态,得到一个长度为O的向量,然后计算向量的样本间标准差为σk的估计值。
作为一种可选的实施例根据隐马尔科夫模型确定基因拷贝数变异CNV包括:确定隐马尔科夫模型的前向概率,其中,前向概率通过如下公式表示:
其中,F(1,Y1)=P(X1|Y1);
确定隐马尔科夫模型的后向概率,其中,后向概率通过如下公式表示:
其中,B(L,ZL)=1,L为观测序列的长度;确定隐马尔科夫模型中每个隐状态的后验概率,其中,后验概率通过如下公式表示:
Ymk=P(Ym=k|X1,...,XL)∞F(m,Ym=k)B(m,Ym=k);
根据前向概率、后向概率、以及后验概率确定基因拷贝数变异CNV。
采用本发明上述实施例,在确定隐马尔科夫模型后,可以计算隐马尔科夫模型的前向概率、后向概率,以及每个隐状态的后验概率,进而根据计算出的前向概率、后向概率、以及后验概率确定基因拷贝数变异CNV所包括的多个观测区域bin。
作为一种可选的实施例,在根据隐马尔科夫模型确定多个观测区域bin中存在预定基因拷贝数变异CNV的异常观测区域之后,该实施例还可以包括:在多个观测区域bin中删除基因拷贝数变异CNV所包含的观测区域bin。
采用本发明上述实施例,在通过隐马尔科夫模型确定体基因拷贝数变异CNV后,可以在多个观测区域bin中删除基因拷贝数变异CNV所包含的观测区域bin,从而可以准确进行NIPT检测,提高胎儿染色体异常判断的准确性,降低检测的假阳性,达到了提高染色体异常检测的准确性的技术效果。
可选地,基因拷贝数变异CNV包括母体基因拷贝数变异CNV、以及种群基因拷贝数变异CNV。
可选地,在基因拷贝数变异CNV包括母体基因拷贝数变异CNV的情况下,确定母体基因拷贝数变异CNV所包含的观测区域bin为第一观测区域,在多个观测区域bin中删除第一观测区域。
可选地,在基因拷贝数变异CNV包括种群基因拷贝数变异CNV的情况下,确定种群基因拷贝数变异CNV所包含的观测区域bin为第二观测区域,在多个观测区域bin中删除第二观测区域。
作为一种可选的实施例,在多个观测区域bin中删除基因拷贝数变异CNV所包含的观测区域bin之后,方法还包括:通过平滑样条法对校正区域中碱基序列reads的数量进行GC校正,其中,校正区域为删除基因拷贝数变异CNV后的观测区域bin。
采用本发明上述实施例,在多个观测区域bin中删除基因拷贝数变异CNV所包含的观测区域bin之后,通过对删除基因拷贝数变异CNV后的观测区域bin之后的校正区域采用平滑样条法进行GC校正,可以尽可能避免干扰误差,提高染色体异常检测的准确性。
需要说明的是,除了三体信号及上面提到的干扰因素之后,很多其它因素也会对UR ratio造成较大的影响,譬如样品上机批次及测序GC-bias等。
对此,可以通过局部多项式回归、权重校正、平滑样条法进行校正。通过计算统计参数,最终选择平滑样条法,该方法能更好的对GC进行校正,尽可能减少误差干扰,有效地判断UR ratio的值是否有统计学意义上的异常。
另外,不同染色体区域有不同的基线覆盖度,尽管GC校正减轻了基线差异,但是远不能消除基线之间的差异。
为了消除基线之间的差异,通过计算全部正常对照样本的GC校正后的bin含量的均值和标准差,构建加权线性回归模型,从而校正不同染色体基线带来的偏差,然后重新计算每条染色体的含量。
作为一种可选的实施例,在通过平滑样条法对校正区域中碱基序列reads的数量进行GC校正之后,该实施例还可以包括:计算经过GC校正后多个校正区域中碱基序列reads的数量的校正均值和校正标准差;根据加权线性回归模型校正样本染色体的基线带来的偏差;确定样本染色体校正后的碱基序列reads的数量。
采用本发明上述实施例,在经过GC校正后,可以计算校正后的多个校正区域中碱基序列reads的数量的校正均值和校正标准差,然后在根据校正均值和校正标准差构建校正均值和校正标准差构建加权线性回归模型,可以根据校正均值和校正标准差构建加权线性回归模型校正样本染色体的基线带来的偏差,确定校正后的样本染色体所包含的碱基序列reads的数量,从而使得出的样本染色体所包含的碱基序列reads的数量更加精确,提高染色体异常检测的准确性。
作为一种可选的实施例,在确定校正后的样本染色体所包含的碱基序列reads的数量之后,该实施例还可以包括:根据校正后的样本染色体所包含的碱基序列reads的数量计算样本染色体的Z值。
采用本发明上述实施例,根据校正后的样本染色体所包含的碱基序列reads的数量计算样本染色体的Z值,可以根据计算出的Z值确定染色体是否存在异常,进而确定胎儿的患病风险。
需要说明的是,Z值,即z-score,也叫标准分数(stand score)是一个数与平均数的差再除以标准差的过程,能够真实的反应一个分数距离平均数的相对标准距离,在统计学中,标准分数是一个观测点或数据点的值高于观测值或测量值的平均值的标准差的符号数。
可选地,每条常染色体的z-score(Z值)计算公式为:Zi=(xi-ui)/σi,其中,xi表示第i号染色体校正后reads百分比;ui表示参考数据库中第i号染色体的read百分比的平均值;σi表示参考数据库中第i号染色体的read百分比的标准误差,i为待检测生物的常染色体的序号,例如,在对人类进行诊断的过程中,i=1,2,...22。
在完成Z值的技术后,可以根据Z值进行常染色体非整倍体判定。
可选地,若Zi>3(i=1,2,...22)/σii,则判定第i号染色体为非整倍体。
根据本发明上述实施例,检测了两万余例样本,成功剔除了1例母亲13号染色体存在大片段重复所造成的胎儿13号染色体三体的假阳性,1例母亲21号染色体存在大片段重复所造成的胎儿21号染色体三体的假阳性,1例母亲20号染色体存在大片段重复所造成的胎儿20号染色体三体的假阳性,及1例母亲10号染色体存在大片段缺失所造成的胎儿10号染色体异常,并且,羊穿验证结果与NIPT结果相同,均报出母体微重复或缺失。
需要说明的是,在附图的流程图虽然示出了逻辑顺序,但是在某些情况下,可以以不同于此处的顺序执行所示出或描述的步骤。
本申请还提供了一种存储介质的实施例,该实施例的存储介质包括存储的程序,其中,在程序运行时控制存储介质所在设备执行本发明实施例的数据处理方法。
本申请还提供了一种处理器的实施例,该实施例的处理器用于运行程序,其中,程序运行时执行本发明实施例的数据处理方法。
根据本发明实施例,还提供了一种数据处理装置实施例,需要说明的是,该数据处理装置可以用于执行本发明实施例中的数据处理方法,本发明实施例中的数据处理方法可以在该数据处理装置中执行。
图2是根据本发明实施例的一种可选的数据处理装置的示意图,如图2所示,该装置包括:获取单元21,用于获取基因序列,其中,基因序列为样本染色体上的基因序列;比对单元23,用于比对基因序列与人类基因组参考序列,确定样本染色体中与人类基因组参考序列唯一匹配的碱基序列reads;统计单元25,用于统计每个观测区域bin中碱基序列reads的数量,其中,观测区域bin为样本染色体按照预定分窗条件进行分窗后得到的多个区域bin;第一确定单元27,用于根据多个观测区域bin中碱基序列reads的数量确定隐马尔科夫模型;第二确定单元29,用于根据隐马尔科夫模型确定多个观测区域bin中存在预定基因拷贝数变异CNV的异常观测区域,其中,预定基因拷贝数变异包括:母体基因拷贝数变异CNV和/或种群基因拷贝数变异CNV。
需要说明的是,该实施例中的获取单元21可以用于执行本申请实施例中的步骤S102,该实施例中的比对单元23可以用于执行本申请实施例中的步骤S104,该实施例中的统计单元25可以用于执行本申请实施例中的步骤S106,该实施例中的第一确定单元27可以用于执行本申请实施例中的步骤S108,该实施例中的第二确定单元29可以用于执行本申请实施例中的步骤S110。上述模块与对应的步骤所实现的示例和应用场景相同,但不限于上述实施例所公开的内容。
根据本发明上述实施例,在获取样本染色体上的基因序列后,将获取的基因序列与预定的人类基因参考序列进行对比,得到样本染色体中与人类基因组参考序列唯一匹配的碱基序列reads,然后按照预定分窗条件对样本染色体分窗,得到多个观测区域bin,并统计每个观测区域bin中碱基序列reads的数量,再根据多个观测区域bin中碱基序列reads的数量确定隐马尔科夫模型,可以根据隐马尔科夫模型确定基因拷贝数变异CNV,从而确定通过隐马尔科夫模型确定染色体异常诊断中由于基因拷贝数变异CNV所产生的干扰因素,解决了现有技术无法确定染色体异常诊断中干扰因素的技术问题,进而在基因序列或人类基因参考序列将确定的基因拷贝数变异CNV去除后,便可准确诊断染色体是否异常,降低假阳性,达到了提高染色体异常检测的准确性的技术效果。
作为一种可选的实施例,隐马尔科夫模型的五元素包括:观测序列、隐状态的序列、隐状态的初始概率、隐状态的转移概率、隐状态的发射概率,第一确定单元包括:第一确定模块,用于确定观测序列{Xm}为:多个观测区域bin中碱基序列reads的数量的序列,m为观测区域bin序号,Xm表示序号为m的观测区域bin中碱基序列reads的数量;第二确定模块,用于确定隐状态的序列{Ym}为:产生不同数目的碱基序列reads所对应的观测区域bin的状态的序列,其中,在碱基序列reads的数目高于预定阈值的情况下,确定观测区域bin的状态为拷贝数增加,用k=1表示;在碱基序列reads的数目等于预定阈值的情况下,确定观测区域bin的状态为拷贝数中性,用k=0表示;在碱基序列reads的数目低于预定阈值的情况下,确定观测区域bin的状态为拷贝数缺失,用k=-1表示;Ym=k,Ym表示序号为m的观测区域bin的状态;第三确定模块,用于确定隐状态的初始概率为:P(Y1=k)=αk,其中,在k=-1的情况下,αk为观测区域bin的状态为拷贝数缺失的概率;在k=0的情况下,αk为观测区域bin的状态为拷贝数中性的概率;在k=1时的情况下,αk为观测区域bin的状态为拷贝数增加的概率;第四确定模块,用于确定隐状态的转移概率{Ym}为:P(Ym+1=km+1|Ym=km)=pjαkm=(1-pj)I(km+1=km),表示在序号为km状态,为Ym的情况下,序号为km+1,状态为Ym+1的概率,其中,I为指示函数,在km+1=km的情况下,I=1,在km+1≠km的情况下,I=1,pj为预设概率值,αkm为序号为m的观测区域bin在状态为k的情况下的概率;第五确定模块,用于确定隐状态的发射概率为:
表示在序号为m,隐状态为Ym=k的情况下,观测区域bin中碱基序列reads的数量为Xm的概率,σk为预定标准差。
作为一种可选的实施例,第二确定单元可以包括:第六确定单元,用于确定隐马尔科夫模型的前向概率,其中,前向概率通过如下公式表示:
其中,F(1,Y1)=P(X1|Y1);第七确定单元,用于确定隐马尔科夫模型的后向概率,其中,后向概率通过如下公式表示:
其中,B(L,ZL)=1,L为观测序列的长度;第八确定单元,用于确定隐马尔科夫模型中每个隐状态的后验概率,其中,后验概率通过如下公式表示:
Ymk=P(Ym=k|X1,...,XL)∞F(m,Ym=k)B(m,Ym=k);
第九确定单元,用于根据前向概率、后向概率、以及后验概率确定基因拷贝数变异CNV。
作为一种可选的实施例,该实施例还可以包括:删除单元,用于在根据隐马尔科夫模型确定基因拷贝数变异CNV之后,在多个观测区域bin中删除基因拷贝数变异CNV所包含的观测区域bin。
作为一种可选的实施例,该实施例还可以包括:GC校正单元,用于在多个观测区域bin中删除基因拷贝数变异CNV所包含的观测区域bin之后,通过平滑样条法对校正区域中碱基序列reads的数量进行GC校正,其中,校正区域为删除基因拷贝数变异CNV后的观测区域bin。
作为一种可选的实施例,该实施例还可以包括:第一计算单元,用于在通过平滑样条法对校正区域中碱基序列reads的数量进行GC校正之后,计算经过GC校正后多个校正区域中碱基序列reads的数量的校正均值和校正标准差;构建单元,用于根据校正均值和校正标准差构建加权线性回归模型;偏差校正单元,用于根据加权线性回归模型校正样本染色体的基线带来的偏差;第三确定单元,用于确定样本染色体校正后的碱基序列reads的数量。
作为一种可选的实施例,该实施例还可以包括:第二计算单元,用于在确定校正后的样本染色体所包含的碱基序列reads的数量之后,根据校正后的样本染色体所包含的碱基序列reads的数量计算样本染色体的Z分数。
上述的装置可以包括处理器和存储器,上述单元均可以作为程序单元存储在存储器中,由处理器执行存储在存储器中的上述程序单元来实现相应的功能。
存储器可能包括计算机可读介质中的非永久性存储器,随机存取存储器(RAM)和/或非易失性内存等形式,如只读存储器(ROM)或闪存(flash RAM),存储器包括至少一个存储芯片。
上述本申请实施例的顺序不代表实施例的优劣。
在本申请的上述实施例中,对各个实施例的描述都各有侧重,某个实施例中没有详述的部分,可以参见其他实施例的相关描述。在本申请所提供的几个实施例中,应该理解到,所揭露的技术内容,可通过其它的方式实现。
其中,以上所描述的装置实施例仅仅是示意性的,例如所述单元的划分,可以为一种逻辑功能划分,实际实现时可以有另外的划分方式,例如多个单元或组件可以结合或者可以集成到另一个系统,或一些特征可以忽略,或不执行。另一点,所显示或讨论的相互之间的耦合或直接耦合或通信连接可以是通过一些接口,单元或模块的间接耦合或通信连接,可以是电性或其它的形式。
另外,在本申请各个实施例中的各功能单元可以集成在一个处理单元中,也可以是各个单元单独物理存在,也可以两个或两个以上单元集成在一个单元中。上述集成的单元既可以采用硬件的形式实现,也可以采用软件功能单元的形式实现。
所述集成的单元如果以软件功能单元的形式实现并作为独立的产品销售或使用时,可以存储在一个计算机可读取存储介质中。基于这样的理解,本申请的技术方案本质上或者说对现有技术做出贡献的部分或者该技术方案的全部或部分可以以软件产品的形式体现出来,该计算机软件产品存储在一个存储介质中,包括若干指令用以使得一台计算机设备(可为个人计算机、服务器或者网络设备等)执行本申请各个实施例所述方法的全部或部分步骤。而前述的存储介质包括:U盘、只读存储器(ROM,Read-Only Memory)、随机存取存储器(RAM,Random Access Memory)、移动硬盘、磁碟或者光盘等各种可以存储程序代码的介质。
以上所述仅是本申请的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本申请原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本申请的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!