一种对组装序列排序的方法及系统与流程
本发明涉及生物信息技术领域,特别是涉及一种对组装序列排序的方法及系统。
背景技术:
dna是生物体遗传信息的主要载体,高质量的基因组参考序列是现代遗传学、分子生物学等现代生物科学的重要基础。因此,基因测序对探索与认识生命本质等基础生物科学研究、人类重要遗传病防治及动植物遗传育种等应用性研究均具有十分重要的意义。真实状态中的细胞核是一个狭小的三维立体空间,直链分子结构的dna会以复杂的卷曲方式位于细胞核内,原一维dna序列被赋予三维空间构象,并导致了大量复杂的基因控制作用方式,因此,简单的一维dna序列信息由于不能提供真实dna空间分布相关的信息,无法满足现有的分析需求。
可结合染色质三维构象技术、高通量测序技术、生物信息分析方法,研究全基因组范围内整个染色质dna在空间位置上的关系,获得高分辨率的染色质三维结构信息。基于染色质三维构象测序数据中染色质片段间的交互强度呈现出随距离衰减的规律,染色质三维构象测序序列可以用于基因组组装,即将杂乱的基因序列组装到染色体水平。
在现有技术中通常是通过生物信息学手段利用染色质三维构象测序数据,实现基因组组装目的的。在此过程中需要通过聚类、排序、定向等步骤,然后,通过生成相应的基因交互组热图来评估基因组装结果,若组装结果好的染色体做出的热图,其交联信号应集中在热图的对角线区域,但是,在实际生成的热图在非对角线区域显示强交互信号,而造成这种现象的主要原因是排序不准确的问题。
技术实现要素:
针对于上述问题,本发明提供一种组装序列排序方法及系统,解决了现有技术中数据排序不准确的问题。
为了实现上述目的,本发明提供了如下技术方案:
一种对组装序列排序的方法,该方法包括:
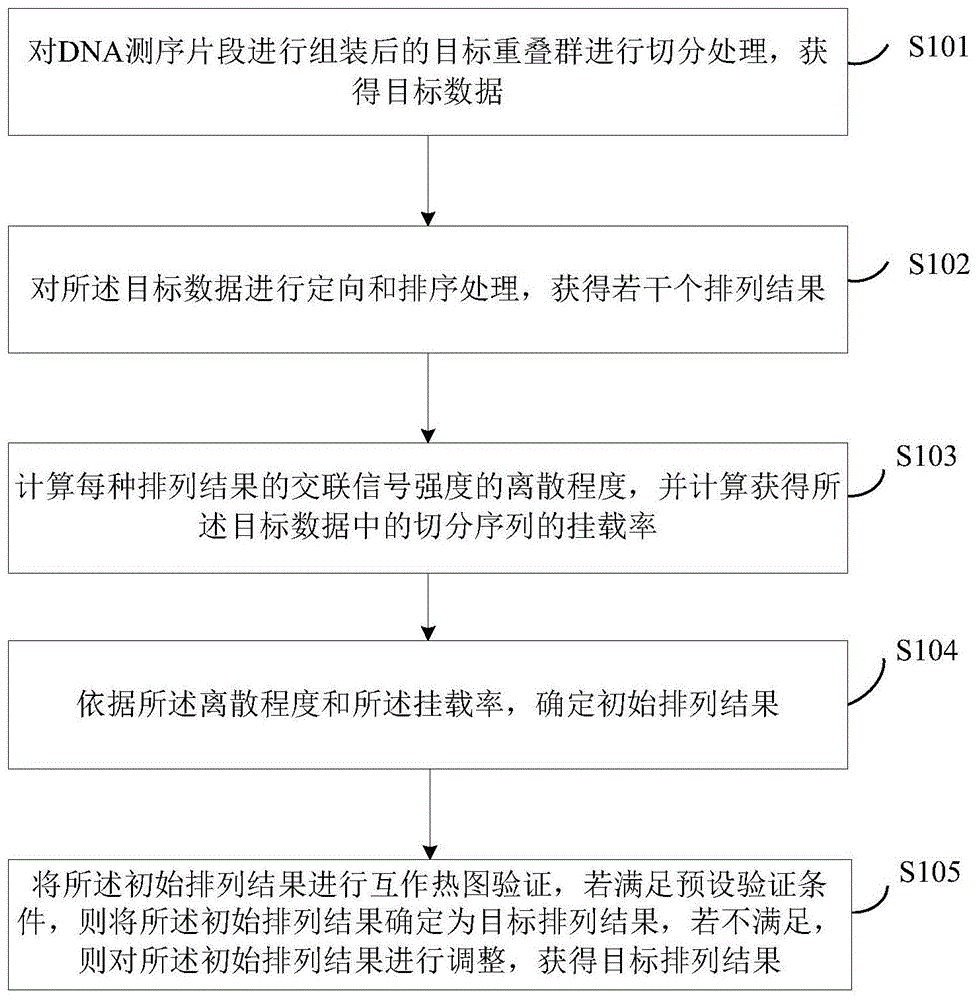
对dna测序片段进行组装后的目标重叠群进行切分处理,获得目标数据;
对所述目标数据进行定向和排序处理,获得若干个排列结果;
计算每种排列结果的交联信号强度的离散程度,并计算获得所述目标数据中的切分序列的挂载率,所述切分序列表征对所述目标重叠群按照预设切分长度进行切分后的序列;
依据所述离散程度和所述挂载率,确定初始排列结果;
将所述初始排列结果进行互作热图验证,若满足预设验证条件,则将所述初始排列结果确定为目标排列,若不满足,则对所述初始排列结果进行调整,获得目标排列。
可选地,所述对dna测序片段进行组装后的目标重叠群进行切分处理,获得目标数据,包括:
将染色质三维构象测序序列比对到所述目标重叠群上,根据所述目标重叠群之间的交联信号强度进行聚类处理,获得若干个聚类组;
对每个所述聚类组中的目标重叠群,按照预设切分长度进行切分处理,获得切分序列。
可选地,所述对dna测序片段进行组装后的目标重叠群进行切分处理,获得目标数据,包括:
对所述目标重叠群按照预设切分长度进行切分处理,获得切分序列;
将染色质三维构象测序序列比对到所述切分序列上,根据每个切分序列之间的交联信号强度对所述切分序列进行聚类,获得若干个聚类组。
可选地,所述对dna测序片段进行组装后的目标重叠群进行切分处理,获得目标数据,包括:
将染色质三维构象测序序列比对到所述目标重叠群上,并对所述目标重叠群按照预设切分长度进行切分处理,获得切分序列;
根据每个切分序列之间的交联信号强度对所述切分序列进行聚类,获得若干个聚类组。
可选地,所述目标重叠群为针对整个基因组的重叠群或者待处理的部分基因组的重叠群。
可选地,该方法包括:
响应于目标重叠群中聚类到目标聚类组的切分序列数量大于预设数量阈值,将所述目标重叠群划分至所述目标聚类组;
若所述目标重叠群中达到预设位置阈值对应的位置连续的切分序列被划分至不同的聚类组,将所述目标重叠群根据切分序列的位置进行切分,并将切分后的重叠群划分至对应的聚类组。
可选地,所述对所述目标数据进行定向和排序处理,获得若干个排列结果,包括:
遍历所有目标重叠群,分别以该重叠群的首尾两个切分序列作为所述重叠群的起始位置,将所述起始位置对应的切分序列记为第一序列;
依据切分序列之间的交联信号强度,依次确定排列在所述第一序列之后的切分序列,获得若干个排列结果。
可选地,所述计算每种排列结果的交联信号强度的离散程度,并计算获得所述目标数据中的切分序列的挂载率,包括:
依据每个排列结果中进行排序的切分序列的数量和切分序列的总数量,计算获得切分序列的挂载率;
依据每个排列结果中任意两个切分序列之间的距离,和所述两个切分序列的交联信号强度,计算获得所述离散程度。
可选地,所述依据所述离散程度和所述挂载率,确定初始排列结果,包括:
根据所述挂载率,确定候选排列结果;
将所述候选排列结果中所述离散程度最小的排列结果,确定为初始排列结果。
一种对组装序列排序的系统,该系统包括:
切分单元,用于对dna测序片段进行组装后的目标重叠群进行切分处理,获得目标数据;
排序单元,用于对所述目标数据进行定向和排序处理,获得若干个排列结果;
计算单元,用于计算每种排列结果的交联信号强度的离散程度,并计算获得所述目标数据中的切分序列的挂载率,所述切分序列表征对所述目标重叠群按照预设切分长度进行切分后的序列;
确定单元,用于依据所述离散程度和所述挂载率,确定初始排列结果;
验证单元,用于将所述初始排列结果进行互作热图验证,若满足预设验证条件,则将所述初始排列结果确定为目标排列,若不满足,则对所述初始排列结果进行调整,获得目标排列。
相较于现有技术,本发明提供了一种组装序列排序方法及系统,对dna测序片段进行组装后的目标重叠群进行切分处理,其中,切分处理可以将重叠群切分成相同大小的切分序列,从而减少由于重叠群长度不同在均一化交联信号强度使的误差,提高聚类准确性。同时,基于切分后的切分序列进行定向和排序,可以实现将排序和定向同时进行,并且基于切分序列进行聚类或者排序,可以对有明显组装错误的重叠组进行纠错。
附图说明
为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据提供的附图获得其他的附图。
图1为本发明实施例提供的一种对组装序列进行排序的方法的流程示意图;
图2为本发明实施例提供的一种染色质三维构象实验流程的示意图;
图3为本发明实施例提供的一种对组装序列进行排序的流程示意图;
图4为本发明提供的一种双子叶植物组装序列排序后互作热图;
图5为本发明实施例提供的四种排序结果对应的互作热图;
图6为本发明提供的另一种双子叶植物组装序列排序后互作热图;
图7为本发明实施例提供的一种组装序列排序系统的结构示意图。
具体实施方式
下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
本发明的说明书和权利要求书及上述附图中的术语“第一”和“第二”等是用于区别不同的对象,而不是用于描述特定的顺序。此外术语“包括”和“具有”以及他们任何变形,意图在于覆盖不排他的包含。例如包含了一系列步骤或单元的过程、方法、系统、产品或设备没有设定于已列出的步骤或单元,而是可包括没有列出的步骤或单元。
在本发明实施例中提供了一种对组装序列排序的方法,在本发明实施例中主要是基于染色质三维构象测序序列对组装序列排序的方法,参见图1,该方法可以包括以下步骤:
s101、对dna测序片段进行组装后的目标重叠群进行切分处理,获得目标数据;
其中,所述切分处理为对所述目标重叠群按照预设切分长度进行切分。该目标重叠群可以为针对整个基因组的重叠群,也可以为待处理的部分基因组的重叠群,例如,某条染色体、或者某条染色体的某个区域对应的部分基因组。
需要说明的是,在步骤s101中需要对重叠群进行切分处理,其中重叠群来源对dna测序片段的组装,而dna测序片段来源于原始的基因组即待通过染色质三维构象测序序列技术进行排序的基因组,其来源本发明不做限制,即采用二代测序或者三代测序获得原始基因组均适用。其中,二代测序为高通量测序,主要使用illumina平台的仪器,一次对几十万到几百万条dna分子进行序列测定,得到的片段读长短。三代测序是以pacbio或oxfordnanopore平台的测序技术,得到的长序列的片段,适用于基因组组装等工作。
为了便于对本发明实施例进行解释说明,下面将本发明实施例常用的一些术语进行解释说明。
reads(dna测序片段):测序得到的一个读段,即一个原始的测序数据,在二代测序中主要是双端成对存在。
序列组装:无论是二代测序中的短reads还是三代测序的长reads,得到的测序数据相较于整个基因组而言仍然是极小的,序列组装就是通过各种算法和方法将这些小片段连接起来,从而得到最初的contig(重叠群)。
contig:根据测序reads得到的重叠信息将存在的重叠片段建立一种组合关系,形成重叠群即contig,是最初版本中没有gap的序列。需要说明的是,测序获得的reads经过拼接可以形成没有gap的contig,contig经过进一步的组装可以形成比contig更长的scaffold。因此,在本发明实施例中原始序列可以是contig,也可以是scaffold。在本发明实施例为了后续描述,将序列组装结果统称为contig。
bin(切分序列):将contig按照相同大小切分成小段序列,每一段序列就成为一个bin。
参考序列(ref):为待通过染色质三维构象测序序列进行组装的序列,包括基因组重叠群、部分基因组重叠群。
交联信号强度:将染色质三维构象测序的原始数据比对到参考序列后,统计成对有效reads的比对位置及数量,利用两个片段酶切位点的数量将有效reads数值进行均一化,得到的数值即为两个片段之间交联信号强度。
在本发明中解决的是基于染色质三维构象测序序列对组装序列排序问题,对于染色质三维构象测序的实验方式本发明不做具体限制,只要是能够实现染色质三维构象测序的实验目的即可,对其中的实验流程、方法、步骤和实验平台等均不做具体限制。
下面以一种染色质三维构象测序实验流程对染色质三维构象测序实验进行说明,请参见图2,该实验流程包括:
s201、通过甲醛交联固定,将细胞内由蛋白质介导的空间上邻近的染色质片段进行共价连接;
s202、限制性内切酶hindiii进行酶切;
s203、将得到的粘性末端用生物素标记;
s204、采用nhel酶连接;
s205、纯化dna并且采用超声波打断,生物素亲和层析,分离生物素化的dna;
s206、加接头进行ngs测序pe150genomecoverage100×。
在本发明实施例中进行了对重叠群的预处理,该预处理表征了对重叠群的聚类和切分处理,但是本发明并不对聚类和切分的先后顺序进行限制,进行该预处理的目的是使得获得的目标数据能够满足预设聚类精度。这是由于现有技术中的聚类的精度相对较低。现有技术中通常是通过凝聚聚类方法(hierarchicalagglomerativeclustering)进行聚类,在聚类之前需要对测序获得的数据进行评估,常用的数据评估方式为:
分别将测序得到的reads比对上参考基因组,选择比对上基因组唯一位置的reads(即uniquemapreads);
对未比对上基因组的reads进行剪切,再进行比对;
最后将两次的uniquemapreads进行合并,并整合双端数据比对的结果,得到paireduniquemapreads;
过滤paireduniquemapreads中酶切位点位于reads比对位置两端的无效数据。
然后利用过滤后的有效染色质三维构象测序数据,将基因组组装到染色体级别,现有技术的相关研究中表明同一染色体上contig之间的信号交联强度要明显高于不同染色体;一条染色体上,距离越近,信号交联强度越高;两条contig之间,通过不同位置的信号交联强度确定相对顺序。
因此在进行分析时,需要计算contig之间交联信号强度,根据交联信号强度进行聚类获得聚类组。而本发明实施例为了提升聚类精度并不是直接对重叠群(contig)进行简单的聚类。具体的,本发明实施例中提供了三种切分处理的方式。
第一种处理方式,即对dna测序片段进行组装后的目标重叠群进行切分处理,获得目标数据,包括:
将染色质三维构象测序序列比对到所述目标重叠群上,根据所述目标重叠群之间的交联信号强度进行聚类处理,获得若干个聚类组;
对每个所述聚类组中的目标重叠群,按照预设切分长度进行切分处理,获得切分序列。
在第一种理方式中得到的目标数据为切分序列,在该方式中可以理解为在原有的聚类的基础上进行了切分处理,其中,目标重叠群就是参考基因组。
第二处理方式,即对dna测序片段进行组装后的目标重叠群进行切分处理,获得目标数据,包括:
对所述目标重叠群按照预设切分长度进行切分处理,获得切分序列;
将染色质三维构象测序序列数据比对到所述切分序列对应级别的参考基因组上,根据每个切分序列之间的交联信号强度对所述切分序列进行聚类,获得若干个聚类组。
在第二种处理的方式中,目标数据为聚类组,在该方式中是先对目标重叠群进行切分,再对切分后的切分序列(bin)进行聚类,其中,切分序列即为参考序列。使用切分序列进行聚类将更准确。这是由于参考序列中的contig长度不一,虽然在计算contig之间的信号强度时有均一化的计算方法,但也不能完全消除各种因素(主要是酶切位点数目的较大差异)对结果的影响,所以将参考基因组切分成相同大小的bin,由于bin的大小相同,假设酶切位点在基因组上是均匀分布的,不同bin之间存在的酶切位点数据差异较小,从而减少由于contig长度不同在均一化交联信号强度时的误差,提高聚类结果的准确性。
在第三种处理方式中,即对dna测序片段进行组装后的目标重叠群进行切分处理,获得目标数据,包括:
将染色质三维构象测序序列比对到所述目标重叠群上,并对所述目标重叠群按照预设切分长度进行切分处理,获得切分序列;
根据每个切分序列之间的交联信号强度对所述切分序列进行聚类,获得若干个聚类组。
该方式中的目标数据也是聚类组,该方式与第二种处理方式的区别主要是将染色质三维构象测序序列进行比对的对象不同,均是先进行切分后进行聚类,方式三除了可以实现上述的使用bin进行聚类更准确以外,还能够在需要调整bin的大小时,可以省略比对过程。
s102、对所述目标数据进行定向和排序处理,获得若干个排列结果。
由于目标数据是进行切分处理后的数据,在对其进行排序和定向处理时,可以根据切分序列之间的交联信号强度进行。但是需要说明的是,步骤s101中的第一种切分处理方式时根据的切分序列交联信号强度进行定向和排序时,该切分序列是将染色质三维构象测序数据比对到了目标重叠群后再进行切分产生的,因此该切分序列中包含了相关的比对信息,因此,在利用交联信号强度作为该步骤的处理依据时,需要考虑该目标数据的具体获得方式,及切分数据中包含的相关数据等,从而选择对应的处理依据,使得处理结果更加精确。
由上述实施例可见,在步骤s101中进行预处理的过程中均实现了切分的处理,然后对目标数据进行了排序,该目标数据表征了经过切分后的数据,可以理解为对每个bin进行了对应的排序和定向,需要说明的是,现有技术中是先排序后定向,而本方案是同时实现了排序和定向,从而获得了多个排列结果。
s103、计算每种排列结果的交联信号强度的离散程度,并计算获得所述目标数据中的切分序列的挂载率;
s104、依据所述离散程度和所述挂载率,确定初始排列结果;
s105、将所述初始排列结果进行互作热图验证,若满足预设验证条件,则将所述初始排列结果确定为目标排列结果,若不满足,则对所述初始排列结果进行调整,获得目标排列结果。
在获得了多个排列结果后,会根据切分片段(bin)的挂载率、离散程度、热图选择最优方案确定最终的目标排列结果。
该过程具体包括:
s301、根据基因组的大小将重叠群(contig)按照预设大小切分成多个bin,得到bin级别的参数ref。
需要说明的是,预设大小表征了切分指标,而该切分指标可以根据实际需求进行设置,通常需要参考染色体的大小,对应的,该预设大小会为染色体大小的千分之一左右,在实际操作过程中,一般会使用100k、200k、500k、1m这4个数值。
s302、将染色质三维构象测序数据比对到上一步得到的ref上,进行质控、过滤和评估。
该步骤实现了对比对后的数据进行清洗的目的,具体的质控、过滤和评估的手段本发明不做限制。
s303、根据bin之间的交联信号强度,对所有的bin进行聚类。
需要说明的是,在该例子中是采用现有技术进行聚类的,但是并不局限于该方法,只要是能够实现根据交联信号强度进行聚类的方法和软件均可,通常为了计算方便会选择凝聚聚类方法,但是上述聚类方法并不是唯一方式,本发明实施例中不对具体的聚类方法进行限制。
当进行聚类时,将有效reads比对的bam文件输入相关聚类软件进行聚类,软件会统计每个bin之间的成对的测序片段(paired-reads),并根据paired-reads数据和bin上的酶切点得到bin之间的交联矩阵,通过层次聚类的算法将bin分配到不同的聚类组(cluster)。
在本发明的实施例中还提供了一种对基因组组装的校正方法,包括:
响应于重叠群中聚类到目标聚类组的切分序列数量大于预设数量阈值,将所述重叠群划分至所述目标聚类组;
若所述重叠群中达到预设位置阈值对应的位置连续的切分序列被划分至不同的聚类组,将所述重叠群根据切分序列的位置进行切分,并将切分后的重叠群划分至对应的聚类组。
在上述实施例的基础上,具体参见步骤s304。
s304、根据一个cluster中bin的数量将contig分到不同cluster:若conitg有≥95%的bin被聚类到一个cluster,那么认为这个contig属于该cluster;若一个contig有>5%的位置连续的bin被分到不同cluster,则将该contig根据bin的位置切分成连续几段,分配到对应的cluster,完成对基因组组装的校正。
其中,预设数量阈值对应上述具体步骤中的95%,预设位置阈值对应步骤s304中的5%,具体的数值的设置需要根据实际情况进行设置,在该实施例中只是举例说明,并不做固定数值限定。
s305、将contig分配到不同cluster后,进行cluster内部排序。
即在聚类完成后需要进行排序的步骤,在本发明实施例中该排序过程中包括了定向过程,具体如:
遍历所有重叠群,分别以该重叠群的首尾两个切分序列作为所述重叠群的起始位置,将所述起始位置对应的切分序列记为第一序列;
依据切分序列之间的交联信号强度,依次确定排列在所述第一序列之后的切分序列,获得若干个排列结果。
需要说明的是,由于在步骤s101中进行切分处理的目标重叠群可以为整个基因组的重叠群,也可以为针对部分基因组的重叠群,对于后者通常是对排序不理想的重叠群进行处理,这时是已知其聚类的,只需要进行切分处理即可,这样在遍历过程中,会对所有重叠群进行遍历,而该重叠群可来自对应的聚类组中。
对应的,会先确定起始位置,即呈现在排列结果中第一个bin。在本发明实施例中需要遍历聚类组中的所有contig,并分别以该contig头尾的两个bin作为该聚类组的起始,这样在确定了第一个bin之后,就会进行后续的排序。
根据第二个bin与第一个bin有最强交联强度,确定第二个bin;
然后依次确定后续的各个bin,使得第三个bin跟第一、第二个bin具有最强交联强度(平均值);第四个bin跟第一、二、三个bin具有最强交联强度(平均值);第n个bin跟第n-1、n-2、n-3个bin具有最强交联强度(平均值)。
得到所有可能的排序后,计算每种排列结果的交联信号强度的离散程度d及聚类组的bin挂载率,即:
依据每个排列结果中进行排序的切分序列的数量和切分序列的总数量,计算获得切分序列的挂载率;
依据每个排列结果中任意两个切分序列之间的距离,和所述两个切分序列的交联信号强度,计算获得所述离散程度。
为了更准确的计算挂载率和离散程度,通常是针对每个排列记过中各个聚类组中的切分序列的相关数据进行计算的。
bin的挂载率=排序中bin的数目/bin总数*100%
d=∑(|bin1-bin2|*signal)
公式中,|bin1-bin2|代表两个bin之间的距离,signal为这两个bin的交联信号强度。
按挂载率对上述的2n种排列结果进行排序,取挂载率前20%的排列结果,并在此条件下取离散程度d最小的为最优排序。其中,该20%表征一个阈值,可以根据排列结果的数量进行具体的设置,也可以为其他数值。
对最优排列结果做热图验证,从热图中可见最优的排序其信号强度更为集中,对角线以外的区域信号强度递减,更符合真实规律,若热图验证结果不理想即无法满足预设验证条件时,可以选择其他排列结果中的一个排列结果,例如挂载率最靠前的排列结果,重新生成热图进行验证。需要说明的是,该预设验证条件可以根据生成热图的软件或算法的精度进行确定,例如,可以为针对目标排列生成的热图中的交联信号全部集中在对角线区域,或者设置一定的偏差阈值,例如,允许生成的热图在非对角线区域显示强交互强度的范围阈值等。
s306、依次完成所有聚类组的内部排序,然后做全基因组热图进行验证。
本发明提供了一种组装序列排序方法,对dna测序片段进行组合后的重叠群进行聚类和切分处理,其中,切分处理可以将重叠群切分成相同大小的切分序列,从而减少由于重叠群长度不同在均一化交联信号强度使的误差,提高聚类准确性。同时,基于切分后的切分序列进行定向和排序,可以实现将排序和定向同时进行,并且基于切分序列进行排序获得的排列结果更加准确,解决了现有技术中需要先排序后定向的问题,使得组装序列排序更加准确。
下面以具体的应用实例对本发明提供的组装序列的排序进行说明。
参见图4,为本发明实施例提供的一种双子叶植物组装序列排序后互作热图,其中,图a为现有技术排序结果,图b为本发明排序结果。
对某双子叶植物使用nanopore平台测序,组装基因组(即前所述参考基因组)大小550mb、contign50580k。通过核型分析手段确定该物种基因组有13条染色体。采用常规方法构建染色质三维构象文库,测了100×的染色质三维构象测序数据,进行染色体组装。
使用现有软件进行染色体聚类和排序。contig挂载率87.19%。参见图4a,以左下角为原点,横纵坐标每一个虚线格依次为13条染色体,交联强度越大在图中即显示颜色越深。结果显示,染色体之间没有大片段的较强的交联信号,即对角线之外的虚线格中无明显深色显示,说明组装结果没有明显错误。除chr03外,其他染色体内部信号分布比较紊乱,即对角线上的虚线格中主要的信号没有聚集在对角线附近,图中可以明显看出深浅交错排布,未呈现出以对角线为中心由深至浅的平缓变化,说明每条染色体内部contig的排序存在着不同程度的问题。
参见图4b,通过本发明的技术方案按500kb进行bin的切分。可选用本发明的技术方案:例如,以现有技术中软件的聚类结果为基础,对每个聚类组中的contig进行切分、排序、计算离散程度及挂载率、确定初始排序结果、互作热图验证,进一步进行全基因组互作热图验证;又如,对该组装基因组切分后重新进行聚类、排序、计算离散程度及挂载率、确定初始排序结果、互作热图验证。结果显示,染色体内部的顺序有了明显改善,信号分布符合随距离衰减的规律,即对角线上的虚线格中交联信号聚集在对角线上,以对角线为中心呈现由深至浅的平缓变化。并且contig挂载率提升到了95.60%,说明本发明的排序算法更接近与真实的结果。
例如对聚类组cluster12中的132个contig(切成了402个bin)按照本申请的方式进行排序,获得了264种排列结果,计算每种排列结果的交联信号值的离散程度及cluster的bin挂载率。
参见表1,列举了4种排序方式的bin挂载率和离散程度d,这4种排序挂载率均在前20%,其中以contig1535_1的排序方式离散程度d最小,所以选择contig1535_1为最优排序。
表1
参见图5,为本发明实施例提供的上述四种排序结果对应的热图,其中图a至d依次对应contig1265_1、contig2507_1、contig490_3、contig1535_1的热图,从热图结果中也可以得出contig1535_1为最优排序,即对角线外的区域无明显交联信号、交联信号聚集在对角线区域并以对角线为中心呈现由深至浅的平缓变化。
在另一双子叶植物,该植物基因组大小657mb,组装的contign505mb。通过核型分析手段确定该物种基因组有29条染色体。采用常规方法构建染色质三维构象文库,利用染色质三维构象测序数据结果进行染色体组装。
参见图6,为本发明另一种双子叶植物基于染色质三维构象测序序列对组装序列排序后互作热图,其中,图a为现有技术组装结果。对于同一个样本来说,在基因组的两个染色体之间,一般不存在很强烈的交联信号,从图6a中可以看出原始基因组中存在着不少contig上的组装错误(在热图非对角线区域显示出高交联信号强度,如黑色方框及箭头所述区域)。然而现有技术中的软件并不能纠正这些错误,而是将contig整体归类到一个cluster。
而采用本发明的技术方案,bin大小为500kb。通过bin去聚类,可以将一条contig上组装错误的bin分开。例如,图6a中黑色箭头所指区域,该区域内显示有对角线外的高交联信号强度(深色),即cluster05与cluster13存在高交联信号强度,说明该基因组组装结果在对应区域存在错误。针对这一个组装错误,可以直接从现有技术软件排序结果中调取cluster05与cluster13的数据,分别对这两个cluster中的contig进行切分、重新进行聚类、排序、计算离散程度及挂载率、确定初始排序结果、互作热图验证。在聚类过程中发现,cluster05上有一个contig10,该contig被切分成了28个bin,其中部分bin15~bin28在cluster05内部没有明显的信号强度,而是和cluster13中的bin具有较高的信号强度,所以这个contig上15~28个bin很有可能是组装错误。在聚类时,将contig10切分成了两部分,bin1-14对应的contig聚类至cluster05、bin15~bin28对应的contig聚类至cluster13。实现了对cluster05中contig10组装结果的校正。对全基因组重叠群的排序及组装结果的校正,可选用本发明的技术方案:例如,对该组装基因组切分后重新进行聚类、排序、计算离散程度及挂载率、确定初始排序结果、互作热图验证。结果如图6b,在染色体之间没有明显的交联信号,即对角线之外的虚线格中无明显深色显示;同时,染色体内部的顺序有了明显改善,信号分布符合随距离衰减的规律,即对角线上的虚线格中交联信号聚集在对角线上(深色),以对角线为中心呈现由深至浅的平缓变化。
因此,通过本发明的实施例不仅可以提高排序结果的准确性,还可以纠正contig组装时的错误。
对应的,参见图7,在本发明实施例中还提供了一种对组装序列排序的系统,该系统包括:
切分单元10,用于对dna测序片段进行组装后的目标重叠群进行切分处理,获得目标数据;
排序单元20,用于对所述目标数据进行定向和排序处理,获得若干个排列结果;
计算单元30,用于计算每种排列结果的交联信号强度的离散程度,并计算获得所述目标数据中的切分序列的挂载率,所述切分序列表征对所述目标重叠群按照预设切分长度进行切分后的序列;
确定单元40,用于依据所述离散程度和所述挂载率,确定初始排列结果;
验证单元50,用于将所述初始排列结果进行互作热图验证,若满足预设验证条件,则将所述初始排列结果确定为目标排列,若不满足,则对所述初始排列结果进行调整,获得目标排列。
本发明提供了一种组装序列排序系统,在切分单元中对dna测序片段进行组合后的重叠群进行聚类和切分处理,其中,切分处理可以将重叠群切分成相同大小的切分序列,从而减少由于重叠群长度不同在均一化交联信号强度使的误差,提高聚类准确性。同时,在排序单元中基于切分后的切分序列进行定向和排序,可以实现将排序和定向同时进行,并且基于切分序列进行聚类或者排序,进行重叠组的局部聚类,在排序过程中能够将切分序列还原,通过分析聚类结果,可以对有明显组装错误的重叠组进行纠错。
在上述实施例的基础上,所述切分单元10包括:
第一聚类子单元,用于将染色质三维构象测序序列比对到所述目标重叠群上,根据所述目标重叠群之间的交联信号强度进行聚类处理,获得若干个聚类组;
第一切分子单元,用于对每个所述聚类组中的目标重叠群,按照预设切分长度进行切分处理,获得切分序列。
在上述实施例的基础上,所述切分单元10包括:
第二切分子单元,用于对所述目标重叠群按照预设切分长度进行切分处理,获得切分序列;
第二聚类子单元,用于将染色质三维构象测序序列比对到所述切分序列对应级别的参考基因组上,根据每个切分序列之间的交联信号强度对所述切分序列进行聚类,获得若干个聚类组。
在上述实施例,所述切分单元10包括:
第三切分子单元,用于将染色质三维构象测序序列数据比对到所述目标重叠群上,并对所述目标重叠群按照预设切分长度进行切分处理,获得切分序列;
第三聚类子单元,用于根据每个切分序列之间的交联信号强度对所述切分序列进行聚类,获得若干个聚类组。
在上述实施例的基础上,所述目标重叠群为针对整个基因组的重叠群或者待处理的部分基因组的重叠群。
在上述实施例的基础上,该系统还包括:
第一划分子单元,用于响应于重叠群中聚类到目标聚类组的切分序列数量大于预设数量阈值,将所述重叠群划分至所述目标聚类组;
第二划分子单元,用于若所述重叠群中达到预设位置阈值对应的位置连续的切分序列被划分至不同的聚类组,将所述重叠群根据切分序列的位置进行切分,并将切分后的重叠群划分至对应的聚类组。
在上述实施例的基础上,所述排序单元20包括:
确定子单元,用于遍历所有目标重叠群,分别以该重叠群的首尾两个切分序列作为所述重叠群的起始位置,将所述起始位置对应的切分序列记为第一序列;
排序子单元,用于依据切分序列之间的交联信号强度,依次确定排列在所述第一序列之后的切分序列,获得若干个排列结果。
在上述实施例的基础上,所述计算单元30包括:
第一计算子单元,用于依据每个排列结果中进行排序的切分序列的数量和切分序列的总数量,计算获得切分序列的挂载率;
第二计算子单元,用于依据每个排列结果中任意两个切分序列之间的距离,和所述两个切分序列的交联信号强度,计算获得所述离散程度。
在上述实施例的基础上,所述确定单元40包括:
第一排序确定子单元,用于根据所述挂载率,确定候选排列结果;
第二排序确定子单元,用于将所述候选排列结果中所述离散程度最小的排列结果,确定为初始排列结果。
本说明书中各个实施例采用递进的方式描述,每个实施例重点说明的都是与其他实施例的不同之处,各个实施例之间相同相似部分互相参见即可。对于实施例公开的装置而言,由于其与实施例公开的方法相对应,所以描述的比较简单,相关之处参见方法部分说明即可。
对所公开的实施例的上述说明,使本领域专业技术人员能够实现或使用本发明。对这些实施例的多种修改对本领域的专业技术人员来说将是显而易见的,本文中所定义的一般原理可以在不脱离本发明的精神或范围的情况下,在其它实施例中实现。因此,本发明将不会被限制于本文所示的这些实施例,而是要符合与本文所公开的原理和新颖特点相一致的最宽的范围。
- 还没有人留言评论。精彩留言会获得点赞!