基因组编辑的精细作图和因果基因鉴定的制作方法
1.本发明领域是分子生物学,更具体地,是用于编辑植物细胞的基因组以鉴定期望性状的因果等位基因或将期望性状精细作图到所述基因组的小区域以进行基因鉴定的方法。
2.以电子方式递交的序列表的引用
3.所述序列表的官方副本经由efs
‑
web作为ascii格式的序列表以电子方式提交,文件名为7826_seqlist.txt,创建于2018年10月23日,且具有154千字节大小,并与本说明书同时提交。包含在所述ascii格式的文件中的序列表是本说明书的一部分并且通过引用以其整体并入本文。
背景技术:
4.植物中的遗传作图是通过使用遗传标记、针对这些标记的群体分离和重组频率的标准遗传原理来定义基因座的连锁关系的方法。精细作图是指对负责期望性状的因果基因或序列元件进行分离的作图过程。这通常是通过在分离的植物材料中使用遗传标记鉴定重组事件来完成的,所述分离的植物材料衍生自在性状表现和相关区域序列单倍型方面不同的亲本。首先,从具有不同目的特征的亲本中产生分离群体(f2、bc1、bc2等)。然后用在亲本之间以跨基因组的有规律的小间隔的多态性遗传标记对该群体进行基因分型,并针对目的性状进行表型分型。标记处的基因型与表型相关联,以鉴定可能控制目的性状的区域。然后使用基于已鉴定的遗传区间的与性状相关(或不相关)的亲本等位基因中的现有标记来鉴定重组事件。通常在较小的区域中制作新的标记,以鉴定信息最丰富的重组事件。一旦鉴定出事件,便从具有这些事件的个体获得表型,以进一步界定区间。这通常需要进行一次或多次迭代,并导致一个或少量的推定控制目的性状的候选基因或序列基序。然后用基因组编辑或转基因测试这些候选基因或序列基序。
5.但是,并非所有的基因组基因座都易受此类方法的影响。例如,某些区域显示出与给定品系或群体的低同源性,或者非共线区域可能会阻止重组的发生。在这种情况下,仍需要一种方法来分离负责期望性状的因果基因或序列元件。
技术实现要素:
6.本文所述的方法涉及产生新的遗传变体,以加速低重组基因组区域中或存在
‑
缺失值(“pav”)阻止重组的情况下或当基于标准图的克隆方法不是最佳方法或无法产生期望结果时的现有遗传作图程序。本文所述的方法还可提供针对靶向区域的验证信息,并可用于完全绕过精细作图的后期阶段,从而缩短了验证基因或区域的时间。在可以在受控环境中完成期望性状的表型分型的情况下,本文所述的方法可以将创建分离群体和进行基因分型以鉴定重组体的时间减少一代。
7.本公开涉及用于鉴定针对期望性状的一个或多个因果基因或遗传基因座的方法,所述方法包括:1)在具有期望性状的植物或植物细胞中的内源基因组基因座中的至少一个
靶位点中引入位点特异性修饰;2)获得具有经修饰的核苷酸序列的植物或植物细胞;3)针对所述位点特异性修饰进行筛选;和4)针对所述期望性状的表型的增加或减少进行筛选。在另一个实施例中,所述方法包括鉴定负责期望性状的因果基因或小区域。
8.本公开还涉及用于鉴定期望性状的因果基因的方法,所述方法包括:1)在植物中的内源基因组基因座中引入至少一个位点特异性修饰;和2)获得具有所述位点特异性修饰的植物;3)针对是否存在所述期望性状对所述植物或所述植物的后代进行筛选,并且4)鉴定所述因果基因。
9.本公开还涉及在基因组基因座中产生新的单倍型的方法,所述方法包括:1)在第一植物中的内源基因组基因座中引入至少一个位点特异性修饰;2)使所述第一植物与第二植物杂交;3)针对所得后代中的所述位点特异性修饰进行筛选;和4)将所述后代的单倍型与其表型相关联,以建立所述位点特异性修饰与所述期望性状之间的因果关系
10.本公开还涉及用于对期望性状精细作图的方法,所述方法包括:1)在植物中的内源基因组基因座中的至少一个靶位点中引入位点特异性修饰或缺失;2)获得具有经修饰的核苷酸序列的植物;3)将所述植物与轮回亲本杂交;和4)针对所述杂交的后代中期望性状的丧失或获得进行筛选。在一个实施例中,位点特异性修饰是缺失。
11.在一个实施例中,所述方法进一步包括在内源基因组基因座中引入至少第二位点特异性修饰,其中与所述内源基因组序列、等位基因或基因组基因座相比,所述位点特异性修饰包含至少一个核酸缺失、插入或多态性。在一些实施例中,所述方法进一步包括选择具有经修饰的核苷酸序列的植物。在一些实施例中,选择的植物表现出期望性状的增加或减少的表型。期望性状包括但不限于对疾病的抗性、种子蛋白或油的浓度、谷粒产量、植物健康、高度、茎秆强度和有害生物抗性。
12.在一些实施例中,内源基因组基因基因座位于已知的qtl内,至少被部分测序,或涵盖随机突变精细作图。内源基因座可以具有低的固有重组频率,为着丝粒区域或包括非共线区域。
13.本文公开的方法可用于通过插入基因组编辑来在区域中产生新的单倍型,其中基因组编辑的变体在可控制性状的关键序列基序中不同。内源基因组基因座可以代表独特单倍型,其不能与同一区间内的其他单倍型重组。由于缺乏同源性,独特单倍型可能无法与其他单倍型重组。
14.在一些实施例中,对目的区域的先验知识(基因组序列、标记性状关联、基因注释或数量性状基因座(“qtl”))指导基因组编辑的设计以靶向特定序列,从而产生用于测试的有用变体。在另一个实施例中,所述方法包括缺失序列区域以产生特定的变体,针对期望性状的分离测试所述特定的变体,并且鉴定因果基因或区域。在一些实施例中,所降低的区域小于初始目的区域。
15.在一个实施例中,位点特异性修饰发生在非编码区、启动子、内含子、非翻译区(“utr”)或编码区中。在一些实施例中,位点特异性修饰在内源编码序列中包含缺失、插入缺失(“indel”)或单核苷酸多态性(“snp”)。
16.在一些实施例中,至少一个位点特异性修饰包含在一个或多个靶位点处引入的至少一个双链断裂。双链断裂或位点特异性修饰可以由核酸酶(例如但不限于talen、大范围核酸酶、锌指核酸酶或crispr相关的核酸酶)诱导。cas9内切核酸酶可以被至少一种指导
rna指导。指导rna可以在内源基因组基因座内的单个或几个特定靶位点处引导位点特异性修饰。
17.附图和序列表说明
18.图1显示了在39kb基因组缺失区域上通过重叠缺失对因果基因的精细作图。
19.图2显示了来自缺失#1和缺失#3的t1种子的蛋白和油含量。
20.图3显示了通过重叠缺失系对大豆高蛋白qtl(qhp20)的精细作图。
21.图4显示了来自williams 82(seq id no:30)和野生大豆(seq id no:31)的glyma.20g850100及其旁系同源物glyma.10g134400(seq id no:38)(包括来自williams 82的321bp插入)的基因组序列比对。
22.图5显示了来自williams 82(seq id no:36)和野生大豆(seq id no:32)的glyma.20g850100及其旁系同源物glyma.10g134400(seq id no:40)的蛋白序列比对。
23.图6显示了glyma.20g850100的高蛋白和低蛋白等位基因的示意图。
24.图7显示了在非共线性片段的区域中,bac序列的组装上的rcg1和rcg1b基因的位置的示意图。
25.图8显示了玉蜀黍中第10号染色体上约3.6mb r基因簇中26个基因的位置的示意图。
26.图9示显示了应用于疾病抗性基因座的实验方案。在这种情况下,轮回亲本易患病,并且可以是优良的育种系。在群体发育过程中产生的遗传物质对疾病具有抗性,包含抗性基因座,所述基因座根据育种阶段以不同的纯度渗入轮回亲本背景。该材料可以是近等基因系(nil)。
27.图10显示了赋予疾病抗性的显性功能获得型等位基因的编辑和筛选方案。
28.图11显示了在赋予对炭疽病茎杆腐烂的抗性的热带系和在目的区域中显示低同源性的b73之间的多个基因组比对。
29.图12显示了赋予炭疽病茎杆腐烂抗性的预测基因模型和目的区域中的预期缺失。
30.图13显示了以双基因作用模式赋予疾病抗性的显性功能获得型等位基因的编辑和筛选方案。
具体实施方式
31.应当理解的是,本文所使用的术语仅用于描述特定实施例的目的,而不旨在是限制性的。当在本说明书和所附权利要求中使用时,单数和单数形式的术语例如“一个/一种(a/an)”以及“该(the)”包括复数指代物,除非上下文中另外明确指明。因此,例如术语“植物(plant)、所述植物(the plant)、或一个植物(a plant)”也包括多个植物;也取决于上下文,使用的术语“植物”也可包括该植物遗传相似或相同的后代;使用的术语“核酸”实际上任选地包括该核酸分子的多个拷贝;同样地,术语“探针”任选地(并且通常)涵盖许多相似或相同的探针分子。除非另有定义,本文所用的所有技术和科学术语具有与本公开所属领域的普通技术人员通常所理解相同的含义,除非另有明确说明。
32.本文提出了编辑植物基因组的方法,以对具有期望性状的增加或减少的表型的植物进行精细作图。
33.本文公开的方法可用于对因果基因、小的基因组区域或染色体区间进行精细作
图。基因组序列和基因模型的准确鉴定可以增加本文公开的方法的成功性,因为它允许精确设计靶向被认为控制性状的基因或序列区域的crispr
‑
cas指导rna。在一些实施例中,可以使用生物信息学鉴定或其他方法来鉴定染色体区间中的候选因果基因,然后设计基因组编辑以在片段或区域中依次地缺失候选基因或其部分,从而缺失或破坏所述因果基因产生期望性状的增加或减少的表型。依次地缺失基因或其部分也可以鉴定控制所述性状的成对基因。本文公开的方法允许解析和鉴定具有许多具有相似或重复区段的基因的区域。如本文所提供的,簇中的基因可以被依次地缺失或成对缺失以确定一个或多个因果基因。
34.术语“等位基因”是指在特定基因座处出现的两个或更多个不同核苷酸序列中的一个。
[0035]“等位基因频率”是指等位基因在个体内、品系内或品系的群体内的基因座处存在的频率(比例或百分比)。例如,对于等位基因“a”,基因型“aa”、“aa”或“aa”的二倍体个体分别具有1.0、0.5或0.0的等位基因频率。可以通过对来自一个品系的个体样本的等位基因频率取平均值来估计该品系内的等位基因频率。类似地,可以通过对组成群体的品系的等位基因频率取平均值来计算该群体内的等位基因频率。对于具有有限数目的个体或品系的群体,等位基因频率可以表示为包含所述等位基因的个体或品系(或任何其他指定的分组)的计数。
[0036]
当等位基因是影响性状表达的dna序列或等位基因的一部分或与其连锁时,该等位基因与该性状“相关”。所述等位基因的存在是所述性状将如何表达的指标。
[0037]“回交”是指杂交体后代借以反复与亲本中之一杂交的方法。在回交方案中,“供体”亲本是指待渗入的具有所需的一种或多种基因、基因座或特定表型的亲本植物。“受体”亲本(使用一次或多次)或“轮回”亲本(使用两次或更多次)是指基因或基因座被渗入到其中的亲本植物。例如,参见ragot,m.等人,(1995)marker
‑
assisted backcrossing:a practical example,in techniques et utilisations des marqueurs moleculaires les colloques[标记辅助回交:实践范例,分子标记技术和应用专题讨论会]第72卷,第45
‑
56页,以及openshaw等人,(1994)marker
‑
assisted selection in backcross breeding,analysis of molecular marker data[回交育种中的标记辅助选择,分子标记数据分析],第41
‑
43页。初始杂交产生f1代;然后术语“bc1”指轮回亲本的第二次使用,“bc2”指轮回亲本的第三次使用等等。
[0038]
如本文所用,术语“因果基因”是指编码推定或有助于表型的基因的任何多核苷酸序列。在一些实施例中,因果基因推定或有助于期望性状。在一些实施例中,因果基因位于已知的qtl或靶向的基因组基因座内。
[0039]
厘摩(“cm”)是重组频率的度量单位。一个cm等于有1%的机会,一个遗传基因座处的标记会由于单代中的交换而与第二基因座处的标记分离。
[0040]
如本文所使用的,术语“染色体区间”是指存在于植物单一染色体上的基因组dna的连续线性跨度。位于单条染色体区间上的遗传元件或基因是物理连锁的。染色体区间的大小没有特别的限制。在一些方面,位于单条染色体区间内的遗传元件是遗传连锁的,通常具有例如小于或等于20cm,或者可替代地,小于或等于10cm的遗传重组距离。也就是说,单条染色体区间内的两个遗传元件以小于或等于20%或10%的频率进行重组。
[0041]
在本申请中,短语“紧密连锁”是指两个连锁的基因座之间的重组以等于或小于约
10%(即,在遗传图谱上分隔不超过10cm)的频率发生。换言之,该紧密连锁的基因座有至少90%的机会发生共分离。当标记基因座显示与期望性状共分离(连锁)的显著概率时,它们在本文公开的实施例中尤其有用。紧密连锁的基因座(例如标记基因座和第二基因座)可以显示10%或更低、优选约9%或更低、还更优选约8%或更低、又更优选约7%或更低、还更优选约6%或更低、又更优选约5%或更低、还更优选约4%或更低、又更优选约3%或更低、以及还更优选约2%或更低的基因座间重组频率。在非常优选的实施例中,相关基因座显示约1%或更低,例如约0.75%或更低、更优选约0.5%或更低、或又更优选约0.25%或更低的重组频率。定位于相同染色体并且具有使得两个基因座之间的重组以小于10%(例如,约9%、8%、7%、6%、5%、4%、3%、2%、1%、0.75%、0.5%、0.25%或更少)的频率发生的距离的所述两个基因座也被认为是彼此“邻近的”。在某些情况下,两个不同的标记可以具有相同的遗传图谱坐标。在这种情况下,所述两个标记彼此非常邻近,以至于二者之间的重组以不可检测的这种低频发生。
[0042]
术语“杂交的”或“杂交”是指有性杂交,并且涉及通过授粉将两种单倍体配子融合以产生二倍体后代(例如细胞、种子或植物)。该术语涵盖一株植物被另一株植物授粉和自交(或自体授粉,例如当花粉和胚珠来自同一植物时)二者。
[0043]
如本文所用,术语“期望性状”是指植物或作物中的期望表型。期望性状可以包括但不限于疾病抗性、改变的谷粒特性、谷粒产量、植物健康、种子蛋白或油浓度、有害生物抗性、非生物或生物胁迫抗性、耐旱性、植物高度或茎杆强度。
[0044]“有利等位基因”是在特定基因座的等位基因,它赋予或有助于农学上期望的表型,例如植物中对疾病增加的抗性,并允许对具有农学上期望的表型的植物的鉴定。标记的有利等位基因是与所述有利表型分离的标记等位基因。
[0045]“遗传图谱”是对给定物种内的一个或多个染色体(或连锁群)上的基因座之间的遗传连锁关系的描述,通常以图表或表格形式描述。对于每个遗传图谱,基因座之间的距离通过所述基因座的等位基因在群体中一起出现的频率(基因座的重组频率)来测量。可以使用dna或蛋白标记或者可观察到的表型来检测等位基因。遗传图谱是用来作图的群体、所用标记的类型以及不同群体之间每个标记的多态性潜力的产物。对于不同的遗传图谱,基因座之间的遗传距离可以是不同的。但是,使用通用标记可以将信息从一张图谱关联到另一张图谱上。本领域的普通技术人员可以使用通用标记位置来鉴定每个单独的遗传图谱上的标记和其他目的基因座的位置。尽管由于例如在不同群体中检测交替重复基因座的标记、在用于排序这些标记的统计学方法上的差异、新的突变或实验室错误,经常在标记顺序上有小的变化,但是基因座顺序在图谱之间不应该改变。
[0046]“遗传图谱定位”是遗传图谱上相对于相同连锁群上的周围遗传标记的定位,其中在给定物种内可以找到指定的标记。
[0047]“遗传作图”是通过使用遗传标记、针对这些标记的群体分离和重组频率的标准遗传原理来定义基因座的连锁关系的方法。“精细作图”是指对负责期望性状的因果基因或序列元件进行分离的过程。这通常是通过在分离的植物材料中使用遗传标记鉴定重组事件来完成的,所述分离的植物材料衍生自在性状表现和相关区域序列单倍型方面不同的亲本。首先,从具有不同目的特征的亲本中产生分离群体(f2、bc1、bc2等)。然后用在亲本之间以跨基因组的有规律的小间隔的多态性遗传标记对该群体进行基因分型,并针对目的性状进
行表型分型。标记处的基因型与表型相关联,以鉴定可能控制目的性状的区域。然后使用基于已鉴定的遗传区间的与性状相关(或不相关)的亲本等位基因中的现有标记来鉴定重组事件。通常会在较小的区域中鉴定出新的标记,这可能有助于找到信息最丰富的重组事件。一旦鉴定出事件,便从具有这些事件的个体获得表型,以进一步界定区间。这通常需要进行一次或多次迭代,并导致一个或少量的推定控制目的性状的候选基因或序列基序。然后可以用基因组编辑或转基因测试候选基因或序列基序。
[0048]“遗传标记”是在群体中多态的核酸,并且所述遗传标记的等位基因可以通过一种或多种分析方法(例如rflp、aflp、同工酶、snp、ssr等)来检测和区分。该术语还指与用作探针的基因组序列(例如核酸)互补的核酸序列。可以通过本领域中已知的方法检测对应于群体成员之间的遗传多态性的标记。这些方法包括,例如基于pcr的序列特异性扩增方法、限制性片段长度多态性检测(rflp)、同功酶标记检测、通过等位基因特异性杂交(ash)进行的多核苷酸多态性检测、植物基因组的扩增可变序列检测、自主序列复制检测、简单重复序列检测(ssr)、单核苷酸多态性检测(snp)、或扩增片段长度多态性检测(aflp)。已知方法也用于检测表达的序列标签(est)和衍生自est序列的ssr标记,以及随机扩增多态性dna(rapd)。
[0049]“遗传重组频率”是两个基因基因座之间的交换事件(重组)的频率。重组频率可以通过遵循减数分裂后的标记和/或性状的分离来观察。“低固有重组频率”是指基于给定区域中的遗传图谱距离而鉴定的重组事件的数量少。
[0050]“单倍型”是个体在多个遗传基因座处的基因型,即等位基因的组合。典型地,由单倍型描述的遗传基因座在物理和遗传上是连锁的,即在同一染色体区段上。术语“单倍型”可指位于特定基因座处的等位基因或指位于沿染色体区段的多个基因座处的等位基因。
[0051]
如本文使用的,关于序列的“异源性”是指该序列源于外来物种,或者,如果源于相同物种的话,则是通过蓄意人为干预从其在组合物和/或基因组基因座中的天然形式进行实质性修饰得到的序列。例如,有效地连接至异源多核苷酸的启动子来自与从其衍生该多核苷酸的物种不同的物种,或者,如果来自相同/类似的物种,那么一方或双方基本上由它们的原来形式和/或基因组基因座修饰得到,或者该启动子不是被有效地连接的多核苷酸的天然启动子。
[0052]
术语“杂交体”是指在至少两个遗传上不同的亲本的杂交之间获得的后代。
[0053]
术语“渗入”是指基因座的期望等位基因从一种遗传背景传递到另一种遗传背景的现象。例如,可以经由相同物种的两个亲本之间的有性杂交将指定基因座处的所需等位基因的渗入传递给至少一个后代,其中所述亲本中的至少一个在其基因组内具有所述所需的等位基因。可替代地,例如等位基因的传递可以通过两个供体基因组之间的重组而发生,例如在融合原生质体中,其中至少其中一个供体原生质体在其基因组中具有所希望的等位基因。所需的等位基因可以,例如通过与表型相关联的标记,在qtl、转基因等处进行检测。在任何情况下,包含所述所需等位基因的后代可以反复与具有所需遗传背景的品系回交并选择所需等位基因,以产生固定在选择的遗传背景中的等位基因。
[0054]
当“渗入”重复两次或更多次时,该方法通常被称为“回交”。
[0055]“品系”或“品种”是一组具有相同亲本的个体,其通常在一定程度上是近交的,并且在大多数基因座处通常是纯合和同质的(同基因的或接近同基因的)。“亚系”是指遗传上
不同于起源于相同祖先的其他类似近交系亚群的近交系亚群。
[0056]
如本文所使用的,术语“连锁”用于描述一种标记基因座与另一种标记基因座或一些其他基因座的相关联程度。分子标记与影响表型的基因座之间的连锁关系以“概率”或“调整的概率”表示。连锁可以表示为所需的限制或范围。例如,在一些实施例中,当任何标记与任何其他标记在单次减数分裂图谱(基于已经进行一轮减数分裂的群体(例如,f2)的遗传图谱)上分隔小于50、40、30、25、20或15个图距单位(或cm)时,所述标记是连锁的(遗传上或物理上)。在一些方面,限定加括号的连锁范围是有利的,例如,在10cm和20cm之间、在10cm和30cm之间、或在10cm和40cm之间。标记与第二基因座的连锁越紧密,标记对第二基因座的指示效果越好。因此,“紧密连锁的基因座”,例如标记基因座和第二基因座显示10%或更低、优选约9%或更低、还更优选约8%或更低、又更优选约7%或更低、还更优选约6%或更低、又更优选约5%或更低、还更优选约4%或更低、又更优选约3%或更低、以及还更优选约2%或更低的基因座间重组频率。在非常优选的实施例中,相关基因座显示约1%或更低,例如约0.75%或更低、更优选约0.5%或更低、或又更优选约0.25%或更低的重组频率。定位于相同染色体并且具有使得两个基因座之间的重组以小于10%(例如,约9%、8%、7%、6%、5%、4%、3%、2%、1%、0.75%、0.5%、0.25%或更少)的频率发生的距离的所述两个基因座也被认为是彼此“邻近”。因为一个cm是显示1%的重组频率的两个标记之间的距离,因此任何标记与紧密相邻(例如,以等于或小于10cm的距离)的任何其他标记紧密连锁(遗传上和物理上)。在相同染色体上的两个紧密连锁的标记可相互定位为9、8、7、6、5、4、3、2、1、0.75、0.5或0.25cm或更近。
[0057]
术语“连锁不平衡”是指遗传基因座或性状(或两者)的非随机分离。在任一情况下,连锁不平衡意味着相关的基因座沿着一段染色体在物理上足够接近,以便它们以高于随机(即非随机)的频率一起分离。显示连锁不平衡的标记被认为是连锁的。连锁的基因座有超过50%的机会(例如约51%至约100%的机会)发生共分离。换句话说,共分离的两个标记具有小于50%(并且根据定义,在相同连锁群上分隔小于50cm)的重组频率。如本文所使用的,连锁可以存在于两个标记之间,或可替代地,标记和影响表型的基因座之间。标记基因座可以与性状“相关联”(连锁)。标记基因座和影响表型性状的基因座的连锁程度例如通过该分子标记与所述表型共分离的统计学概率(例如,f统计或lod评分)进行测量。
[0058]
连锁不平衡最常见地用量度r2评估,所述量度r2使用以下文献中的公式计算:hill,w.g.和robertson,a,theor.appl.genet.[理论和应用遗传学]38:226
‑
231(1968)。当r2=1时,两个标记基因座间存在完全的连锁不平衡,意味着所述标记还未进行重组分离并且具有相同的等位基因频率。所述r2值依赖于所使用的群体。r2值大于1/3显示了用于定位的足够强的连锁不平衡(ardlie等人,nature reviews genetics[遗传学自然评论]3:299
‑
309(2002))。因此,当成对标记基因座间的r2值大于或等于0.33、0.4、0.5、0.6、0.7、0.8、0.9、或1.0时,等位基因处于连锁不平衡。
[0059]
如本文所使用的,“连锁平衡”描述其中两个标记独立地分离的情况,即,在后代中随机分配。显示连锁平衡的标记被认为是不连锁的(无论它们是否位于相同染色体上)。
[0060]“基因座”是在染色体上的位置,例如,核苷酸、基因、序列或标记所处的位置。基因座在植物基因组(“内源基因组基因座”)中对于植物可以是内源的。
[0061]“优势对数(lod)值”或“lod评分”(risch,science[科学]255:803
‑
804(1992))用
于遗传区间定位以描述两个标记基因座之间的连锁程度。两个标记间lod评分为三指示连锁概率比无连锁的概率高1000倍,而lod评分为二指示连锁概率比无连锁的概率高100倍。大于或等于二的lod评分可以用于检测连锁。lod评分还可以用于在“数量性状基因座”作图中显示标记基因座和数量性状之间的关联强度。在这一情况下,lod评分大小取决于所述标记基因座与影响所述数量性状的基因座之间的紧密度,以及所述数量性状效应的大小。
[0062]“标记”是发现遗传或物理图谱上的位置或发现标记与性状基因座(影响性状的基因座)之间的连锁的方式。标记所检测的位置可通过检测多态性等位基因及其遗传定位而获知,或通过对已经进行物理标测的序列进行杂交、序列匹配或扩增而获知。标记可以是dna标记(检测dna多态性)、蛋白(检测编码的多肽的变异)或简单遗传的表型(诸如“腊质”表型)。可从基因组核苷酸序列或从表达的核苷酸序列(例如,从剪接的rna或cdna)开发dna标记。根据dna标记技术,所述标记由侧接于所述基因座的互补性引物和/或与所述基因座处的多态性等位基因杂交的互补性探针组成。dna标记或遗传标记还可用于描述该染色体自身上的基因、dna序列或核苷酸(而非用于检测该基因或dna序列的组分),并且其通常在该dna标记与人遗传学中的特定性状相关联时使用(例如乳腺癌标记)。术语标记基因座是该标记所检测的基因座(基因、序列或核苷酸)。
[0063]
检测群体成员之间的遗传多态性的标记在本领域中是公认的。标记可由其所检测的多态性的类型以及用于检测所述多态性的标记技术所定义。标记类型包括但不限于:限制性片段长度多态性检测(rflp)、同功酶标记检测、随机扩增的多态性dna(rapd)、扩增片段长度多态性检测(aflp)、简单重复序列检测(ssr)、植物基因组的扩增可变序列检测、自主序列复制检测、或单核苷酸多态性检测(snp)。snp可通过,例如,通过dna测序、基于pcr的序列特异性扩增方法、通过等位基因特异性杂交(ash)进行的多核苷酸多态性检测、动态等位基因特异性杂交(dash)、分子信标、微阵列杂交、寡核苷酸连接酶分析、flap内切核酸酶、5’内切核酸酶、引物延伸、单链构象多态性(sscp)或温度梯度凝胶电泳(tgge)进行检测。dna测序(诸如焦磷酸测序技术)具有能够检测组成单倍型的一系列连锁snp等位基因的优点。单倍型倾向于比snp更具信息性(检测更高水平的多态性)。
[0064]“标记等位基因”,可替代地“标记基因座的等位基因”可以指群体中标记基因座处发现的多个多态性核苷酸序列中之一。
[0065]“标记辅助选择”(或mas)是基于标记基因型选择个体植物的方法。
[0066]“标记单倍型”是指标记基因座处的等位基因的组合。
[0067]“标记基因座”是物种基因组中的特定染色体定位,在该定位处可发现特异标记。标记基因座可用于追踪第二连锁基因座(例如影响表型性状表达的连锁基因座)的存在。例如,标记基因座能够用于监控在遗传上或物理上连锁的基因座处的等位基因的分离。
[0068]
如上所述,当鉴定连锁基因座时,术语“分子标记”可以用于指遗传标记,或其用作参照点的编码产物(例如,蛋白)。标记能够来源于基因组核苷酸序列或来源于表达的核苷酸序列(例如来源于剪接的rna、cdna等),或来源于编码的多肽。该术语也指与标记序列互补或与其侧接的核酸序列,如用作探针或能够扩增所述标记序列的引物对的核酸。“分子标记探针”是可以用于鉴定标记基因座存在与否的核酸序列或分子,例如与标记基因座序列互补的核酸探针。可替代地,在某些方面,标记探针是指能够区别存在于标记基因座处的特定等位基因的任何类型(即基因型)的探针。当核酸,例如根据沃森
‑
克里克碱基配对原则,
在溶液中特异性杂交时,所述核酸是“互补的”。当位于插入缺失区域,例如本文所述的非共线区域时,本文所述标记中的一些也称为杂交标记。这是因为,根据定义,所述插入区域是关于无所述插入的植物的多态性。因此,该标记仅需要指示所述插入缺失区域是否存在。任何合适的标记检测技术都可以用于鉴定此类杂交标记,例如在本文提供的实例中使用snp技术。
[0069]
基因组的“物理图谱”是显示染色体dna上可鉴定的标志(包括基因、标记等)的线性顺序的图谱。然而,与遗传图谱相比,标志间的距离是绝对的(例如以碱基对或者分离的和重叠的连续基因片段测量)并且不基于基因重组(所述基因重组可在不同的群体中有所变化)。
[0070]“植物”可为整个植株、其任何部分、或来源于植物的细胞或组织培养物。因此,术语“植物”可以指以下中的任一项:整个植株、植物组成或器官(例如叶片、茎、根等)、植物组织、种子、植物细胞、和/或以上各项的后代。植物细胞是植物的细胞,其取自植物或来源于取自植物的细胞培养物。
[0071]“多态性”是群体内两个或更多个个体之间的dna中的变异。多态性在群体中优选地具有至少1%的频率。有用的多态性可以包括单核苷酸多态性(snp)、简单重复序列(ssr)、或插入/缺失多态性(本文中也称为“插入缺失”)。
[0072]“后代植物”是通过两个植物间的杂交所产生的植物。
[0073]
术语“数量性状基因座”或“qtl”是指在至少一种遗传背景下(例如在至少一个育种群体中),与数量表型性状的差异表达关联的dna区域。qtl的区域涵盖或紧密地连锁于影响所考虑的性状的一个或多个基因。“qtl的等位基因”可以包含在连续的基因组区域或连锁群中的多个基因或其他遗传因子,例如单倍型。qtl的等位基因可以表示在指定窗口内的单倍型,其中所述窗口是可以用一组的一个或多个多态性标志物定义和追踪的连续的基因组区域。单倍型可以指定被窗口内的每一标志物的等位基因的独特指纹定义。
[0074]“轮回亲本”是指在渐渗方案中用于多个回交的亲本:从具有不期望背景的供体向具有更期望遗传背景的优良系转移期望性状的过程。
[0075]“参考序列”或“共有序列”是用作序列比对基础的定义的序列。通过对在该基因座处的多个品系进行测序,在序列比对程序(例如sequencher)中比对这些核苷酸序列并且然后获得所述比对的最通用核苷酸序列来获得phm标记的参考序列。见于这些个体序列中的多态性标注于所述共有序列中。参考序列通常并非任何个体dna序列的精确复制,而是代表可用序列的混合并且用于设计针对该序列内的多态性的引物和探针。
[0076]
在“相斥”相连锁(“repulsion”phase linkage)中,目的基因座处的“有利”等位基因与邻近的标记基因座处的“不利”等位基因物理上连锁,并且两个“有利”等位基因不被一起继承(即,两个基因座是在不同的同源染色体上彼此“异相”)。
[0077]
本文公开的实施例可用于任何植物物种,包括但不限于单子叶植物和双子叶植物。目的植物的实例包括但不限于玉米(corn,zea mays),芸苔属(brassica)物种(例如,甘蓝型油菜(b.napus)、芜菁(b.rapa)、芥菜(b.juncea))(特别是可用作种子油来源的那些芸苔属物种),苜蓿(紫花苜蓿(medicago sativa)),水稻(rice,oryza sativa),黑麦(rye,secale cereale),高粱(sorghum,sorghum bico,or,sorghum vulgare),粟(例如,珍珠粟(pearl millet,pennisetum glaucum)、黍(proso millet,panicum miliaceum)、谷子
(foxtail millet,setaria italica)、龙爪稷(finger millet,eleusine coracana)),向日葵(sunflower,helianthus annuus),红花(safflower,carthamus tinctorius),小麦(wheat,triticum aestivum),大豆(soybean,glycinle max),烟草(tobacco,nicotiana tabacum),马铃薯(potato,solanum tuberosum),花生(peanut,arachis hypogaea),棉花(海岛棉(gossypium barbadense)、陆地棉(gossypium hirsutum)),甘薯(番薯(ipomoea batatas)),木薯(cassava,manihot esculenta),咖啡(咖啡属(coffea)物种),椰子(coconut,cocos nucifera),菠萝(pineapple,ananas comosus),柑橘树(柑橘属(citrus)物种),可可(cocoa,theobroma cacao),茶树(tea,camellia sinensis),香蕉(芭蕉属(musa)物种),鳄梨(avocado,persea americana),无花果(fig,ficus casica),番石榴(guava,psidium guajava),芒果(mango,mangifera indica),橄榄(olive,olea europaea),木瓜(番木瓜(carica papaya))),腰果(cashew,anacardium occidentale),澳洲坚果(macadamia,macadamia integrifolia),巴旦杏(almond,prunus amygdalus),甜菜(sugar beets,beta vulgaris),甘蔗(甘蔗属(saccharum)物种),燕麦,大麦,蔬菜,观赏植物和针叶树。
[0078]
蔬菜包括番茄(tomatoes,lycopersicon esculentum)、莴苣(例如,莴苣(lactuca sativa))、青豆(菜豆(phaseolus vulgaris))、利马豆(lima bean,phaseolus limensis)、豌豆(香豌豆属(lathyrus)物种)和黄瓜属的成员例如黄瓜(cucumber,c.sativus)、香瓜(cantaloupe,c.cantalupensis)和甜瓜(musk melon,c.melo)。观赏植物包括杜鹃(杜鹃花属(rhododendron)物种)、绣球花(hydrangea,macrophylla hydrangea)、木槿(hibiscus,hibiscus rosasanensis)、玫瑰(蔷薇属(rosa)物种)、郁金香(郁金香属(tulipa)物种)、水仙(水仙属(narcissus)物种)、矮牵牛(petunias,petunia hybrida)、康乃馨(carnation,dianthus caryophyllus)、一品红(poinsettia,euphorbia pulcherrima)和菊花。可以用于实践实施例的针叶树包括(例如)松树如火炬松(loblolly pine,pinus taeda)、湿地松(slash pine,pinus elliotii)、西黄松(ponderosa pine,pinus ponderosa)、黑松(lodgepole pine,pinus contorta)和辐射松(monterey pine,pinus radiata);花旗松(douglas
‑
fir,pseudotsuga menziesii);西方铁杉(western hemlock,tsuga canadensis);北美云杉(sitka spruce,picea glauca);红杉(redwood,sequoia sempervirens);枞树(true firs),如银杉(胶冷杉(ables amabilis))和胶枞(香脂冷杉(abies balsamea));以及雪松,如西方红雪松(北美乔柏(thuja plicata))和阿拉斯加黄雪松(黄扁柏(chamaecyparis nootkatensis))。所述实施例的植物包括作物植物(例如玉米、苜蓿、向日葵、芸苔属、大豆、棉花、红花、花生、高粱、小麦、粟、烟草等),如玉米和大豆植物。
[0079]
草坪草包括但不限于:一年生早熟禾(annual bluegrass,poa annua);一年生黑麦草(黑麦草(lolium multiflorum));加拿大早熟禾(canada bluegrass,poa compressa);紫羊茅(chewing’s fescue,festuca rubra);细弱翦股颖(colonial bentgrass,agrostis tenuis);匍匐翦股颖(creeping bentgrass,agrostis palustris);沙生冰草(crested wheatgrass,agropyron desertorum);扁穗冰草(fairway wheatgrass,agropyron cristatum);硬羊茅(长叶羊茅(festuca longifolia));草地早熟禾(kentucky bluegrass,poa pratensis);鸭茅(orchardgrass,dactylis glomerata);多
年生黑麦草(perennial ryegrass,lolium perenne);红狐茅(紫羊茅(festuca rubra));小糠草(redtop,agrostis alba);粗茎早熟禾(rough bluegrass,poa trivialis);羊茅(sheepfescue,festuca ovina);无芒雀麦(smooth bromegrass,bromus inermis);高羊茅(tallfescue,festuca arundinacea);梯牧草(timothy,phleum pratense);绒毛剪股颖(velvet bentgrass,agrostis canina);碱茅(weeping alkaligrass,puccinelliadistans);蓝茎冰草(western wheatgrass,agropyron smithii);狗牙根(狗牙根属(cynodon)物种);圣奥古斯丁草(st.augustine grass,stenotaphrumm secundatum);结缕草(结缕属(zoysia)物种);百喜草(bahia grass,paspalum notatum);地毯草(carpet grass,axonopus affinis);假俭草(centipede grass,eremochloa ophiuroides);隐花狼尾草(kikuyu grass,pennisetum clandesinum);海滨雀稗(seashore paspalum,paspalum vaginatum);格兰马草(blue gramma,bouteloua gracilis);野牛草(buffalo grass,buchloe dactyloids);垂穗草(sideoats gramma,bouteloua curtipendula)。
[0080]
目的植物包括提供目的种子的谷物类植物、油料种子植物和豆科植物。目的种子包括谷物种子,例如玉米、小麦、大麦、水稻、高粱、黑麦、粟等。油料种子植物包括棉花、大豆、红花、向日葵、芸苔属、玉蜀黍、苜蓿、棕榈、椰子、亚麻、蓖麻、橄榄等。豆科植物包括豆类和豌豆。豆类包括瓜耳豆、槐豆、胡芦巴、大豆、四季豆、豇豆、绿豆、利马豆、蚕豆、小扁豆、鹰嘴豆等。
[0081]
遗传定位
[0082]
已经认识到,在相当一些情况下可在生物体的基因组中定位与特定性状相关联的特定遗传基因座。植物育种人员可以有利地使用分子标记通过检测标记等位基因来鉴定所需的个体,所述标记等位基因显示与所需表型共分离的统计学上显著的概率,表现为连锁不平衡。通过鉴定与目的性状共分离的分子标记或分子标记簇,所述植物育种人员能够通过选择合适的分子标记等位基因(称为标记辅助选择的方法)来迅速选择所需表型。
[0083]
多种方法可用于检测与目的性状共分离的分子标记或分子标记簇。这些方法的基本理念是检测具有显著不同平均表型的可替代的基因型(或等位基因)的标记。因此,比较标记基因座之间的可替代的基因型(或等位基因)之间的差异大小或所述差异的显著性水平。推断性状基因位于最靠近具有最大相关性的基因型差异的一个或多个标记的位置。这样两种用于检测目的性状基因座的方法是:1)基于群体的关联分析和2)传统的连锁分析。
[0084]
在基于群体的关联分析中,从具有多个创始者(例如优良育种品系)的已有群体中获得品系。基于群体的关联分析依靠连锁不平衡(ld)和下列观点:在非结构化的群体中,在如此多代随机交配之后,仅有控制目的性状的基因和与这些基因紧密连锁的标记之间的关联将保留下来。实际上,大多数已有群体具有群体亚结构。因此,通过把个体分配到群体中,使用得自跨基因组无规分布的标记的数据,采用结构化关联方法有助于控制群体结构,从而最小化由于单个群体(也称为亚群体)内的群体结构带来的不平衡。对于在亚群体中每个品系,将表型值与每个标记基因座处的基因型(等位基因)进行比较。显著的标记
‑
性状关联指示标记基因座和涉及该性状表达的一个或多个基因座接近。
[0085]
传统的连锁分析基于相同的原理;然而,连锁不平衡通过从少量建立者创建群体而生成。选择创建者以最大化结构化群体内的多态性水平,并且评估多态性位点与给定表
型的共分离水平。已经使用大量统计学方法来鉴定显著的标记
‑
性状关联。一种此类方法是区间作图方法(lander和botstein,genetics[遗传学]121:185
‑
199(1989),其中针对控制目的性状的基因位于该位置的概率来测试沿遗传图谱(例如以1cm的区间)的许多位置中的每一个位置。基因型/表型数据用于计算每个测试位置的lod评分(概率比率的对数)。当lod评分大于阈值时,存在控制目的性状的基因位于遗传图谱上的该定位处的显著证据(将位于两个特定标记基因座之间)。
[0086]
标记和连锁关系
[0087]
连锁的常见量度是性状共分离的频率。这可以以共分离百分比(重组频率)表示,或以厘摩(cm)表示。cm是遗传重组频率的量度单位。一个cm等于有1%的机会,一个遗传基因座处的性状会由于单代中的交换而与另一个基因座处的性状分离(意味着这些性状总共有99%的机会发生分离)。由于染色体距离与性状之间的交换事件的频率大致成正比,因此存在与重组频率相关联的近似物理距离。
[0088]
标记基因座本身是性状,并且在分离期间能够通过跟踪该标记基因座、根据标准连锁分析对其进行评估。因此,一个cm等于有1%的机会,一个标记基因座会由于单代中的交换而与另一个基因座分离。
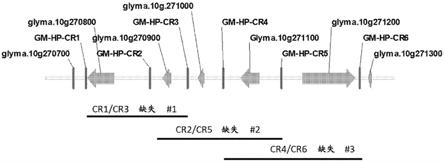
[0089]
标记距离控制目的性状的基因越近,则该标记作为所述所需性状的指示越有效和有利。紧密连锁的基因座显示约10%或更低、优选约9%或更低、还更优选约8%或更低、又更优选约7%或更低、还更优选约6%或更低、又更优选约5%或更低、还更优选约4%或更低、又更优选约3%或更低、以及还更优选约2%或更低的基因座间杂交频率。在高度优选的实施例中,相关基因座(例如标记基因座和靶基因座)显示约1%或更低、例如约0.75%或更低、更优选地约0.5%或更低、或又更优选地约0.25%或更低的重组频率。因此,所述基因座分开距离为约10cm、9cm、8cm、7cm、6cm、5cm、4cm、3cm、2cm、1cm、0.75cm、0.5cm或0.25cm或更低。换言之,定位于相同染色体并且具有使得两个基因座之间的重组以小于10%(例如,约9%、8%、7%、6%、5%、4%、3%、2%、1%、0.75%、0.5%、0.25%或更少)的频率发生的距离的所述两个基因座被认为是彼此“邻近的”。
[0090]
尽管特定的标记等位基因可以与期望性状的增加或减少的表型共分离,重要的是注意所述标记基因座不一定引起所述期望性状表型的表达。例如,该标记多核苷酸序列是负责表型的基因的一部分(例如,是基因可读框的一部分)不是必需条件。特异标记等位基因与性状之间的关联性,是由于在所述等位基因所起源的植物品系中,所述标记等位基因和所述等位基因之间的初始“偶联”相连锁。最后通过反复重组,所述标记和遗传基因座之间的交换事件能够改变这种取向。由于这个原因,所述有利的标记等位基因可以根据存在于具有有利性状的亲本中的用于创建分离群体的连锁相发生改变。这不改变可以使用所述标记来监测表型分离的事实。它仅仅改变在给定分离群体中哪个标记等位基因被认为是有利的。
[0091]
标记辅助选择
[0092]
分子标记可以用于多种植物育种应用(如参见staub等人,(1996)hortscience[园艺科学]31:729
‑
741;tanksley(1983)plant molecular biology reporter.[植物分子生物学导报]1:3
‑
8)。受关注的主要领域之一是使用标记辅助选择增加回交和基因渗入的效率。展示出与影响所需表型性状的基因座连锁的分子标记为在植物群体中选择性状提供了
有用工具。在表型难以测定的情况下尤其如此。由于dna标记测定比田间表型分析更省力、更便宜并且占用的物理空间更小,可测定更大的群体,增加了发现具有从供体品系移动至受体品系的靶区段的重组体的概率。连锁越紧密,标记越有用,这是因为重组不太可能发生于所述标记和引起该性状的基因之间,所述重组可导致假阳性。由于需要双重组事件,侧接标记减少了假阳性选择发生的概率。理想情况是基因本身具有标记,使得标记和基因之间的重组不能发生。此类标记称为“完美标记”。
[0093]
当基因通过标记辅助选择渗入时,不仅引入了基因而且引入了侧接区域(gepts.(2002).crop sci[作物科学];42:1780
‑
1790)。这称为“连锁累赘”。在供体植物与受体植物极不相关的情况下,这些侧接区域携带可以编码农艺学上不需要的性状的另外的基因。即便与优良植物品系回交多个周期后,该“连锁累赘”也可能导致产量下降或其他负面农艺学特征。这有时也称为“产量累赘”。侧接区域的大小可以通过另外的回交而减小,虽然这并不总是成功的,因为育种人员不能控制该区域或重组断点的大小(young等人,(1998)genetics[遗传学]120:579
‑
585)。在由于低同源性、低重组频率或非共线性而导致不成功的作图的情况下,本文公开的方法提供了传统作图的替代策略。在经典育种中,通常只是偶然地,选择了有助于减小供体区段大小的重组(tanksley等人,(1989).biotechnology[生物技术]7:257
‑
264)。即使在20次此类型的回交后,可以预期找到相当大的仍然与所述基因连锁的供体染色体碎片被选择。然而如果使用标记的话,就可能选取那些在受关注的基因附近经历了重组的稀有个体。在150株回交植物中,有95%的机会,至少一株植物将经历该基因的1cm(基于单次减数分裂图距)内的杂交。标记使得能够明确鉴定这些个体。使用300株植物的一次另外的回交,在该基因另一例的1cm单次减数分裂图距内有95%的杂交概率,从而产生在基于单次减数分裂图距的小于2cm的靶基因附近的区段。这用标记可以在两代中实现,而不用标记时则需要平均100代(参见tanksley等人,同上)。当基因的确切定位已知时,围绕该基因的侧接标记可用于在不同的群体大小中对重组进行选择。例如,在更小的群体中,预期重组可以进一步远离该基因,因此需要更远端的侧接标记来检测该重组。
[0094]
实施标记辅助选择的主要组成是:(i)限定在其中标记
‑
性状关联性将被测定的群体,其可以是分离群体、或随机的或结构化的群体;(ii)监测多态性标记相对于该性状的分离或关联性,并使用统计学方法确定连锁或关联性;(iii)基于统计学分析的结果限定一组所需标记,以及(iv)使用和/或外推该信息至当前的育种种质组中,以使得能够作出基于标记的选择决定。本公开中描述的标记,以及其他标记类型,例如ssr和flp,可以用于标记辅助选择方案中。
[0095]
ssr可以被定义为长度6bp或更小的串联重复dna的相对较短序列(tautz(1989)nucleic acid research[核酸研究]17:6463
‑
6471;wang等人(1994)theoretical and applied genetics[理论和应用遗传学],88:1
‑
6)。多态性由于重复单元数目的变化而产生,这可能是由于dna复制过程中的滑移引起的(levinson和gutman(1987)mol biol evol[分子生物学与进化]4:203
‑
221)。重复长度的变化可以通过设计pcr引物至保守的非重复侧翼区域来检测(weber和may(1989)am j hum genet.[美国人类遗传学]44:388
‑
396)。因为ssr是多等位基因的、共显性的、可再生的、并适合于高通量自动化,所以非常适合作图和标记辅助选择(rafalski等人,(1996)generating and using dna markers in plants[在植物中生成和使用dna标记].在:non
‑
mammalian genomic analysis:a practical guide
[非哺乳动物基因组分析:实用指南].学术出版社(academic press),第75
‑
135页中)。
[0096]
可以产生各种类型的ssr标记,并且可以通过扩增产物的凝胶电泳获得ssr谱。标记基因型的评分基于扩增片段的大小。也可以生成各种类型的flp标记。最常见地,使用扩增引物来生成片段长度多态性。除了通过引物扩增的区域通常不是高度重复的区域之外,这样的flp标记在许多方面与ssr标记相似。通常由于插入或缺失(“indel”),扩增区域或扩增子在种质间仍具有足够的可变性,使得由扩增引物产生的片段能够在多态性个体中被区分,并且已知此类插入缺失常常发生于植物中(evans等人plos one[公共科学图书馆
·
综合](2013).8(11):e79192)。
[0097]
snp标记检测单碱基对核苷酸取代。在所有分子标记类型中,snp是最丰富的,因此具有提供最高遗传图谱分辨率的潜力(plos one[公共科学图书馆
·
综合](2013).8(11):e79192)。由于snp不需要大量的dna并且测定的自动化可以是直接的,所以可以以所谓的“超高通量”方式,以甚至比ssr更高的通量水平测定snp。snp也有可能成为相对低成本的系统。这三个因素一起使得将snp用于标记辅助选择中具有高度的吸引力。可利用如下几种方法用于snp基因分型,包括但不限于:杂交、引物延伸、寡核苷酸连接、核酸酶切割、微测序和编码球(coded sphere)。如下文献中已经对这些方法进行了综述:gut(2001)hum mutat[人类基因突变]17,第475
‑
492页;shi(2001)clin chem[临床化学]47,第164
‑
172页;kwok(2000)pharmacogenomics[药物基因组学]1,第95
‑
100页;以及bhattramakki和rafalski(2001),discovery and application of single nucleotide polymorphism markers in plants[单核苷酸多态性标记在植物中的发现与应用],在:r.j.henry编辑,plant genotyping:the dna fingerprinting of plants,cabi publishing,wallingford[植物基因分型:植物的dna指纹识别,cabi出版社,瓦林福德]中。广泛的可商购的技术利用这些和其他方法来检测snp,所述可商购的技术包括:masscode.tm.(凯杰公司(qiagen))、(第三波技术公司(third wave technologies))和invader(美国应用生物系统公司(applied biosystems))、(美国应用生物系统公司)以及(依诺米那公司(illumina))。
[0098]
可以使用序列内或跨连锁序列的许多snp来描述任何特定基因型的单倍型(ching等人,(2002),bmc genet.[bmc遗传学]3:19,gupta等人,2001,rafalski(2002b),plant science[植物科学]162:329
‑
333)。单倍型可以比单个snp更具信息性,并且可以更详细地描述任何特定的基因型。例如,单一的snp可能是具有早熟的特定品系或品种的等位基因`t`,但在用于轮回亲本的植物育种群体中也可能出现等位基因`t`。在这种情况下,单倍型(例如连锁的snp标记处的等位基因的组合)可能更具信息性。一旦将唯一单倍型分配给供体染色体区域,该单倍型可以用于该群体或其任何亚群中以确定个体是否具有特定的基因。参见,例如,wo 2003054229。使用本领域普通技术人员已知的自动化高通量标记检测平台使得该方法高效且有效。
[0099]
除上述的ssr、flp和snp外,其他类型的分子标记也被广泛使用,包括但不限于:表达的序列标签(est)、源自est序列的ssr标记、随机扩增的多态性dna(rapd)和其他基于核酸的标记。
[0100]
同工酶谱和连锁形态特征在某些情况下也可以间接用作标记。尽管它们不直接检测dna差异,但它们往往受到特定遗传差异的影响。然而,检测dna变异的标记比同工酶或形态学标记多得多且更多态(tanksley(1983)plant molecular biology reporter[植物分子生物学导报]1:3
‑
8)。
[0101]
序列比对或重叠群还可以用于发现本文所列特异标记的上游或下游的序列。然后使用接近于本文所述的标记的这些新序列来发现和开发功能上等效的标记。例如,对不同的物理和/或遗传图谱进行比对以定位未在本公开中描述但位于相似区域内的等效标记。这些图谱可能在植物物种内,或者甚至跨越与植物进行遗传上或物理上比对的其他物种,例如玉蜀黍、稻、小麦或大麦。在一些实施例中,通过基因编辑对新序列进行修饰或缺失以进行精细作图或因果基因鉴定。
[0102]
一般来说,标记辅助选择使用多态性标记,这些标记已被鉴定为具有与期望性状表型共分离的显著可能性。推测这样的标记被作图在提供植物中期望性状的表型的一个或多个基因附近,并且被认为是期望性状的指示物或标记。测试植物中标记中所需的等位基因的存在,并且预期在一个或多个基因座处含有所需基因型的植物将所需基因型连同所需表型一起转移至其后代。因此,可以通过检测一个或多个标记等位基因来选择具有期望性状的增加或减少的表型的植物,并且此外,还可以选择来自这些植物的后代植物。因此,获得在给定染色体区域中含有所需基因型的植物,并且然后与另一植物杂交。然后使用一种或多种标记对这种杂交的后代进行基因型评估,并且然后选择在给定染色体区域中具有相同基因型的后代植物。
[0103]
基因编辑
[0104]
修饰或改变内源基因组dna的方法是本领域已知的。在一些方面,提供了用于修饰天然存在的多核苷酸或整合的转基因序列(包括调节元件、编码序列和非编码序列)的方法和组合物。这些方法和组合物在将核酸靶向至基因组中的预先工程化的靶识别序列中是有用的。多核苷酸的修饰可以例如通过将单链或双链断裂(“dsb”)引入dna分子中来完成。
[0105]
由双链断裂诱导剂(例如在多核苷酸链中切割磷酸二酯键的内切核酸酶)诱导的双链断裂可导致dna修复机制的诱导,包括非同源末端连接途径以及同源重组。内切核酸酶包括一系列不同的酶,包括限制性内切核酸酶(参见例如roberts等人,(2003)nucleic acids res[核酸研究]1:418
‑
20),roberts等人,(2003)nucleic acids res[核酸研究]31:1805
‑
12,和belfort等人,(2002)在mobile dna[运动dna]ii,第761
‑
783页,编辑craigie等人,(asm出版社,华盛顿特区)),大范围核酸酶(参见例如wo 2009/114321;gao等人(2010)plant journal[植物杂志]1:176
‑
187),tal效应子核酸酶或talen(参见例如us 20110145940,christian,m.,t.cermak,等人2010.targeting dna double
‑
strand breaks with tal effector nucleases[用tal效应子核酸酶靶向dna双链断裂].genetics[遗传学]186(2):757
‑
61和boch等人,(2009),science[科学]326(5959):1509
‑
12),锌指核酸酶(参见例如kim,y.g.,j.cha,等人(1996).“hybrid restriction enzymes:zinc finger fusions to foki cleavage[杂交限制性内切酶:锌指与foki融合蛋白的切割]”)和crispr
‑
cas内切核酸酶(参见例如2007年3月1日公开的wo 2007/025097)。
[0106]
一旦在基因组中诱导了双链断裂,则细胞dna修复机制被激活以修复断裂。有两种dna修复途径。一种被称为非同源末端连接(nhej)途径(bleuyard等人,(2006)dna repair
[dna修复]5:1
‑
12),另一种被称为同源
‑
定向修复(hdr)。染色体的结构完整性典型地通过nhej来保存,但是缺失、插入或其他重排(如染色体易位)是可能的(siebert和puchta,2002 plant cell[植物细胞]14:1121
‑
31;pacher等人,2007 genetics[遗传学]175:21
‑
9)。hdr途径是修复双链dna断裂的另一种细胞机制,并且包括同源重组(hr)和单链退火(ssa)(lieber.2010 annu.rev.biochem.[生物化学年鉴]79:181
‑
211)。
[0107]
除了双链断裂诱导剂,还可以实现位点特异性碱基转化以工程化一个或多个核苷酸变化,从而在基因组中创建一个或多个本文所述的位点特异性修饰。这些包括例如,由c
·
g至t
·
a或a
·
t至g
·
c碱基编辑脱氨酶介导的位点特异性碱基编辑(gaudelli等人,programmable base editing of a
·
t to g
·
c in genomic dna without dna cleavage[在无dna切割时基因组dna中a
·
t至g
·
c的可编程碱基编辑].
″
nature[自然](2017);nishida等人“targeted nucleotide editing using hybrid prokaryotic and vertebrate adaptive immune systems[使用杂交体原核和脊椎动物适应性免疫系统进行靶向核苷酸编辑].”science[科学]353(6305)(2016);komor等人“programmable editing of a target base in genomic dna without double
‑
stranded dna cleavage[在无双链dna切割时基因组dna中靶碱基的可编程编辑].”nature[自然]533(7603)(2016):420
‑
4)。位点特异性修饰还可以包括核苷酸或一个以上核苷酸的缺失。
[0108]
在一些实施例中,可以通过在所需改变附近的基因组中在限定位置诱导双链断裂(dsb)来促进基因编辑。在一些实施例中,可以将dsb的引入与多核苷酸修饰模板的引入组合。
[0109]
可以通过本领域已知的任何方法将多核苷酸修饰模板引入细胞中,所述方法例如但不限于瞬时引入方法、转染、电穿孔、显微注射、颗粒介导的递送、局部施用、晶须介导的递送、经由细胞穿透肽的递送或介孔二氧化硅纳米颗粒(msn)介导的直接递送。
[0110]“经修饰的核苷酸”、“经编辑的核苷酸”或“基因组编辑”是指当与其非修饰的核苷酸序列相比时,包含至少一个改变的目的核苷酸序列。此类改变包括,例如:(i)至少一个核苷酸的替代、(ii)至少一个核苷酸的缺失、(iii)至少一个核苷酸的插入、或(iv)(i)
‑
(iii)的任何组合。“经编辑的细胞”或“经编辑的植物细胞”是指与在基因组序列中不包含至少一个改变的对照细胞或植物细胞相比,在基因组序列中包含所述改变的细胞。
[0111]
如本文所用,术语“多核苷酸修饰模板”或“修饰模板”是指当与待编辑的靶核苷酸序列相比时,包含至少一个核苷酸修饰的多核苷酸。核苷酸修饰可以是至少一个核苷酸取代、添加或缺失。任选地,多核苷酸修饰模板可以进一步包含位于至少一个核苷酸修饰侧翼的同源核苷酸序列,其中侧翼同源核苷酸序列为待编辑的希望的核苷酸序列提供了充足同源性。
[0112]
编辑组合有dsb和修饰模板的基因组序列的过程通常包括:向宿主细胞提供识别染色体序列中的靶序列的dsb诱导剂或编码dsb诱导剂的核酸,其中dsb诱导剂能够诱导基因组序列中的dsb;并且提供与待编辑的核苷酸序列相比时包含至少一个核苷酸变化的至少一个多核苷酸修饰模板。内切核酸酶可以通过本领域已知的任何方法提供给细胞,所述方法例如但不限于瞬时引入方法、转染、显微注射、和/或局部施用、或间接经由重组构建体。内切核酸酶可以作为蛋白或作为指导多核苷酸复合物直接提供给细胞或经由重组构建体间接提供。使用本领域已知的任何方法,可以瞬时地将内切核酸酶引入细胞中,或可以将
内切核酸酶并入宿主细胞的基因组中。在crispr
‑
cas系统的情况下,如wo 2016073433中所述的,可以用细胞穿透肽(cpp)促进内切核酸酶和/或指导多核苷酸摄入进细胞。
[0113]
如本文所用,“基因组区域”是指细胞基因组中染色体的区段。在一个实施例中,基因组区域包括存在于靶位点任一例上的细胞的基因组中的染色体的区段,或者可替代地,还包含靶位点的一部分。基因组区域可以包含至少5
‑
10、5
‑
15、5
‑
20、5
‑
25、5
‑
30、5
‑
35、5
‑
40、5
‑
45、5
‑
50、5
‑
55、5
‑
60、5
‑
65、5
‑
70、5
‑
75、5
‑
80、5
‑
85、5
‑
90、5
‑
95、5
‑
100、5
‑
200、5
‑
300、5
‑
400、5
‑
500、5
‑
600、5
‑
700、5
‑
800、5
‑
900、5
‑
1000、5
‑
1100、5
‑
1200、5
‑
1300、5
‑
1400、5
‑
1500、5
‑
1600、5
‑
1700、5
‑
1800、5
‑
1900、5
‑
2000、5
‑
2100、5
‑
2200、5
‑
2300、5
‑
2400、5
‑
2500、5
‑
2600、5
‑
2700、5
‑
2800、5
‑
2900、5
‑
3000、5
‑
3100或更多个碱基,这样使得基因组区域具有足够的同源性以与相应的同源区域进行同源重组。
[0114]
内切核酸酶是在多核苷酸链内切割磷酸二酯键的酶。内切核酸酶包括限制性内切核酸酶,其在特异性位点处切割dna而不损坏碱基;并且包括大范围核酸酶,也称为归巢内切核酸酶(he酶),其相似于限制性内切核酸酶,在特异性识别位点处结合并且切割,然而对于大范围核酸酶,识别位点典型地更长,约18bp或更长(于2012年3月22日提交的专利申请pct/us 12/30061)。基于保守的序列基序将大范围核酸酶分类为四个家族,所述家族是laglidadg、giy
‑
yig、h
‑
n
‑
h、和his
‑
cys box家族。这些基序参与金属离子的配位和磷酸二酯键的水解。he酶的显著之处在于它们的长识别位点,并且还在于耐受其dna底物中的一些序列多态性。对于大范围核酸酶的命名约定相似于对其他限制性内切核酸酶的约定。大范围核酸酶还分别表征为针对由独立的orf、内含子、和内含肽编码的酶的前缀f
‑
、i
‑
、或pi
‑
。在重组过程中的一个步骤涉及在识别位点处或在所述识别位点附近的多核苷酸切割。可以将切割活性用于产生双链断裂。对于位点特异性重组酶和它们的识别位点的综述,参见,sauer(1994)curr op biotechnol[生物技术新见]5:521
‑
7;以及sadowski(1993)faseb[美国实验生物学学会联合会杂志]7:760
‑
7。在一些实例中,重组酶来自整合酶(integrase)或解离酶(resolvase)家族。
[0115]
锌指核酸酶(zfn)是由锌指dna结合结构域和双链
‑
断裂
‑
诱导剂结构域组成的工程化双链断裂诱导剂。识别位点特异性由锌指结构域赋予,所述锌指结构域典型地包含两个、三个、或四个锌指,例如具有c2h2结构,然而其他锌指结构是已知的并且已经被工程化。锌指结构域适于设计特异性结合所选择的多核苷酸识别序列的多肽。zfn包括连接至非特异性内切核酸酶结构域(例如来自iis型内切核酸酶例如foki的核酸酶结构域)的工程化dna结合锌指结构域。额外的功能性可以融合到锌指结合结构域中,所述额外的功能性包括转录激活子结构域、转录阻遏物结构域、和甲基化酶。在一些实例中,核酸酶结构域的二聚化是切割活性所需的。每个锌指在靶dna中识别三个连续的碱基对。例如,3指结构域识别9个连续核苷酸的序列,由于所述核酸酶的二聚化需要,因此两组锌指三联体用于结合18个核苷酸的识别序列。
[0116]
本文中术语“cas基因”是指在细菌系统中通常与侧翼crispr基因座偶联、缔合或接近或在邻近处的基因。术语“cas基因”,“crispr相关的(cas)基因”在本文中可互换地使用。本文的术语“cas内切核酸酶”是指由cas基因编码的蛋白或蛋白复合物。当与适合的多核苷酸组分复合时,本文公开的cas内切核酸酶能够识别、结合特异性dna靶序列的全部或部分、并任选地使特异性dna靶序列的全部或部分产生切口或切割特异性dna靶序列的全部
或部分。本文描述的cas内切核酸酶包含一个或多个核酸酶结构域。本披露的cas内切核酸酶包括具有hnh或hnh
‑
样核酸酶结构域和/或ruvc或ruvc
‑
样核酸酶结构域的那些。本披露的cas内切核酸酶可以包括cas9蛋白、cpf1蛋白、c2c1蛋白、c2c2蛋白、c2c3蛋白、cas3、cas5、cas7、cas8、cas10或这些的复合物。
[0117]
如本文所用的,术语“指导多核苷酸/cas内切核酸酶复合物”、“指导多核苷酸/cas内切核酸酶系统”、“指导多核苷酸/cas复合物”、“指导多核苷酸/cas系统”、“指导cas系统”在本文中可互换地使用,并且是指能够形成复合物的至少一种指导多核苷酸和至少一种cas内切核酸酶,其中所述指导多核苷酸/cas内切核酸酶复合物可以将cas内切核酸酶引导至dna靶位点,使cas内切核酸酶能够识别、结合到、并任选地使dna靶位点产生切口或切割(引入单链或双链断裂)dna靶位点。本文中指导多核苷酸/cas内切核酸酶复合物可以包含四种已知的crispr系统(horvath和barrangou,2010,science[科学]327:167
‑
170)(例如i型、ii型或iii型crispr系统)中任一种的一种或多种cas蛋白和一种或多种合适的多核苷酸组分。cas内切核酸酶在靶序列处解开dna双链体并任选地切割至少一条dna链,如通过由与cas蛋白复合的多核苷酸(例如但不限于crrna或指导rna)识别靶序列所介导的。如果正确的前间隔子邻近基序(pam)位于或相邻于dna靶序列的3’末端,则通过cas内切核酸酶对靶序列进行的此类识别和切割典型地会发生。替代性地,本文中的cas蛋白可能缺乏dna切割或切口活性,但是当与合适的rna组分复合时,仍然可以特异性结合dna靶序列。
[0118]
指导多核苷酸/cas内切核酸酶复合物可以切割dna靶序列的一条或两条链。可以切割dna靶序列的两条链的指导多核苷酸/cas内切核酸酶复合物通常包含具有处于功能状态的所有其内切核酸酶结构域的cas蛋白(例如野生型内切核酸酶结构域或其变体在每个内切核酸酶结构域中保留一些或全部活性)。因此,在cas蛋白的每个内切核酸酶结构域中保留一些或全部活性的野生型cas蛋白(例如,本文披露的cas9蛋白)或其变体是可以切割dna靶序列的两条链的cas内切核酸酶的合适实例。包含功能性ruvc和hnh核酸酶结构域的cas9蛋白是可以切割dna靶序列的两条链的cas蛋白的实例。可以切割dna靶序列的一条链的指导多核苷酸/cas内切核酸酶复合物可以在本文中表征为具有切口酶活性(例如,部分切割能力)。cas切口酶通常包含一个功能性内切核酸酶结构域,该结构域允许cas仅切割dna靶序列的一条链(即,形成切口)。例如,cas9切口酶可以包含(i)突变的、功能失调的ruvc结构域和(ii)功能性hnh结构域(例如野生型hnh结构域)。作为另一个实例,cas9切口酶可以包含(i)功能性ruvc结构域(例如野生型ruvc结构域)和(ii)突变的功能失调的hnh结构域。适用于本文使用的cas9切口酶的非限制性实例是已知的。
[0119]
可以使用一对cas9切口酶来增加dna靶向的特异性。一般来说,这可以通过提供两个cas9切口酶来进行,这两个cas9切口酶通过与具有不同引导序列的rna组分缔合,在希望靶向的区域的相反链上在dna序列附近进行靶向和切口。每个dna链的这样的附近切割产生双链断裂(即,具有单链突出端的dsb),其然后被识别为非同源末端连接(nhej)(倾向于产生导致突变的不完美修复)或同源重组(hr)的底物。在这些实施例中的每个切口可以,例如,彼此分离至少约5、10、15、20、30、40、50、60、70、80、90、或100(或在5与100之间的任何整数)个碱基。本文中的一种或两种cas9切口酶蛋白可以用于cas9切口酶对。例如,可以使用具有突变的ruvc结构域但具有功能性hnh结构域的cas9切口酶(即,cas9 hnh+/ruvc
‑
)(例如,酿脓链球菌cas9 hnh+/ruvc
‑
)。通过使用本文中的合适的rna组分(具有将每个切口酶
靶向每个特异性dna位点的指导rna序列),将每个cas9切口酶(例如,cas9 hnh+/ruvc
‑
)引导到彼此邻近(分离多达100个碱基对)的特定的dna位点。
[0120]
cas蛋白可以是包含一个或多个异源蛋白结构域(例如除cas蛋白之外的1、2、3或更多个结构域)的融合蛋白的一部分。这样的融合蛋白可以包含任何另外的蛋白序列,以及任选地在任何两个结构域之间(例如在cas和第一异源结构域之间)的连接体序列。可以与本文中的cas蛋白融合的蛋白结构域的实例包括但不限于表位标签(例如,组氨酸[his]、v5、flag、流感血球凝集素[ha]、myc、vsv
‑
g、硫氧还蛋白[trx]);报告子(例如谷胱甘肽
‑5‑
转移酶[gst]、辣根过氧化物酶[hrp]、氯霉素乙酰转移酶[cat]、β
‑
半乳糖苷酶、β
‑
葡萄糖醛酸酶[gus]、荧光素酶、绿色荧光蛋白[gfp]、hcred、dsred、青色荧光蛋白[cfp]、黄色荧光蛋白[yfp]、蓝色荧光蛋白[bfp]);以及具有一个或多个以下活性的结构域:甲基化酶活性、脱甲基酶活性、转录激活活性(例如,vp16或vp64)、转录抑制活性、转录释放因子活性、组蛋白修饰活性、rna切割活性和核酸结合活性。cas蛋白还可以与结合dna分子或其他分子的蛋白融合,例如麦芽糖结合蛋白(mbp)、s
‑
标签、lex a dna结合结构域(dbd)、gal4a dna结合结构域和单纯疱疹病毒(hsv)vp16。关于cas蛋白的更多实例,参见2016年5月12日提交的pct专利申请pct/us 16/32073和2016年5月12日提交的pct/us 16/32028(均通过引用并入本文)。
[0121]
在某些实施例中指导多核苷酸/cas内切核酸酶复合物可以结合dna靶位点序列,但不切割在靶位点序列处的任何链。这样的复合物可以包含其中所有核酸酶结构域都是突变的、功能失调的cas蛋白。例如,可以结合到dna靶位点序列但在靶位点序列处不切割任何链的本文的cas9蛋白可以包含突变的、功能失调的ruvc结构域和突变的、功能失调的hnh结构域。结合但不切割靶dna序列的本文中的cas蛋白可以用于调节基因表达,例如,在该情况下,cas蛋白可以与转录因子(或其部分)融合(例如抑制子或激活子,例如本文披露的那些中的任一种)。在其他方面,可以将失活的cas蛋白与具有内切核酸酶活性的另一种蛋白例如fok i内切核酸酶融合。
[0122]
本文中的cas内切核酸酶基因可以编码ii型cas9内切核酸酶,例如但不限于wo 2007/025097的seq id no:462、474、489、494、499、505和518中列出的cas9基因,并通过引用并入本文。在另一个实施例中,cas内切核酸酶基因是微生物或优化的cas9内切核酸酶基因。cas内切核酸酶基因可以可操作地连接至cas密码子区域上游的sv40核靶向信号和cas密码子区域下游的二分型vird2核定位信号(tinland等人,(1992)proc.natl.acad.sci.usa[美国国家科学院院刊]89:7442
‑
6)。
[0123]
其他cas内切核酸酶系统已经在pct专利申请pct/us 16/32073和pct/us 16/32028中描述,将这两个申请通过引用并入本文中。
[0124]
本文中的“cas9”(以前称为cas5、csnl、或csx12)是指与cr核苷酸和tracr核苷酸或与单指导多核苷酸形成复合物的ii型crispr系统的cas内切核酸酶,其用于特异性识别和切割dna靶序列的全部或部分。cas9蛋白包含ruvc核酸酶结构域和hnh(h
‑
n
‑
h)核酸酶结构域,它们各自可以在靶序列处切割单个dna链(两个结构域的协同作用导致dna双链切割,而一个结构域的活性导致一个切口)。通常,ruvc结构域包含亚结构域i、ii和iii,其中结构域i位于cas9的n
‑
末端附近,并且亚结构域ii和iii位于蛋白的中间,即位于hnh结构域的侧翼(hsu等人,cell[细胞],157:1262
‑
1278)。ii型crispr系统包括利用与至少一种多核苷酸
16/32028,以及zetsche b等人2015.cell[细胞]163,1013)并在特定位置切割靶dna的内切核酸酶。应当理解的是,基于本文所述的使用指导的cas系统的方法和实施例,现在人们可以定制这些方法这样使得它们可以利用任何指导的内切核酸酶系统。
[0131]
如本文所用的,术语“指导多核苷酸”涉及可以与cas内切核酸酶形成复合物的多核苷酸序列,并且使得cas内切核酸酶能够识别、结合并任选地切割dna靶位点。指导多核苷酸可以是单分子或双分子。指导多核苷酸序列可以是rna序列、dna序列或其组合(rna
‑
dna组合序列)。任选地,指导多核苷酸可以包含至少一种核苷酸、磷酸二酯键或连接修饰,例如但不限于锁核酸(lna)、5
‑
甲基dc、2,6
‑
二氨基嘌呤、2
’‑
氟代a、2
’‑
氟代u、2
’‑
o
‑
甲基rna、硫代磷酸酯键、与胆固醇分子的连接、与聚乙二醇分子的连接、与间隔子18(六乙二醇链)分子的连接、或导致环化的5’至3’共价连接。仅仅包含核糖核酸的指导多核苷酸也被称为“指导rna”或“grna”(还参见美国专利申请us 2015
‑
0082478 a1和us 2015
‑
0059010 a1,两者均通过引用以其全文并入本文)。
[0132]
指导多核苷酸可以是包含cr核苷酸序列和tracr核苷酸序列的双分子(也称为双链体指导多核苷酸)。cr核苷酸包括可以与靶dna中的核苷酸序列杂交的第一个核苷酸序列区域(称为可变靶向结构域或vt结构域)和作为cas内切核酸酶识别(cer)结构域的一部分的第二核苷酸序列(也称为tracr配对序列)。tracr配对序列可以沿互补区域与tracr核苷酸杂交,并一起形成cas内切核酸酶识别结构域或cer结构域。cer结构域能够与cas内切核酸酶多肽相互作用。双链体指导多核苷酸的cr核苷酸和tracr核苷酸可以是rna、dna和/或rna
‑
dna组合序列。在一些实施例中,双链体指导多核苷酸的cr核苷酸分子被称为“crdna”(当由dna核苷酸的连续延伸构成时)或“crrna”(当由rna核苷酸的连续延伸构成时)或“crdna
‑
rna”(当由dna和rna核苷酸的组合构成时)。所述cr核苷酸可以包含天然存在于细菌和古细菌中的crna的片段。可以存在于本文披露的cr核苷酸中的细菌和古细菌中天然存在的crna片段的大小可以是但不限于2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20或更多个核苷酸。在一些实施例中,tracr核苷酸被称为“tracrrna”(当由rna核苷酸的连续延伸构成时)或“tracrdna”(当由dna核苷酸的连续延伸构成时)或“tracrdna
‑
rna”(当由dna和rna核苷酸的组合构成时)。在一个实施例中,指导rna/cas9内切核酸酶复合物的rna是包含双链体crrna
‑
tracrrna的双链体化的rna。
[0133]
在5
’‑
至
‑3’
方向上,tracrrna(反式激活crispr rna)包含(i)与crispr ii型crrna的重复区退火的序列和(ii)含茎环的部分(deltcheva等人,nature[自然]471:602
‑
607)。双链体指导多核苷酸可以与cas内切核酸酶形成复合物,其中所述指导多核苷酸/cas内切核酸酶复合物(还称作指导多核苷酸/cas内切核酸酶系统)可以将该cas内切核酸酶引导至基因组靶位点,使该cas内切核酸酶能够识别、结合到靶位点中并任选地使靶位点产生切口或切割(向靶位点中引入单链或双链断裂)。(还参见2015年3月19日公开的美国专利申请us 20150082478a1和us 20150059010a1,两者均特此通过引用以其全文并入)。
[0134]
单指导多核苷酸可以与cas内切核酸酶形成复合物,其中所述指导多核苷酸/cas内切核酸酶复合物(还称为指导多核苷酸/cas内切核酸酶系统)可以将cas内切核酸酶引导至基因组靶位点,使所述cas内切核酸酶能够识别、结合靶位点、并任选地使靶位点产生切口或切割靶位点(引入单链或双链断裂)。(也参见美国专利申请us 20150082478 a1和us 20150059010 a1,两者均通过引用以其全文并入。)
[0135]
术语“可变靶向结构域”或“vt结构域”在本文中可互换使用,并且包括可以与双链dna靶位点的一条链(核苷酸序列)杂交(互补)的核苷酸序列。第一核苷酸序列结构域(vt结构域)与靶序列之间的互补百分比可以为至少50%、51%、52%、53%、54%、55%、56%、57%、58%、59%、60%、61%、62%、63%、63%、65%、66%、67%、68%、69%、70%、71%、72%、73%、74%、75%、76%、77%、78%、79%、80%、81%、82%、83%、84%、85%、86%、87%、88%、89%、90%、91%、92%、93%、94%、95%、96%、97%、98%、99%或100%。可变靶向结构域可以是至少12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29或30个核苷酸长度。在一些实施例中,可变靶向结构域包含12至30个核苷酸的连续延伸。可变靶向域可以由dna序列、rna序列、修饰的dna序列、修饰的rna序列或其任何组合构成。
[0136]
术语(指导多核苷酸的)“cas内切核酸酶识别结构域”或“cer结构域”在本文中可互换地使用,并且包括与cas内切核酸酶多肽相互作用的核苷酸序列。cer结构域包含tracr核苷酸配对序列,随后是tracr核苷酸序列。cer结构域可以由dna序列、rna序列、修饰的dna序列、修饰的rna序列(参见例如us 20150059010a1,其通过援引以其全文并入本文)或其任何组合构成。
[0137]
术语指导rna,crrna或tracrrna的“功能片段”、“功能上等效的片段”和“功能等效片段”在本文中可互换地使用,并且分别意指本披露的指导rna、crrna或tracrrna的一部分或子序列,其中分别保留用作指导rna、crrna或tracrrna的能力。
[0138]
术语指导rna、crrna或tracrrna(分别地)的“功能性变体”、“功能上等效的变体”和“功能等效变体”在本文中可互换地使用,并且分别意指本披露的指导rna、crrna或tracrrna的变体,其中分别保留用作指导rna、crrna或tracrrna的能力。
[0139]
术语“单指导rna”和“sgrna”在本文中可互换使用,并涉及两个rna分子的合成融合,其中包含可变靶向结构域(与tracrrna杂交的tracr配对序列连接)的crrna(crispr rna)与tracrrna(反式激活crispr rna)融合。单指导rna可以包含可与ii型cas内切核酸酶形成复合物的ii型crispr/cas系统的crrna或crrna片段和tracrrna或tracrrna片段,其中所述指导rna/cas内切核酸酶复合物可以将cas内切核酸酶引导至dna靶位点,使得cas内切核酸酶能够识别、结合dna靶位点、并任选地使dna靶位点产生切口或切割(引入单链或双链断裂)dna靶位点。
[0140]
术语“指导rna/cas内切核酸酶复合物”、“指导rna/cas内切核酸酶系统”、“指导rna/cas复合物”、“指导rna/cas系统”、“grna/cas复合物”、“grna/cas系统”、“rna
‑
指导的内切核酸酶”,“rgen”在本文中可互换地使用并且意指至少一种rna组分和至少一种能够形成复合物的cas内切核酸酶,其中所述指导rna//cas内切核酸酶复合物可以将cas内切核酸酶引导至dna靶位点,使cas内切核酸酶能够识别、结合dna靶位点并任选地使dna靶位点产生切口或切割(引入单链或双链断裂)dna靶位点。本文中的指导rna/cas内切核酸酶复合物可以包含四种已知的crispr系统(horvath和barrangou,2010,science[科学]327:167
‑
170)(诸如i型、ii型或iii型crispr系统)中任一种的一种或多种cas蛋白和一种或多种合适的rna组分。指导rna/cas内切核酸酶复合物可以包括ii型cas9内切核酸酶和至少一种rna组分(例如,crrna和tracrrna、或grna)。(也参见美国专利申请us 2015
‑
0082478 a1和us 2015
‑
0059010 a1,两者均通过引用以其全文并入)。
[0141]
使用在本领域已知的任何方法(例如,但不限于,粒子轰击、农杆菌转化或局部施
用),可以将作为单链多核苷酸或双链的多核苷酸的指导多核苷酸瞬时地引入细胞。指导多核苷酸还可以通过引入(通过,诸如但不限于粒子轰击或农杆菌转化等方法)包含编码指导多核苷酸的异源核酸片段的重组dna分子被间接引入细胞,所述重组dna分子可操作地连接于能够在所述细胞转录所述指导rna的特异性启动子。特异性启动子可以是但不限于rna聚合酶iii启动子,其允许具有精确定义的未修饰的5’末端和3’末端的rna转录(dicarlo等人,nuclelc acids res.[核酸研究]41:4336
‑
4343;ma等人,mol.ther.nucleic acids[分子治疗
‑
核酸]3:e161),如在wo 2016025131中所述,其通过援引以其全文并入本文。
[0142]
术语“靶位点”、“靶序列”、“靶位点序列”、“靶dna”、“靶基因座”、“基因组靶位点”、“基因组靶序列”、“基因组靶基因座”和“前间区”在本文中可互换地使用,并且意指多核苷酸序列,例包括但不限于,在细胞的染色体、附加体,或基因组中的任何其他dna分子(包括染色体dna、叶绿体dna、线粒体dna、质粒dna)中的核苷酸序列,在所述序列处指导多核苷酸/cas内切核酸酶复合物可以进行识别、结合并任选地产生切口或进行切割。靶位点可以是细胞的基因组中的内源位点,或者替代性地,靶位点对于该细胞可以是异源的并且从而不是天然存在于细胞的基因组中,或者与在自然界发生的位置相比,可以在异质基因组位置中找到靶位点。如本文所用,术语“内源性靶序列”和“天然靶序列”在本文中可互换使用,是指对细胞基因组来说是内源的或天然的靶序列。细胞包括但不限于人、非人、动物、细菌、真菌、昆虫、酵母、非常规酵母和植物细胞,以及通过本文所述的方法产生的植物和种子。“人工靶位点”或“人工靶序列”在本文中可互换使用,并且是指已经引入细胞的基因组中的靶序列。这种人工靶序列可以在序列上与细胞的基因组中的内源或天然靶序列相同,但是位于细胞的基因组中的不同位置(即,非内源的或非天然的位置)处。
[0143]“改变的靶位点”、“改变的靶序列”、“修饰的靶位点”、“修饰的靶序列”在本文中可互换使用,并且是指如本文公开的靶序列,当与非改变的靶序列相比时,所述靶序列包含至少一个改变。此类“改变”包括,例如:(i)至少一个核苷酸的替代、(ii)至少一个核苷酸的缺失、(iii)至少一个核苷酸的插入、或(iv)(i)
‑
(iii)的任何组合。
[0144]
靶dna序列(靶位点)的长度可以变化,并且包括例如为至少12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30或更多个核苷酸长度的靶位点。还有可能靶位点可以是回文的,即,一条链上的序列与在互补链上以相反方向的读取相同。切口/切割位点可以在靶序列内,或者切口/切割位点可以在靶序列之外。在另一种变异中,切割可以发生在彼此正好相对的核苷酸位置处,以产生平端切割,或者在其他情况下,切口可以交错以产生单链突出端,也称为“粘性端”,其可以是5’突出端抑或3’突出端。还可以使用基因组靶位点的活性变体。此类活性变体可以包含与给定靶位点至少65%、70%、75%、80%、85%、90%、91%、92%、93%、94%、95%、96%、97%、98%、99%或更高的序列同一性,其中所述活性变体保留生物活性,因此能够被cas内切核酸酶识别和切割。测量由内切核酸酶引起的靶位点的单链或双链断裂的测定是本领域已知的,并且通常测量试剂在含有识别位点的dna底物上的总体活性和特异性。
[0145]
本文中的“前间区序列邻近基序”(pam)指与由本文所述的指导多核苷酸/cas内切核酸酶系统识别的(靶向的)靶序列(前间区序列)邻近的短核苷酸序列。如果靶dna序列后面不是pam序列,则cas内切核酸酶可能无法成功识别所述靶dna序列。本文中的pam的序列和长度可以取决于所使用的cas蛋白或cas蛋白复合物而不同。所述pam序列可以是任何长
度,但典型地是1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19或20个核苷酸长度。
[0146]
术语“靶向”、“基因靶向”和“dna靶向”在本文中可互换地使用。本文中的dna靶向可能是在特异性的dna序列(例如细胞的染色体或质粒)中特异性引入敲除、编辑、或敲入。通常,本文中可以通过在具有与合适的多核苷酸组分缔合的内切核酸酶的细胞中的特异性dna序列处切割一条或两条链来进行dna靶向。这种dna切割,如果是双链断裂(dsb),可以促进nhej或hdr过程,这可能导致靶位点处的修饰。
[0147]
本文的靶向方法能以例如在该方法中靶向两个或更多个dna靶位点的这样的方式进行。这种方法可以任选地被表征为多重方法。在某些实施例中,可以同时靶向两个、三个、四个、五个、六个、七个、八个、九个、十个或更多个靶位点。多重方法典型地通过本文的靶向方法进行,其中提供了多个不同的rna组分,每一个被设计成将指导多核苷酸/cas内切核酸酶复合物引导到唯一的dna靶位点。
[0148]
术语“敲除”、“基因敲除”和“基因敲除”在本文中可互换使用。本文所用的敲除表示已经通过用cas蛋白进行靶向使得细胞的dna序列部分或完全无效;例如,这种dna序列在敲除之前可能已编码氨基酸序列,或可能已具有调节功能(例如启动子)。可以通过插入缺失(通过nhej在靶dna序列中插入或缺失核苷酸碱基),或通过特异性去除在靶向位点处或其附近处降低或完全破坏序列功能的序列来产生敲除。
[0149]
指导多核苷酸/cas内切核酸酶系统可以与共同递送的多核苷酸修饰模板组合使用以允许编辑(修饰)目的基因组核苷酸序列。(也参见美国专利申请us 2015
‑
0082478 a1和wo 2015/026886 a1,两者均通过引用以其全文并入)。
[0150]
术语“敲入”、“基因敲入”、“基因插入”和“遗传敲入”在本文中可互换地使用。敲入代表通过用cas蛋白靶向在细胞中的特异性dna序列处进行的dna序列的替换或插入(通过hr,其中还使用合适的供体dna多核苷酸)。敲入的实例包括但不限于异源氨基酸编码序列在基因的编码区中的特异性插入,或转录调节元件在遗传基因座中的特异性插入。
[0151]
可以采用不同方法和组合物来获得细胞或生物,所述细胞或生物具有插入到针对cas内切核酸酶的靶位点中的目的多核苷酸。此类方法可以采用同源重组以提供目的多核苷酸在靶位点处的整合。在提供的一种方法中,在供体dna构建体中将目的多核苷酸提供至生物细胞。如本文所用的,“供体dna”是包括待插入到cas内切核酸酶的靶位点的目的多核苷酸的dna构建体。供体dna构建体可进一步包含位于目的多核苷酸侧翼的同源的第一区域和第二区域。供体dna的同源的第一区域和第二区域分别与存在于细胞或生物基因组的靶位点中或位于所述靶位点侧翼的第一和第二基因组区域具有同源性。“同源”意指dna序列是相似的。例如,在供体dna上发现的“与基因组区域同源的区域”是与细胞或生物体基因组中给定的“基因组序列”具有类似序列的dna的区域。同源的区域可以具有足以促进在切割的靶位点处的同源重组的任何长度。例如,同源的区域的长度可以包括至少5
‑
10、5
‑
15、5
‑
20、5
‑
25、5
‑
30、5
‑
35、5
‑
40、5
‑
45、5
‑
50、5
‑
55、5
‑
60、5
‑
65、5
‑
70、5
‑
75、5
‑
80、5
‑
85、5
‑
90、5
‑
95、5
‑
100、5
‑
200、5
‑
300、5
‑
400、5
‑
500、5
‑
600、5
‑
700、5
‑
800、5
‑
900、5
‑
1000、5
‑
1100、5
‑
1200、5
‑
1300、5
‑
1400、5
‑
1500、5
‑
1600、5
‑
1700、5
‑
1800、5
‑
1900、5
‑
2000、5
‑
2100、5
‑
2200、5
‑
2300、5
‑
2400、5
‑
2500、5
‑
2600、5
‑
2700、5
‑
2800、5
‑
2900、5
‑
3000、5
‑
3100或更多个碱基,这样使得同源的区域具有足够的同源性以与相应的基因组区域进行同源重组。“足够的同源性”表示两个多核苷酸序列具有足够的结构相似性以充当同源重组反应的底物。结构相似性包
括每个多核苷酸片段的总长度以及多核苷酸的序列相似性。序列相似性可以通过在序列的整个长度上的百分比序列同一性和/或通过包含局部相似性(例如具有100%序列同一性的连续核苷酸)的保守区域以及在序列长度的一部分上的百分比序列同一性来描述。
[0152]
相对于参考序列(主题序列),“序列同一性百分比(%)”被确定为在比对序列并引入空位(如果需要)以实现最大百分比序列同一性后,并且不考虑作为序列同一性的一部分的任何氨基酸保守取代,候选序列(查询序列)中与参考序列中的相应氨基酸残基或核苷酸相同的氨基酸残基或核苷酸的百分比。用于确定序列同一性百分比目的而进行的比对能以本领域技术范围内的各种方式实现,例如,使用公共可用的计算机软件,例如blast、blast
‑
2。本领域的技术人员可以确定用于比对序列的适当参数,包括在进行比较的序列的全长度上实现最大比对所需的任何算法。为了确定两种氨基酸序列或两种核酸序列的同一性百分比,出于最佳比较目的,将序列进行比对。两个序列之间的同一性百分比是序列共有的相同位置的数目的函数(例如,查询序列的同一性百分比=查询序列和主题序列之间的相同位置的数目/查询序列的位置总数(例如,重叠位置)
×
100)。
[0153]
由靶标和供体多核苷酸共享的同源性或序列同一性的量可以变化,并且包括总长度和/或在约1
‑
20bp、20
‑
50bp、50
‑
100bp、75
‑
150bp、100
‑
250bp、150
‑
300bp、200
‑
400bp、250
‑
500bp、300
‑
600bp、350
‑
750bp、400
‑
800bp、450
‑
900bp、500
‑
1000bp、600
‑
1250bp、700
‑
1500bp、800
‑
1750bp、900
‑
2000bp、1
‑
2.5kb、1.5
‑
3kb、2
‑
4kb、2.5
‑
5kb、3
‑
6kb、3.5
‑
7kb、4
‑
8kb、5
‑
10kb,或多达并包括靶位点的总长度的范围内具有单位整数值的区域。这些范围包括所述范围内的每个整数,例如1
‑
20bp的范围包括1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19和20bp。同源性的量也可以通过两个多核苷酸的完整比对长度上的序列同一性百分比来描述,该序列同一性百分比包括约至少50%、55%、60%、65%、70%、71%、72%、73%、74%、75%、76%、77%、78%、79%、80%、81%、82%、83%、84%、85%、86%、87%、88%、89%、90%、91%、92%、93%、94%、95%、96%、97%、98%、99%或100%的序列同一性百分比。足够的同源性包括多核苷酸长度、总体百分比序列同一性,和任选地连续核苷酸的保守区域或局部百分比序列同一性的任何组合,例如,足够的同源性可以被描述为与靶基因座的区域具有至少80%序列同一性的75
‑
150bp的区域。还可以通过用来在高严格条件下特异性杂交的两个多核苷酸的预测能力来描述足够的同源性,参见例如sambrook等人,(1989)molecular cloning:a laboratory manual[分子克隆:实验室手册](cold spring harbor laboratory press,ny[纽约冷泉港实验室出版社]);current protocols in molecular biology[分子生物学现代方案],ausubel等人,编辑(1994)current protocols[实验室指南](greene publishing associates,inc.[格林出版合伙公司]和john wiley&sons,inc.[约翰威利父子公司]);以及tijssen(1993)laboratory techniques in blochem istry and molecular biology
‑‑
hybridization with nucleic acid probes[生物化学和分子生物学中的实验室技术
‑‑
与核酸探针杂交](elsevier[爱思唯尔出版社],纽约)。
[0154]
在给定的基因组区域和在供体dna上发现的相应的同源的区域之间的结构相似性可以是允许同源重组发生的任何程度的序列同一性。例如,由供体dna的“同源的区域”和生物体基因组的“基因组区域”共享的同源性或序列同一性的量可以是至少50%、55%、60%、65%、70%、75%、80%、81%、82%、83%、84%、85%、86%、87%、88%、89%、90%、91%、
92%、93%、94%、95%、96%、97%、98%、99%或100%序列同一性,这样使得序列进行同源重组。
[0155]
供体dna上的同源的区域可以与靶位点侧翼的任何序列具有同源性。虽然在一些实施例中,同源的区域与紧邻靶位点侧翼的基因组序列共享显著的序列同源性,但是应当认识到同源的区域可以被设计为与可能更靠近靶位点的5’或3’的区域具有足够的同源性。在又其他实施例中,同源的区域还可以与靶位点的片段以及下游基因组区域具有同源性。在一个实施例中,第一同源的区域进一步包含靶位点中的第一片段,并且第二同源的区域包含靶位点中的第二片段,其中第一片段和第二片段不同。
[0156]
如本文所用的,“同源重组”包括在同源的位点处的两个dna分子之间的dna片段的交换。同源重组的频率受多个因素影响。不同的生物体相对于同源重组的量和同源与非同源重组的相对比例而变化。通常,同源区域的长度会影响同源重组事件的频率:同源区域越长,频率越高。为观察同源重组而需要的同源区的长度也是随物种而异的。在许多情况下,已经利用了至少5kb的同源性,但已经观察到具有仅25
‑
50bp的同源性的同源重组。参见,例如,singer等人,(1982)cell[细胞]31:25
‑
33;shen和huang,(1986)genetics[遗传学]112:441
‑
57;watt等人,(1985)proc.natl.acad.sci.usa[美国国家科学院院刊]82:4768
‑
72,sugawara和haber,(1992)mol cell biol[分子细胞生物学]12:563
‑
75,rubnitz和subramani,(1984)mol cell biol[分子细胞生物学]4:2253
‑
8;ayares等人,(1986)proc.natl.acad.sci.usa[美国国家科学院院刊]83:5199
‑
203;liskay等人,(1987)genetics[遗传学]115:161
‑
7。
[0157]
同源
‑
定向修复(hdr)是在细胞中用来修复双链dna和单链dna断裂的机制。同源
‑
定向修复包括同源重组(hr)和单链退火(ssa)(lieber.2010annu.rev.biochem[生物化学年鉴].79:181
‑
211)。hdr的最常见形式称为同源重组(hr),其在供体和受体dna之间具有最长的序列同源性要求。hdr的其他形式包括单链退火(ssa)和断裂诱导的复制,并且这些需要相对于hr更短的序列同源性。缺口(单链断裂)处的同源
‑
定向修复可以经由与在双链断裂处的hdr不同的机制发生(davis和maizels.(2014)pnas[美国科学院院报](0027
‑
8424),111(10),第e924
‑
e932页)。
[0158]
植物细胞的基因组的改变,例如通过同源重组(hr),其是对于遗传工程而言的有力工具。已经证明了在植物中(halfter等人,(1992)mol gen genet[分子和普通遗传学]231:186
‑
93)和昆虫中(dray和gloor,1997,genetics[遗传学]147:689
‑
99)的同源重组。在其他生物体中也可以实现同源重组。例如,在寄生原生动物利什曼原虫中,至少需要150
‑
200bp的同源性进行同源重组(papadopoulou和dumas,(1997)nucleic acids res[核酸研究]25:4278
‑
86)。在丝状真菌构巢曲霉中,已经用仅50bp侧翼同源性实现基因置换(chaveroche等人,(2000)nuclelc acids res[核酸研究]28:e97)。在纤毛虫嗜热四膜虫中也已经证明了靶向基因置换(gaertig等人,(1994)nucleic acids res[核酸研究]22:5391
‑
8)。在哺乳动物中,使用可以在培养基中生长、转化、选择、和引入小鼠胚胎中的多能胚胎干细胞系(es),同源重组在小鼠中已经是最成功的(watson等人,(1992)recombinant dna[重组dnal,第2版,(scientific american books distributed by wh freeman&co.[由wh freeman&co.公司发行的科学美国人图书])。
[0159]
易错dna修复机制可以在双链断裂位点处产生突变。非同源末端连接(nhej)途径
是用来将断裂的末端结合在一起的最常见的修复机制(bleuyard等人,(2006)dna repair[dna修复]5:1
‑
12)。染色体的结构完整性通常通过修复来保持,但是缺失、插入、或其他重排是可能的。一个双链断裂的两个末端是nhej最普遍的底物(kirik等人,(2000)emboj[欧洲分子生物学学会杂志]19:5562
‑
6),然而如果发生两种不同的双链断裂,则来自不同断裂的游离端可以被连接,并且导致染色体缺失(siebert和puchta,(2002)plant cell[植物细胞]14:1121
‑
31),或在不同的染色体之间的染色体易位(pacher等人,(2007)genetics[遗传学]175:21
‑
9)。
[0160]
可以通过本领域已知的任何手段引入供体dna。可以通过本领域已知的任何转化方法(包括,例如农杆菌介导的转化或生物射弹粒子轰击)提供供体dna。供体dna可以瞬时地存在于细胞中,或可以经由病毒复制子引入。在cas内切核酸酶和靶位点的存在下,供体dna被插入到转化植物的基因组中。(参见指南语言)
[0161]
指导rna/cas内切核酸酶系统的另外的用途已进行了描述(参见美国专利申请us 2015
‑
0082478 a1、wo 2015/026886 a1、us 2015
‑
0059010 a1、美国申请62/023246,和美国申请62/036,652,将其全部通过引用并入本文),并包括,但不限于修饰或取代目的核苷酸序列(如调节元件)、目的多核苷酸插入、基因敲除、基因敲入、剪接位点的修饰和/或引入交替剪接位点、编码目的蛋白的核苷酸序列的修饰、氨基酸和/或蛋白融合物、以及通过在目的基因中表达反向重复序列引起的基因沉默。
[0162]
实例
[0163]
提供以下实例以说明但不限制所附权利要求书。应当理解,本文所述实例和实施例仅用于说明目的,并且本领域的技术人员将认识到在不脱离本文公开的实施例的情况下可改变多种试剂或参数。
[0164]
实例1.来自大豆中快速中子诱变的高蛋白突变体中因果基因的精细作图
[0165]
蛋白是大豆种子中最有价值的组分。从快速中子突变体群体中鉴定出一种高蛋白/低油突变体系(pol)(bolon等人2011phenotypic and genomic analysis of a fast neutron mutant population resource in soybean[大豆中快速中子突变群体资源的表型和基因组分析].plant physiol[植物生理学]156:240
‑
253)。pol突变体被作图到第10号染色体上的39kb缺失,其中包含三个可能的候选基因。然而,由于缺失区域中没有重组,因此未鉴定出因果基因。crispr/cas9用于在该区域中创建三个重叠缺失,以鉴定引起高蛋白/低油含量的因果基因(图1)。
[0166]
如表1所示,设计了六种靶向目的区域中特定位点的指导rna(grna)。该区域的基因组序列示于seq id no:27。通过转化将每对grna和cas9递送至大豆。基于变体的分子分析,鉴定出具有杂合的cr1/cr3缺失#1和cr4/cr6缺失#3的t0植物。来自自交的t0植物的t1种子以纯合缺失、杂合缺失和野生型的1∶2∶1分离。
[0167]
表1.指导rna设计为在目的区域产生缺失
[0168]
[0169][0170]
t1种子蛋白和油含量通过单种子nir确定,如前所述(roesler等人2016,plant physiol.[植物生理学]171(2):878
‑
93)。与来自cr4/cr6缺失#3系的t1种子和野生型平均值相比,来自cr1/cr3缺失#1系的t1种子表现出蛋白含量的增加和油含量的降低,表明cr1/cr3缺失#1系中的缺失片段包含高蛋白/低油的因果基因(图2)。缺失#1区的序列分析鉴定了两个潜在基因,glyma.10g270800和glyma.10g270900。由于最初的快速中子po1突变体中未缺失glyma.10g270800基因,因此第二个glyma.10270900很可能是高蛋白含量的因果基因。glyma.10g 270800编码网状内皮素样蛋白,其可能在调节内质网中油和蛋白的生物合成中起重要作用。为了验证glyma.10g270900是高蛋白表型的因果基因,在glyma.10g270800的外显子1中设计了指导rna(gm
‑
ret
‑
cr1,表1中的seq id no:17)以敲除网状内皮素样蛋白。如果网状内皮素样敲除系表现出高蛋白表型,这将证明网状内皮素样蛋白参与调节大豆种子中的蛋白和油含量。通过crispr/cas9敲除优良大豆中的网状内皮素样基因有望增加种子蛋白含量。
[0171]
实例2.大豆高蛋白qtl(qhp20)的精细作图
[0172]
鉴于大豆中蛋白含量的重要性,与高蛋白含量相关的数量性状基因座(qtl)已广泛作图。通过多重作图研究检测到第20号染色体上的一个主要高蛋白qtl(qhp20),并且显示出对种子蛋白和油含量的一致影响(chung等人2003crop sci[作物科学]43:1053
‑
1067;nichols等人2006crop sci[作物科学]46:834
‑
839;bolon等人2010bmc plant biology[bmc植物生物学]10:41;hwang等人2014 bmc genomics[bmc基因组学]15:1)。qhp20被作图到2.4mb的区间,并且由于所述区域的重组率较低,因此无法进一步前进。使用crispr/cas9技术,创建了一系列重叠缺失系以对qhp20精细作图。靶向qhp20区域内特定位点的指导rna被设计为在qhp20 qtl区域内产生重叠丢失。当与cas9一起递送至高蛋白供体时,这些指导物有望产生约700kb至1.4mbp的基因组缺失(表2)。选择具有缺失的t0植物并进行基因分型以验证预期缺失的发生。t0植物可以在单个或两个染色体上被编辑,因此在被编辑的基因座处分别是半合子或纯合子。在t1种子上进行表型分析,例如种子中的蛋白和油含量,以鉴定可改变种子蛋白含量的目的子区域。通过与使用近等基因系的传统qtl作图相同的作图技术,可以通过crispr/cas9创建的重叠缺失线来对qtl作图。表4列出了缺失系的可能的蛋白表型和qtl的位置。例如,如果cr40/cr42和cr41/cr44缺失系均显示蛋白含量降低,而cr43/cr45缺失系显示蛋白无变化,则将qhp20定义为cr41和cr42之间的区间(参见图3)。如果需要,可以设计另一轮指导rna,以进一步缩小子区域中的候选基因。鉴定出候选基因后,可以通过另外的编辑实验(例如移码敲除或精确的区段丢失/替换)来确认基因的功能(参见表3)。
[0173]
表2.设计用于在qhp20区域中产生缺失的指导rna
[0174][0175][0176]
表3.基于重叠缺失系的蛋白表型的qhp20基因编辑精细作图的预期结果
[0177][0178]
实例3.通过基因组编辑验证qhp20 qtl
[0179]
基于高蛋白系和低蛋白系的基因组序列分析,一个候选基因glyma.20g085100(seq id no:36)已被确定为qhp20区域中针对高蛋白表型的潜在因果基因。与高蛋白野生大豆基因组序列和大豆旁系同源glyma.10g134400(seq id no:40)相比,来自优良低蛋白系(包括williams82)的glyma.20g085100在第4外显子中包含321bp插入,这可能是优良大豆中高蛋白表型丧失的潜在因果突变(见图4)。在所有优良低蛋白系中都发现了321bp插入,但在高蛋白danbaekkong和野生大豆系中却没有。glyma.20g850100编码cct(constans、co
‑
like和tocl)结构域蛋白。cct结构域蛋白在调节开花时间方面起着重要作用,其中对稻、玉蜀黍和其他谷物作物的形态性状和胁迫耐性具有多效性作用(yipu li和mingliang xu,2017,cct family genes in cereal crops:a current overview[谷物农作物中的cct家族基因:最新概述].the crop journals[作物杂志]449
‑
458)。cct结构域蛋白在大豆中的功能尚不清楚。321bp片段插入cct结构域的中间,并产生新的开放阅读框,其产生完全不
同的88个氨基酸的c末端(参见图5)。cct结构域蛋白的破坏可能是无功能的,从而导致优良大豆中的低蛋白含量(参见图6)。为了验证插入是低蛋白的因果突变,设计了一对指导rna gm
‑
cct
‑
cr2(seq id no:25)和cr3(seq id no:26),以缺失优良大豆中的插入(表3)。从优良系中去除321bp插入应该恢复cct结构域蛋白的功能并增加种子蛋白含量。此外,单个指导rna gm
‑
cct cr1(seq id no:24)靶向glyma.20g850100的外显子2,以敲除基因功能。将这种带有cas9的grna引入高蛋白系应该减少种子中的蛋白含量。
[0180]
实例4.对玉蜀黍中具有两个因果基因的疾病qtl作图
[0181]
通过考虑rcg1(由us 8,062,847b2(通过引用并入本文)的seq id no:1编码的seq id no:3)和rcg1b(由us 8,053,631b2(通过引用并入本文)的seq id no:245编码的seq id no:246)来举例说明使用此方法的示例,其是nlr基因对,其中两个基因都是对导致玉米炭疽病茎秆腐烂的半活体营养性病原体禾生炭疽病菌(colletotrichum graminicola)的显著抗性所需要的。这两个基因在罕见的大(约300kb)非共线片段上相距约250kb,其中缺少所述片段的材料无法进行重组(图7;还参见seq id no:137和us 8,062,847b2的图9(a
‑
b),其通过引用并入本文)。编辑精细作图方法用于创建编辑,一旦通过生物信息学分析鉴定了来自供体的抗性基因序列基序,所述编辑可独立地缺失rcg1基因组序列(3445bp)和rcg1b基因组序列(43637bp)。
[0182]
精细作图受到作图亲本之间缺乏同源性的挑战
[0183]
目的区域对应于来自抗性供体系的约500kb片段,由左和右标记定界。作为北美种质的实例,在抗性供体和b73之间的大规模序列比对揭示了目的区域中的低水平同源性以及边界上共线性的逐渐丧失(图11)。共线性是指以保守顺序连续的同源片段。这一发现表明,考虑到序列同源性是减数分裂交换事件发生的先决条件之一,进一步精细作图以缩小目的区域的是无用的。
[0184]
基于crispr的精细作图策略以阐明区间
[0185]
这里提供了另一种方法来进一步缩小目的区域并鉴定因果基因。指导rna被设计为在目的区域中产生大的缺失(表4)。这些缺失与目的区域的功能注释一起,提供了鉴定因果基因的工具。在这个实例中,产生了缺失,其包含因果基因中的每个或两个或无(图12)。
[0186]
基于显性/隐性特征和功能作用模式的丧失/获得,设计了一种实验方案以进一步对目的区间作图(图9)。在群体发展和qtl作图过程中,抗性等位基因有望以显性方式表现。如图10所示,可能会出现功能显性和获得的情况。
[0187]
使用此策略,在精细作图过程中会产生及疾病抗性近等基因系(nil),并用于在渗入区域内创建具有选定缺失的变异体。缺失涵盖整个目的区域和目的区域内的区域的子集。缺失可涵盖或可不涵盖预测以编码基因的区域。缺失可涵盖一个或几个预测的基因。在该实例中的缺失范围为约125kbp至约500kbp。
[0188]
设计了一系列靶向目的区域内特定位点的指导rna对。当与cas9组合递送到细胞中时,这些指导物预期产生基因组缺失。在t0,选择经编辑的植物并进行基因分型以验证预期缺失的发生。t0植物可以在单个或两个染色体上被编辑,因此在被编辑的基因座处分别是半合子或纯合子/杂合子。为了鉴定涵盖因果基因座的编辑,交配方案包括将t0植物与群体中使用的易感病的亲本杂交。在t1时,再次对植物进行基因分型,以验证经编辑的等位基因的孟德尔分离。预计t1植物都含有一个拷贝的易感亲本等位基因和一个拷贝的抗性nil
等位基因或经编辑的等位基因。
[0189]
预期抗性等位基因将是显性的,并且大多数t1植物预期表现出疾病抗性表型,除了特别含有涵盖因果基因座的缺失的经编辑的植物(其对疾病应该是易感的(或抗性较低))外(参见图10)。
[0190]
使用这种筛选方案,对表现出易感表型相比于抗性表型的t1植物进行进一步的测序和比较,以鉴定因果关系区域或基因。
[0191]
在此实例中,两个基因提供了对炭疽病茎秆腐烂的抗性:rcg1b和rcg1。该方法提供了阐明这种作用方式的手段(图13)。
[0192]
在此描述的方法允许进一步阐明复杂区域,其中多于一个蛋白编码基因在参与qtl的贡献,或者通过重组分离簇中的基因是极其困难的(参见图8)。该组装来自第10号染色体短臂上的已知疾病抗性基因簇(“r基因簇”),并且包含约26个彼此相似程度不同的基因,且都非常接近。缺失所述基因或由重组限定的其中的子集允许分离因果基因。
[0193]
表4.指导rna被设计用于在目的炭疽病茎秆腐烂抗性qtl区域中产生缺失。
[0194][0195][0196]
实例5.玉蜀黍qtl的精细作图方案
[0197]
开发群体以鉴定有助于期望性状的染色体qtl。抗性供体是包含期望性状的不同来源,与待改良的优良种质相比,具有较大的效应大小。经良好表征的温带系用作轮回亲本。最初的qtl发现是在具有200个体的测试杂交群体((不同来源的系x温带系)x测试系)中完成。在该群体中发现了显著的qtl,作图到单个区间。然后在相同的群体或使用相同来源和新优良系的其他群体(不同的系x优良近交系)中验证此效果。然后选择验证群体或原始群体进行重组筛选,以寻找该区域的重组体,并开发在整个qtl区间内具有供体片段的nil。
[0198]
精细作图受到作图亲本之间缺乏同源性的挑战
[0199]
通过在单个或多个位置使用重组体和田间表型分型,可以将qtl精细作图到染色体上的小遗传区间。精细作图进一步将区间缩小到一个较小的区域,该区域的两侧是可以唯一地作图到来自优良系的已知连续序列的标记。在不同的抗性供体中,该目的区域对应于该物理区间。
[0200]
尽管筛选了许多重组体,但预计该区域内不会回收任何重组体,从而阻止了目的区间的进一步缩小。
[0201]
确定了完整的不同抗性供体基因组序列。标记数据表明,在目的区间内,优良序列并不相同,但通常认为这两个近交系具有共线性。以不同抗性供体基因组为参考,将优良基因组的10kb片段对齐并分配给它们的最佳匹配位置。预计大多数片段会与在不同抗性供体中的它们的同源区域对齐,并显示出与优良系的高水平同线性,但一些片预期被倒置、重排或仅部分对齐,这表明两个基因组之间存在较大的结构差异。另外,也预期观察到在优良系中很少匹配至没有匹配的区域,这表明一些区域对于不同抗性供体基因组是独特的。在目的区域内这可能是显而易见的。还检查了另外的自交系,并预期它们会显示出相似的模式。总而言之,这些观察结果表明,不同抗性供体中目的区域可以与其他近交系共享非常低水平的序列同源性。
[0202]
序列同源性是减数分裂交换事件发生的先决条件之一。预期结果表明,在精细作图过程中,目的区域中没有重组事件。预期结果表明,通过筛选另外的后代进一步追求这种方法不太可能产生有用的重组体。
[0203]
基于crispr的精细作图策略以阐明区间
[0204]
基于显性/隐性特征和功能作用模式的丧失/获得,设计了一种实验方案以进一步对目的区间作图(图9)。在群体发展和qtl作图过程中,抗性等位基因有望以显性或半显性方式表现。如图10所示,可能会出现功能显性和获得的情况。
[0205]
使用此策略,在精细作图过程中会产生及疾病抗性近等基因系(nil),并用于在渗入区域内创建具有选定缺失的变异体。缺失可以涵盖整个目的区域或目的区域内的区域的子集。这些较小的缺失可以涵盖靶向区域,例如基因丰富的区域,或包含疾病抗性基因簇的区域,或主要结构变异的区域,或基因表达较高的区域。这些缺失的范围可以从kbp到几个mbp。这些缺失可以被设计为重叠或不重叠。
[0206]
设计了一系列靶向目的区域内特定位点的指导rna对。当与cas9组合递送到细胞中时,这些指导物预期产生基因组缺失。在t0,选择经编辑的植物并进行基因分型以验证预期缺失的发生。t0植物可以在单个或两个染色体上被编辑,因此在被编辑的基因座处分别是半合子或纯合子/杂合子。为了鉴定涵盖因果基因座的编辑,交配方案包括将t0植物与群体中使用的易感病的亲本杂交。在t1时,再次对植物进行基因分型,以验证经编辑的等位基因的孟德尔分离。预计t1植物都含有一个拷贝的易感亲本等位基因和一个拷贝的抗性nil等位基因或经编辑的等位基因。
[0207]
预期抗性等位基因将是显性或半显性的,并且大多数t1植物预期表现出疾病抗性表型,除了特别含有涵盖因果基因座的缺失的经编辑的植物(其对疾病应该是易感的(或抗性较低))外(参见图10)。
[0208]
使用这种筛选方案,对表现出易感表型相比于抗性表型的t1植物进行进一步的测
序和比较,以鉴定因果关系区域或基因。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1