一种化合物、抗菌整理液及其制备方法和应用与流程
一种化合物、抗菌整理液及其制备方法和应用
1.本发明是申请日为2019年03月11日、申请号为201910181723.9、发明名称为《一种化合物、抗菌整理液及其制备方法和应用》的分案申请。
技术领域
2.本发明涉及化合物技术领域,尤其是涉及一种化合物、抗菌整理液及其制备方法和应用。
背景技术:
3.天然纤维(棉、麻、丝、毛等)作为使用量最大的纺织材料之一。其中,棉纺织行业在国民经济发展历程中扮演重要角色,近年棉纱的生产能力达到全球50%。棉纺织品具有柔软、透气、吸湿以及穿着舒适性好等特点而为人们所喜爱。但是,一些致病性细菌容易在棉纺织品表面迅速滋生,形成菌斑或微生物膜,在一定范围内引起疾病或者疾病传染,极大的影响了棉织物的使用性和美观程度。因此,围绕其抗菌功能的改性成为提升棉纺织品等天然纤维广泛使用性的关键。
4.天然纤维的表面改性可以通过在其表面修饰化合物来实现。目前,国内外纺织品改性最常用的工艺技术是后整理工艺。后整理工艺相对简单、便于规模化生产。后整理工艺是对成型织物进行功能性处理,一般采用涂层或浸渍功能整理剂的方式。天然纤维的抗菌后整理工艺,主要采用涂层或浸渍抗菌整理剂,使得天然纤维表面吸附或者以化学键合的方式引入抗菌整理剂,以获得一定程度上的抗菌性能。
5.抗菌剂的抗菌性以及其与纤维的结合能力是决定其能否广泛应用的关键因素。
6.基于三聚氯氰的抗菌剂因具有良好的水洗稳定性(cn106928158a,journal of industrial and engineering chemistry,2017,46,p373-378)。然而目前已报道的三聚氯氰抗菌剂所用的两性离子抗菌基团主要以抑菌为主,其抗菌效果并不理想。而三聚氯氰卤胺类抗菌材料,其抗菌活性持久性欠佳,需要再生处理(cn106632259a),而强氧化再生易造成织物损伤(text research journal,2008,78,p60-72;材料导报,2012,26,p89-94)。
7.有鉴于此,特提出本发明。
技术实现要素:
8.本发明的第一目的在于提供一种化合物,该化合物具有抗菌以及能与纤维/合成高分子材料化学键合的特性,同时对纤维/合成高分子材料的力学性能没有负面影响,相比现有的三聚氯氰抗菌整理液具有更好的抗菌防霉和防螨效果,采用更低的使用剂量即可获得持久高效的防霉防螨纺织品,成本更低廉,同时还可以改善织物的柔软性和抗静电性。
9.本发明的第二目的在于提供上述化合物的制备方法,该制备方法路线简单、每一步的反应过程简单,仅涉及取代反应,无需芳烃等剧毒类试剂,操作更安全。
10.本发明的第三目的在于提供上述化合物的应用,其用于抗菌或制备抗菌剂,比现有产品具有显著优势,例如与纤维的键合力更强,抗菌耐洗性更佳、成本更低廉,使用更方
便,整理过程无废水废气排放。
11.本发明的第四目的在于提供一种纺织品的抗菌整理方法,该方法具有流程简单,容易实现自动化等优点。
12.为了实现以上目的,本发明提供了以下技术方案:
13.一种化合物,具有通式(i)的结构:
[0014][0015]
其中,
[0016]
所述r1为卤素,例如cl、br或i;
[0017]
所述r2为卤素(如cl、br或i)、氢、杂原子取代或未取代的的c
1-8
的烃基;
[0018]
x为nh、o、s、杂原子取代或未取代的c
1-18
的烃基;
[0019]
所述r3含有季铵盐基团和季磷盐基团中的至少一个。
[0020]
r2选c
1-8
烷基时,可以为直链烷基或支链烷基,可以是未取代的或被一个或多个杂原子取代的,如c
1-8
直链或支链烷基,例如甲基、乙基、丙基或异丙基等,或以上被一个或多个杂原子(如n、p、s、o等)取代的。
[0021]
r2选c
1-8
环烷基时,可以选环丙基及其取代物、环丁基及其取代物、环戊基及其取代物或环己基及其取代物。
[0022]
r2选c
1-8
烯烃基时,可以选ch2=ch-(ch2)
n-,n=1-6,如1,2,3,4,5或6,或以上基团中氢被取代后的基团。
[0023]
r2选芳烃基时,可以选c6h5ch=ch-、c6h5ch=ch-ch
2-或c6h
5-ch2ch=ch-ch
2-,或以上基团中氢被取代后的基团,或含有杂原子的芳香体系,例如吡啶、呋喃或噻吩等。
[0024]
化合物(i)具有以下特点:
[0025]
(1)卤素取代的三嗪化合物能与纤维/合成高分子表面的-oh、-nh2活性基团发生亲核取代反应,从而牢固地结合在纤维/合成高分子织物上,耐洗涤。
[0026]
(2)r3含有季铵盐基团和季磷盐基团中的至少一个,这些基团都具有良好的杀菌效果,并且相比两性离子抑菌剂(两性离子为抑菌机理)具有更高的抗菌防霉防螨活性(季铵盐和季鏻盐为杀菌作用机理),采用更低的使用剂量即可获得持久高效的防霉防螨纺织品,成本更低廉,同时还可以改善织物的柔软性和抗静电性。
[0027]
综上,具有通式(i)的结构的化合物满足织物抗菌整理的必要条件,并且性能突出,因此可用作抗菌或制备抗菌剂,应用在纺织品、医药、食品和农业等领域,实现物料的长效抗菌,但本发明的用途不限于上述领域。
[0028]
经检测,本发明提供的化合物的优异性能具体表现为:
[0029]
采用更低的使用剂量即可获得广谱、持久和高效的抗菌防霉防螨纺织品,成本更低廉,同时还可以改善织物的柔软性和抗静电性。在对织物本身的力学性能、透湿透气性和
色泽不产生负面影响的前提下,具有更好的抗菌防霉和防螨效果,与纤维的键合力更强,抗菌耐洗性更佳、成本更低廉,使用更方便,整理工艺简单、无废水废气排放,容易实现自动化等优点。
[0030]
本发明的部分实施例中,所述r3为
[0031]
y为卤素,优选br、cl或i,
[0032]
r4为杂原子取代或未取代的c
1-18
的烃基,优选c
1-18
烷基,可以为直链烷基或支链烷基,可以是未取代的或被一个或多个杂原子取代的,如c
1-18
直链或支链烷基,例如甲基、乙基、丙基或异丙基等,或以上被一个或多个杂原子(如n、p、s、o等)取代的;
[0033]
r5为杂原子取代或未取代的c
0-18
的烃基,优选c
1-18
烷基可以为直链烷基或支链烷基,可以是未取代的或被一个或多个杂原子取代的,如c
1-18
直链或支链烷基,例如甲基、乙基、丙基或异丙基等,或以上被一个或多个杂原子(如n、p、s、o等)取代的。
[0034]
中的季铵盐环可以是三元环、四元环、五元环、六元环或七元环,并不限于吡咯环、吡唑环、咪唑环、唑环、哒嗪环、嘧啶环或吡嗪环,或含以上基团的稠杂环。
[0035]
r5为c0时,是指直接与的环连接,即化合物(i)的通式为:
[0036]
[0037]
本发明的部分实施例中,所述r3为
[0038]
本发明的部分实施例中,所述r3为
[0039]
z为n或p,
[0040]
r6为杂原子取代或未取代的c
1-18
的烃基,优选c
1-18
烷基,例如-(ch2)
n-,n=1-18(如1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17或18),或者-(ch2)
n-的同分异构体;
[0041]
r7为杂原子取代或未取代的c
1-18
的烃基,优选c
1-18
烷基、苯环或取代的苯环;c
1-18
烷基如-(ch2)
n-,n=1-18(如1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17或18),或者-(ch2)
n-的同分异构体;
[0042]
r8为杂原子取代或未取代的c
1-18
的烃基,优选c
1-18
烷基、苯环或取代的苯环;c
1-18
烷基如-(ch2)
n-,n=1-18(如1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17或18),或者-(ch2)
n-的同分异构体;
[0043]
r9为杂原子取代或未取代的c
1-18
的烃基,优选c
1-18
烷基、苯环或取代的苯环;c
1-18
烷基如-(ch2)
n-,n=1-18(如1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17或18),或者-(ch2)
n-的同分异构体。
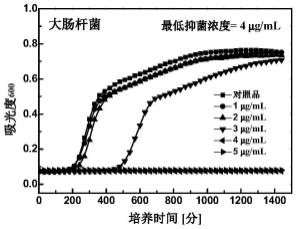
[0044]
其中,r7与r8为相同或不同的基团,例如同为甲基、乙基、异丙基、正丁基、正戊基或环己基。
[0045]
本发明的部分实施例中,化合物(i)的结构式为:
[0046][0047]
a=0~17(例如0、1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16或17),b=0~17(例如0、1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16或17)。
[0048]
本发明的部分实施例中,所述r3为
[0049]
r6为杂原子取代或未取代的c
1-18
的烃基,优选c
1-18
烷基,例如-(ch2)
n-,n=1-18(如1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17或18),或者-(ch2)
n-的同分异构体;
[0050]
r7为杂原子取代或未取代的c
1-18
的烃基,优选c
1-18
烷基、苯环或取代的苯环;c
1-18
烷基如-(ch2)
n-,n=1-18(如1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17或18),或者-(ch2)
n-的同分异构体;
[0051]
r9为杂原子取代或未取代的c
1-18
的烃基,优选c
1-18
烷基,c
1-18
烷基如-(ch2)
n-,n=1-18(如1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17或18),或者-(ch2)
n-的同分异构体;
[0052]r10
为杂原子取代或未取代的c
1-18
的烃基,优选c
1-18
烷基,c
1-18
烷基如-(ch2)
n-,n=1-18(如1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17或18),或者-(ch2)
n-的同分异构体;
[0053]r11
为杂原子取代或未取代的c
1-18
的烃基,优选c
1-18
烷基,c
1-18
烷基如-(ch2)
n-,n=1-18(如1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17或18),或者-(ch2)
n-的同分异构体。
[0054]
本发明的部分实施例中,r7、r8和r9为相同基团,或不同基团。
[0055]
本发明的部分实施例中,所述r3为
[0056]
r6为-(ch2)
n-,n为正整数,优选1~18,更优选2~18,如2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17或18;
[0057]
r7为苯环或取代的苯环,-(ch2)
n-,n为正整数,优选1、2、3或6~18;
[0058]
r8为苯环或取代的苯环,-(ch2)
n-,n为正整数,优选1、2、3或6~18;
[0059]
r9为苯环或取代的苯环,-(ch2)
n-,n为正整数,优选1、2、3或6~18。
[0060]
此时,三嗪环与季铵盐之间的桥接键x优选为nh、o或s。
[0061]
本发明的部分实施例中,r7、r8和r9为相同基团,或为不同基团。
[0062]
本发明的部分实施例中,化合物(i)为以下中的一种:
[0063]
[0064]
其中,至少包含以下类型:
[0065][0066]
本发明化合物(i)的制备方法主要是:
[0067]
将具有通式(ii)结构的化合物与hx-r3反应,得到所述化合物(i);
[0068]
x1为卤素(例如cl、br或i);
[0069]
优选地,通式(ii)结构的化合物与hx-r3的反应在路易斯碱作用下进行,所述路易斯碱优选碱金属或碱土金属的无机碱或有机碱。
[0070]
以上反应主要利用通式(ii)结构的化合物与hx-r3之间的亲核取代反应,反应所需的溶剂以及助剂/催化剂选用利于亲核试剂进攻或促使三嗪环形成正离子的化合物。合成不同r3的化合物(i)时,最优的反应条件有区别。
[0071]
其中,hx-r3主要由叔胺/叔膦与卤代烃反应后生产的季铵盐/季磷盐。
[0072]
优选地,通式(ii)结构的化合物与hx-r3的反应在路易斯碱和有机溶剂作用下进行,路易斯碱可以促使三嗪环形成正离子,路易斯碱的类型通常根据依据hx-r3亲油亲水性而定。若偏亲水性,则可以选择无机碱,无机碱可选用碱金属或碱土金属的强碱、碱金属或碱土金属的碳酸盐,碱金属或碱土金属的碳酸氢盐等中的一种或多种组合物;若采用有机溶剂,则通常选用有机碱,例如n,n-二异丙基乙胺、三乙胺、三甲胺、三丁胺中的一种或多种
组合物。
[0073]
优化地,所述无机碱类选自氢氧化钠、氢氧化钾、氢氧化镁、氢氧化钙、碳酸钾、碳酸氢钾、碳酸钠和碳酸氢钠的一种或多种混合物。
[0074]
优化地,所述有机叔胺类选自n,n-二异丙基乙胺、三甲胺、三乙胺、n,n-二甲基正辛胺、n,n-二甲基苯胺、n,n-二甲基十二胺、n,n-二甲基十二胺、n,n-二甲基十六胺、n,n-二甲基十八胺、n,n-二甲基癸胺、甲基丙烯酸二甲基氨基乙酯和甲基丙烯酸二乙基氨基乙酯的一种或多种混合物。
[0075]
所述r3为时,方法分两步进行:
[0076]
由与y-r4反应生成hx-r3;
[0077]
将具有通式(ii)结构的化合物与hx-r3反应,得到所述化合物(i)。
[0078]
其中,可以是杂原子(o、n、s)取代的吡啶、呋喃、咪唑、吡嗪等。例如,4-巯甲基吡啶,2,6-二甲基-4-氨基吡啶,1-(3-氨丙基)咪唑、4-(羟甲基)咪唑、2-吡嗪基乙硫醇、2-吡嗪甲醇或组胺,但不限于以上列举。
[0079]
所述化合物的结构式为:
[0080]
时,制备方法分两步进行:
[0081]
由与y-(ch2)bch3反应生成hx-r3;
[0082]
将具有通式(ii)结构的化合物与hx-r3反应,得到所述化合物(i)。
[0083]
可以是n,n-二甲基乙醇胺、n,n-二甲基乙二胺、n,n-二乙基乙醇胺、n,n-二异丙基乙醇胺、n,n-二正丁基乙醇胺、n,n-二正戊基乙醇胺或n,n-二环己基乙醇胺,但不限于以上列举。
[0084]
所述r3为时,制备方法分三步进行:
[0085]
由和y-r9反应生成
[0086]
由与hx-r
10-q反应生成hx-r3;q为卤素,优选br、cl或i;x优选o、s,nh,如果x选nh,反应前-nh2得先进行保护,反应完后再做脱保护处理;
[0087]
将具有通式(ii)结构的化合物与hx-r3反应,得到所述化合物(i)。
[0088]
所述r3为时,制备方法分两步进行:
[0089]
由与y-r
6-hx反应生成hx-r3;
[0090]
将具有通式(ii)结构的化合物与hx-r3反应,得到所述化合物(i)。
[0091]
为了更清楚地描述本发明,下文整理了本发明涉及的几类化合物的合成原理(下文仅以三聚氯氰为例,三聚氯氰的取代物为反应物的合成路线与三聚氯氰相同,仅仅将“三聚氯氰”替换为“三聚氯氰的取代物”即可)。
[0092]
咪唑类季铵盐合成原理为:
[0093][0094]
双季铵盐的合成原理为:
[0095][0096]
其中,n为0-17的自然数,n优选0、1、3、5、7、9、11、13、15或17。
[0097]
季鏻盐的合成原理为:
[0098][0099]
如上文所述,本发明的化合物可用于抗菌,尤其是织物的抗菌整理。
[0100]
本发明提供了一种优选的抗菌整理液及其制备方法,包括上述的化合物(i);
[0101]
还包括ph调节剂以及表面活性剂;
[0102]
优选地,所述ph调节剂为路易斯碱,优选碳酸钠、碳酸氢钠、碳酸钾、碳酸氢钾、三乙胺、三甲胺和三丁胺中的一种或多种;
[0103]
优选地,所述表面活性剂包括硬脂酸、十二烷基苯磺酸钠、十六烷基磺酸钠、十八烷基磺酸钠、十六烷基羧酸钠、十二烷基硫酸钠、十六烷基硫酸钠、十二烷基羧酸钠、卵磷脂和脂肪酸甘油酯中的一种或多种。
[0104]
优选地,所述溶剂优选为水、二甲基亚砜、氯仿、醚、酮、酯、腈、酰胺和芳香化合物
中的一种或几种混合物。
[0105]
优选地,所述醚选自四氢呋喃、1,4-二氧六环和乙二醇二甲醚中的一种或几种混合物;
[0106]
优选地,所述酮选自丙酮、丁酮、环己酮、苯乙酮和佛尔酮中的一种或几种混合物;
[0107]
优选地,所述芳香化合物选自甲苯、吡啶、咪唑中的一种或几种混合物;
[0108]
优选地,所述酯选自乙酸乙酯、乙酸正丁酯、乙酸正丙酯、甲酸乙酯、甲酸甲酯中的一种或几种混合物;
[0109]
优选地,所述腈选自乙腈、丙腈、苯甲腈中的一种或几种混合物;
[0110]
优选地,所述酰胺选自n,n-二甲基乙酰胺、n,n-二甲基甲酰胺和n,n-二甲基吡咯烷酮中的一种或几种混合物;
[0111]
优选地,所述抗菌整理液还包括助溶剂,所述助溶剂优选为水、二甲基亚砜、醚、酮、酯、腈、酰胺和芳香化合物中的一种或几种混合物。
[0112]
优选地,所述助溶剂中的醚选自四氢呋喃、1,4-二氧六环,乙二醇二甲醚中的一种或几种混合物;
[0113]
优选地,所述助溶剂中的酮选自丙酮、丁酮、环己酮、苯乙酮、佛尔酮中的一种或几种混合物;
[0114]
优选地,所述助溶剂中的芳香化合物选自甲苯、吡啶、咪唑中的一种或几种混合物;
[0115]
优选地,所述助溶剂中的酯选自乙酸乙酯、乙酸正丁酯、乙酸正丙酯、甲酸乙酯、甲酸甲酯中的一种或几种混合物;
[0116]
优选地,所述助溶剂中的腈选自乙腈、丙腈、苯甲腈;
[0117]
优选地,所述助溶剂中的酰胺类选自n,n-二甲基乙酰胺、n,n-二甲基甲酰胺和n,n-二甲基吡咯烷酮中的一种或几种混合物。
[0118]
优选地,所述抗菌整理液中所述化合物(i)的含量为0.05~10wt%,优选0.1~5wt%;
[0119]
优选地,所述抗菌整理液的ph为8~11,优选8,8.5,9,9.5,10,10.5或11;
[0120]
优选地,所述抗菌整理液中表面活性剂的含量为0.01~2wt%,优选0.05~0.08wt%。
[0121]
优选地,所述抗菌整理液中助溶剂的含量为0~12wt%,优选0.1~10wt%。
[0122]
纺织品的抗菌整理方法,包括下列步骤:
[0123]
将纺织品在上文所述的抗菌整理液中浸泡,之后加热定型;
[0124]
所述浸泡时间优选1s~60min,优选1s,5s,10s,20s,30s,60s,5min,10min,15min,20min,30min或60min;
[0125]
所述浸泡时所述抗菌整理液的重量为所述纺织品的5~50倍,进一步优选10,15,20,25或30倍;
[0126]
优选地,所述加热的温度为60~150℃,优选80℃,85℃,90℃,95℃,100℃或105℃。
[0127]
综上,与现有技术相比,本发明达到了以下技术效果:
[0128]
(1)提供了一种可用于纺织品抗菌整理的化合物,可与材料表面的羟基、氨基等基
团发生化学反应,牢固的键合到材料表面;在对织物力学性能、透湿透气性和色泽等物理性能不产生负面影响前提下,赋予材料表面广谱、高效、持久抗菌防霉防螨活性;
[0129]
(2)产品无溶出,无毒无皮肤刺激性;
[0130]
(3)采用通用的浸-扎-烘干工艺、喷-甩-烘干工艺,进行整理,整理工艺无废水和废气产生;
[0131]
(4)整理液可以高浓度甚至纯品贮存运输,更环保更节能。
附图说明
[0132]
为了更清楚地说明本发明具体实施方式或现有技术中的技术方案,下面将对具体实施方式或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图是本发明的一些实施方式,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
[0133]
图1为实施例2所得化合物e整理棉纺织品后的抗菌结果;
[0134]
图2为实施例11所得化合物e处理棉布(1.0wt%)后的抗菌、防霉杀螨结果;
[0135]
图3为实施例19所得化合物e不同浓度下的抗大肠杆菌结果;
[0136]
图4为实施例19所得化合物e不同浓度下的抗金黄色葡萄球菌结果;
[0137]
图5为实施例19所得化合物e不同浓度整理棉布后的抗菌结果;
[0138]
图6为实施例1所得化合物e与两性离子在1.0wt%浓度下的抗菌效果对比;
[0139]
图7为实施例1所得化合物e抗菌整理棉布的红外光谱分析结果;
[0140]
图8为实施例1所得化合物e抗菌整理棉布的x射线光电子能谱分析结果。
具体实施方式
[0141]
下面将结合附图和具体实施方式对本发明的技术方案进行清楚、完整地描述,但是本领域技术人员将会理解,下列所描述的实施例是本发明一部分实施例,而不是全部的实施例,仅用于说明本发明,而不应视为限制本发明的范围。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。实施例中未注明具体条件者,按照常规条件或制造商建议的条件进行。所用试剂或仪器未注明生产厂商者,均为可以通过市售购买获得的常规产品。
[0142]
下文实施例1至16的制备过程分两步进行,反应路线如下:
[0143]
a+b
→
中间体i
[0144]
中间体i+d
→
e。
[0145]
实施例中反应物a、b、中间体i、反应物d和产物e的结构式分别如表1和2所示。
[0146]
表1
[0147]
[0148]
[0149][0150]
表2
[0151]
[0152][0153][0154]
每个实施例的具体反应条件如下。
[0155]
实施例1
[0156]
在圆底烧瓶中加入n,n-二甲基乙醇胺(dmea,即化合物a)1.35g(15mmol)和溴代辛烷(化合物b)4.35g(22.5mmol),置于45℃的油浴锅中反应2h,量取12ml丙酮作为溶剂加入圆底烧瓶中,接着回流12h;反应结束后,60℃条件下减压旋干溶剂,得白色固体,用无水石油醚洗涤几次,真空烘箱中烘干,得到中间体i,保存于瓶中待用,该中间体的产率为92%,核磁共振数据为:1h nmr(600mhz,d2o)δ3.92(tt,j=4.9,2.2hz,2h),3.41
–
3.33(m,2h),3.29
–
3.22(m,2h),3.01(s,6h),1.66(ddd,j=15.0,9.4,5.6hz,2h),1.32
–
1.10(m,10h),0.75(t,j=6.8hz,3h)。
[0157]
取中间体i,1.42g(5mmol)和碳酸酸钠0.8g溶于10ml去离子水配置成溶液加到滴液漏斗中待用;称量1.2g(4.9mmol)三聚氯氰(即化合物d)溶于30ml四氢呋喃并加入到圆底烧瓶中,在40℃下搅拌5min;将滴液漏斗中的溶液滴入到圆底烧瓶中,控制滴加速度为30min,滴加完后接着反应30min,tlc监控反应;反应结束后,用二氯甲烷萃取三次,取有机相用无水硫酸钠干燥,旋干得到白色固体,丙酮多次洗涤纯化,得到产物(即化合物e),该产物的产率为89%,核磁共振数据为:1h nmr(600mhz,dmso-d6)3.80(t,j=5.0hz,2h),3.42
–
3.35(m,2h),3.37
–
3.31(m,2h),3.08(s,6h),1.70
–
1.61(m,2h),1.32
–
1.19(m,10h),0.85(t,j=7.0hz,3h)。
[0158]
实施例2
[0159]
在圆底烧瓶中加入n,n-二甲基乙醇胺(dmea,即化合物a)1.35g(15mmol)和化合物b(溴代十八烷)9.13g(22.5mmol),置于35℃的油浴锅中反应5h,量取20ml乙醇作为溶剂加入圆底烧瓶中,接着回流8h;反应结束后,40℃条件下减压旋干溶剂,得白色固体,用无水乙醚洗涤几次,真空烘箱中烘干,得到中间体(季铵盐醇,即中间体i)保存于瓶中待用,该中间体的产率为92%,核磁共振数据为:1h nmr(600mhz,d2o)δ3.92(tt,j=4.9,2.2hz,2h),3.41
–
3.33(m,2h),3.29
–
3.22(m,2h),3.01(s,6h),1.66(ddd,j=15.0,9.4,5.6hz,2h),1.32
–
1.10(m,30h),0.75(t,j=6.8hz,3h)。
[0160]
取中间体(季铵盐醇)4.22g(10mmol)和n,n-二异丙基乙胺1.3g(10mmol)溶于20ml二氯甲烷中,加入滴液漏斗中;称量1.91g(10mmol)三聚氯氰(即化合物d)溶于50ml二氯甲烷置于圆底烧瓶中,在30℃下中搅拌30min;将滴液漏斗中的溶液滴入圆底烧瓶,滴速为1h,滴加完后反应过夜,tlc监控反应;反应结束后,快速柱层析分离得到纯的产物,该产物的产率为76%,核磁共振数据为:1h nmr(600mhz,dmso-d6)3.80(t,j=5.0hz,2h),3.42
–
3.35(m,2h),3.37
–
3.31(m,2h),3.08(s,6h),1.70
–
1.61(m,2h),1.32
–
1.19(m,30h),0.85(t,j=7.0hz,3h)。
[0161]
实施例3
[0162]
在圆底烧瓶中加入n,n-二甲基乙二胺(化合物a)0.89g和溴甲烷(化合物b)1.44g,置于65℃恒温油浴中反应1h;量取20ml丙醇作为溶剂加入圆底烧瓶中,接着继续反应15h;反应结束后,40℃条件下减压旋干溶剂,得白色固体,用四氢呋喃洗涤几次,真空烘箱中烘干,得到中间体i(季铵盐胺)保存于瓶中待用,该中间体的产率为95%,核磁共振数据为:1h nmr(600mhz,d2o)3.52(t,2h),3.30(s,9h),3.09(t,2h)。
[0163]
取中间体i(季铵盐胺)1.83g和n,n-二异丙基乙胺2.6g溶于20ml二氯甲烷中,加入滴液漏斗中;称量2.0g三聚氯氰(cc)溶于30ml二氯甲烷置于圆底烧瓶中,在室温搅拌
15min;将滴液漏斗中的溶液滴入圆底烧瓶,滴速为2h,滴加完后反应1h,tlc监控反应;反应结束后,真空减压除去溶剂得到产物,所述产物的产率83%,核磁共振数据为:1h nmr(600mhz,chloroform-d)3.47-3.52(m,4h),3.31(s,9h)。
[0164]
实施例4
[0165]
在圆底烧瓶中加入n,n-二甲基十八烷醇胺(化合物a)3.12g(10mmol)和溴甲烷(化合物b)1.45g(15mmol),置于65℃的油浴锅中反应2h,量取12ml异丙醇作为溶剂加入圆底烧瓶中,接着回流12h;反应结束后,60℃条件下减压旋干溶剂,得白色固体,用无水石油醚洗涤几次,真空烘箱中烘干,得到中间体i,保存于瓶中待用,该中间体的产率为89%,核磁共振数据为:1h nmr(600mhz,d2o)3.62(t,2h),2.83(s,9h),2.2(t,2h),1.3
–
1.58(m,32h)。
[0166]
取中间体i(季铵盐醇)3.98g和n,n-二异丙基乙胺2.6g溶于20ml四氢呋喃中,加入滴液漏斗中;称量2.0g三聚氯氰(cc)溶于30ml四氢呋喃置于圆底烧瓶中,在室温搅拌15min;将滴液漏斗中的溶液滴入圆底烧瓶,滴速为2h,滴加完后反应1h,tlc监控反应;反应结束后,真空减压除去溶剂得到产物,所述产物的产率78%,核磁共振数据为:1h nmr(600mhz,chloroform-d)3.82(t,2h),2.83(s,9h),2.2(t,2h),1.3
–
1.58(m,32h)。
[0167]
实施例5
[0168]
在圆底烧瓶中加入n,n-二甲基甲醇胺(化合物a)0.75g(10mmol)和溴甲烷(化合物b)1.45g(15mmol),置于45℃的油浴锅中反应2h,量取12ml甲醇作为溶剂加入圆底烧瓶中,接着回流12h;反应结束后,60℃条件下减压旋干溶剂,得白色固体,用无水石油醚洗涤几次,真空烘箱中烘干,得到中间体i,保存于瓶中待用,该中间体的产率为97%,核磁共振数据为:1h nmr(600mhz,d2o)5.42(s,2h),3.30(s,9h)。
[0169]
取中间体i,0.85g(5mmol)和碳酸酸钠0.8g溶于10ml去离子水配置成溶液加到滴液漏斗中待用;称量1.2g(4.9mmol)三聚氯氰(即化合物d)溶于30ml四氢呋喃并加入到圆底烧瓶中,在40℃下搅拌5min;将滴液漏斗中的溶液滴入到圆底烧瓶中,控制滴加速度为30min,滴加完后接着反应30min,tlc监控反应;反应结束后,用二氯甲烷萃取三次,取有机相用无水硫酸钠干燥,旋干得到白色固体,丙酮多次洗涤纯化,得到产物(即化合物e),该产物的产率为89%,核磁共振数据为:1h nmr(600mhz,dmso-d6)5.48(s,2h),3.30(s,9h)。
[0170]
实施例6
[0171]
在圆底烧瓶中加入化合物a1.03g和化合物b1.94g,置于85℃恒温油浴中反应0.5h;量取20ml丙醇作为溶剂加入圆底烧瓶中,接着继续反应18h;反应结束后,70℃条件下减压旋干溶剂,得白色固体,用四氢呋喃洗涤几次,真空烘箱中烘干,得到中间体i(季铵盐胺)保存于瓶中待用,该中间体的产率为91%,核磁共振数据为:1h nmr(600mhz,d2o)3.52(t,2h),3.30(s,6h),3.22(m,2h),1.71(m,2h)1.26-1.32(m,10h)0.82(s,3h),0.78(t,3h)。
[0172]
取中间体i(季铵盐醇)1.83g和n,n-二异丙基乙胺2.6g溶于20ml二氯甲烷中,加入滴液漏斗中;称量2.0g三聚氯氰(cc)溶于30ml二氯甲烷置于圆底烧瓶中,在室温搅拌15min;将滴液漏斗中的溶液滴入圆底烧瓶,滴速为2h,滴加完后反应1h,tlc监控反应;反应结束后,真空减压除去溶剂,石油醚洗涤得到产物,所述产物的产率83%,核磁共振数据为:1h nmr(600mhz,chloroform-d)3.58(t,2h),3.30(s,6h),3.22(m,2h),1.71(m,2h)1.26-1.32(m,10h)0.95(s,3h),0.78(t,3h)。
[0173]
实施例7
[0174]
在圆底烧瓶中加入化合物a 1.26g和化合物b 2.78g,置于35℃恒温油浴中反应8h;量取20ml丙醇作为溶剂加入圆底烧瓶中,接着继续反应18h;反应结束后,40℃条件下减压旋干溶剂,得白色固体,用四氢呋喃洗涤几次,真空烘箱中烘干,得到中间体i(季铵盐胺)保存于瓶中待用,该中间体的产率为91%,核磁共振数据为:1h nmr(600mhz,d2o)3.52(t,2h),3.30(s,6h),3.22(m,2h),1.71(m,2h)1.26-1.32(m,10h)0.82(s,3h),0.78(t,3h)。
[0175]
取中间体i(氨基季铵盐)4.03g和n,n-二异丙基乙胺2.6g溶于20ml二氯甲烷中,加入滴液漏斗中;称量2.0g三聚氯氰(cc)溶于30ml二氯甲烷置于圆底烧瓶中,在室温搅拌15min;将滴液漏斗中的溶液滴入圆底烧瓶,滴速为2h,滴加完后反应1h,tlc监控反应;反应结束后,真空减压除去溶剂,石油醚洗涤得到产物,所述产物的产率83%,核磁共振数据为:1h nmr(600mhz,chloroform-d)8.92(s,1h),7.92(d,1h),7.75(d,1h),7.01(s,n-h,1h),5.01(t,2h),4.04(t,2h),3.35(t,2h),2.55(m,2h),2.39(s,3h),2.01(m,2h),1.26-1.29(m,22h),0.88(t,3h)。
[0176]
实施例8
[0177]
在圆底烧瓶中加入化合物a 1.23g和化合物b 2.20g,置于80℃恒温油浴中反应1h;量取20ml异丙醇作为溶剂加入圆底烧瓶中,接着继续反应18h;反应结束后,40℃条件下减压旋干溶剂,得白色固体,用四氢呋喃洗涤几次,真空烘箱中烘干,得到中间体i(季铵盐醇)保存于瓶中待用,该中间体的产率为93%,核磁共振数据为:1h nmr(600mhz,d2o)3.52(t,2h),3.30(s,6h),3.22(m,2h),2.39(s,3h),1.71(m,2h)1.26-1.32(m,10h)0.92(s,3h),0.88(t,3h)。
[0178]
取中间体i(季铵盐醇)3.43g和n,n-二异丙基乙胺2.6g溶于20ml二氯甲烷中,加入滴液漏斗中;称量2.0g化合物c溶于30ml二氯甲烷置于圆底烧瓶中,在室温搅拌15min;将滴液漏斗中的溶液滴入圆底烧瓶,滴速为1h,滴加完后反应30min,tlc监控反应;反应结束后,真空减压除去溶剂,石油醚洗涤多次得到产物,所述产物的产率88%,核磁共振数据为:1h nmr(600mhz,chloroform-d)8.90(d,2h),7.67(d,2h),5.01(t,2h),3.63(t,2h),2.87(t,2h),2.01(m,2h),1.26-1.29(m,14h),0.88(t,3h)。
[0179]
实施例9
[0180]
在圆底烧瓶中加入化合物a 1.09g和化合物b1.42g,置于80℃恒温油浴中反应1h;量取20ml异丙醇作为溶剂加入圆底烧瓶中,接着继续反应18h;反应结束后,40℃条件下减压旋干溶剂,得白色固体,用四氢呋喃洗涤几次,真空烘箱中烘干,得到中间体i(季铵盐胺)保存于瓶中待用,该中间体的产率为93%,核磁共振数据为:1h nmr(600mhz,d2o)3.52(t,2h),3.30(s,6h),3.22(m,2h),2.39(s,3h),1.71(m,2h)1.26-1.32(m,10h)0.92(s,3h),0.88(t,3h)。
[0181]
取中间体i(季铵盐胺)2.51g和n,n-二异丙基乙胺2.6g溶于20ml二氯甲烷中,加入滴液漏斗中;称量2.0g化合物c溶于30ml二氯甲烷置于圆底烧瓶中,在冰浴中搅拌15min;将滴液漏斗中的溶液滴入圆底烧瓶,滴速为1h,滴加完后反应30min,tlc监控反应;反应结束后,真空减压除去溶剂,石油醚洗涤多次得到产物,所述产物的产率88%,核磁共振数据为:1h nmr(600mhz,chloroform-d)8.95(d,2h),8.61(s,1h),7.80(d,2h),4.38(s,3h),4.31(s,2h)。
[0182]
实施例10
[0183]
在圆底烧瓶中加入化合物a 1.12g和化合物b1.94g,置于70℃油浴锅中反应1h;量取20ml异丙醇作为溶剂加入圆底烧瓶中,接着继续反应18h;反应结束后,40℃条件下减压旋干溶剂,得白色固体,用四氢呋喃洗涤几次,真空烘箱中烘干,得到中间体i(季铵盐胺)保存于瓶中待用,该中间体的产率为93%。
[0184]
取中间体i 3.06g和碳酸酸钠0.8g溶于10ml去离子水配置成溶液加到滴液漏斗中待用;称量1.2g(4.9mmol)三聚氯氰(即化合物d)溶于30ml四氢呋喃并加入到圆底烧瓶中,在40℃下搅拌5min;将滴液漏斗中的溶液滴入到圆底烧瓶中,控制滴加速度为30min,滴加完后接着反应30min,tlc监控反应;反应结束后,用二氯甲烷萃取三次,取有机相用无水硫酸钠干燥,旋干得到白色固体,丙酮多次洗涤纯化,得到产物(即化合物e),该产物的产率为89%,核磁共振数据为:1h nmr(600mhz,chloroform-d)8.92(s,1h),7.92(s,1h),5.01(m,2h),2.98(d,2h),2.81(d,2h),2.01(m,2h),1.26-1.29(m,10h),0.88(t,3h)。
[0185]
实施例11
[0186]
在圆底烧瓶中加入化合物a 1.12g和化合物b 333.39g,置于75℃恒温油浴中反应1h;量取30ml二氯甲烷和丙醇(1:3)作为溶剂加入圆底烧瓶中,接着继续反应24h;反应结束后,60℃条件下减压旋干溶剂,得白色固体,用四氢呋喃洗涤几次,真空烘箱中烘干,得到中间体i(季铵盐醇)保存于瓶中待用,该中间体的产率为87%,核磁共振数据为:1h nmr(600mhz,d2o)8.92(s,1h),7.92(d,4h),4.80(m,2h),4.37(m,5h),3.63(t,2h),1.26-1.95(m,32),0.97(d,2h)。
[0187]
取中间体i(季铵盐醇)4.45g和n,n-二异丙基乙胺2.6g溶于20ml二氯甲烷中,加入滴液漏斗中;称量2.0g三聚氯氰(cc)溶于30ml二氯甲烷置于圆底烧瓶中,在室温搅拌15min;将滴液漏斗中的溶液滴入圆底烧瓶,滴速为2h,滴加完后反应1h,tlc监控反应;反应结束后,真空减压除去溶剂,石油醚洗涤得到产物,所述产物的产率83%,核磁共振数据为:1h nmr(600mhz,chloroform-d)8.92(s,1h),7.92(d,4h),4.80(m,2h),4.37(m,5h),3.72(t,2h),1.26-1.95(m,32),0.97(d,2h)。
[0188]
实施例12
[0189]
在圆底烧瓶中加入化合物a 1.12g和化合物b 0.95g,置于65℃恒温油浴中反应1h;量取20ml二氯甲烷作为溶剂加入圆底烧瓶中,接着继续反应18h;反应结束后,40℃条件下减压旋干溶剂,得白色固体,用四氢呋喃洗涤几次,真空烘箱中烘干,得到中间体i(季铵盐醇)保存于瓶中待用,该中间体的产率为91%,核磁共振数据为:1h nmr(600mhz,d2o)8.92(s,1h),7.92(d,4h),4.33-4.37(m,5h),3.63(t,2h)。
[0190]
取中间体i(季铵盐醇)2.08g和n,n-二异丙基乙胺2.6g溶于20ml二氯甲烷中,加入滴液漏斗中;称量2.0g三聚氯氰(cc)溶于30ml二氯甲烷置于圆底烧瓶中,在室温搅拌15min;将滴液漏斗中的溶液滴入圆底烧瓶,滴速为2h,滴加完后反应1h,tlc监控反应;反应结束后,真空减压除去溶剂,石油醚洗涤得到产物,所述产物的产率83%,核磁共振数据为:1h nmr(600mhz,chloroform-d)8.92(s,1h),7.92(d,4h),7.75(d,1h),4.58(t,2h),4.49(t,2h),4.36(s,3h),2.39(s,3h)。
[0191]
实施例13
[0192]
在圆底烧瓶中加入化合物a 3.36g和化合物b 0.95g,置于65℃恒温油浴中反应1h;量取20ml异丙醇作为溶剂加入圆底烧瓶中,接着继续反应24h;反应结束后,60℃条件下
减压旋干溶剂,得白色固体,用四氢呋喃洗涤几次,真空烘箱中烘干,得到中间体i(季铵盐醇)保存于瓶中待用,该中间体的产率为91%,核磁共振数据为:1hnmr(600mhz,d2o)8.92(s,1h),7.92(d,4h),4.33-4.37(m,5h),3.63(t,2h),1.26-1.95(m,32)。
[0193]
取中间体i(季铵盐醇)4.32g和n,n-二异丙基乙胺2.6g溶于20ml二氯甲烷中,加入滴液漏斗中;称量2.0g三聚氯氰(cc)溶于30ml二氯甲烷置于圆底烧瓶中,在室温搅拌15min;将滴液漏斗中的溶液滴入圆底烧瓶,滴速为2h,滴加完后反应1h,tlc监控反应;反应结束后,真空减压除去溶剂,石油醚洗涤得到产物,所述产物的产率83%,核磁共振数据为:1h nmr(600mhz,chloroform-d)8.92(s,1h),7.92(d,4h),4.33-4.37(m,5h),3.75(t,2h),1.26-1.95(m,32)。
[0194]
实施例14
[0195]
在圆底烧瓶中加入化合物a 1.26g和化合物b 0.95g,置于65℃中反应2h,量取20ml丙醇作为溶剂加入圆底烧瓶中,接着继续反应18h;反应结束后,40℃条件下减压旋干溶剂,得白色固体,用四氢呋喃洗涤几次,真空烘箱中烘干,得到中间体i(季铵盐胺)保存于瓶中待用,该中间体的产率为91%,核磁共振数据为:1h nmr(600mhz,d2o)8.92(s,2h),7.75(s,6h),4.36(s,2h)。
[0196]
取中间体i(季铵盐醇)2.21g和n,n-二异丙基乙胺2.6g溶于20ml二氯甲烷中,加入滴液漏斗中;称量2.0g三聚氯氰(cc)溶于30ml二氯甲烷置于圆底烧瓶中,在室温搅拌15min;将滴液漏斗中的溶液滴入圆底烧瓶,滴速为2h,滴加完后反应1h,tlc监控反应;反应结束后,真空减压除去溶剂,石油醚洗涤得到产物,所述产物的产率83%,核磁共振数据为:1h nmr(600mhz,chloroform-d)8.98(s,1h),,7.92(d,1h)7.76(d,1h),4.36(s,3h),4.04(t,2h),2.68(t,2h),2.58(m,2h)。
[0197]
实施例15
[0198]
在圆底烧瓶中加入化合物a 1.09g和化合物b 0.95g,置于80℃恒温油浴中反应1h;量取20ml乙醇作为溶剂加入圆底烧瓶中,接着继续反应18h;反应结束后,40℃条件下减压旋干溶剂,得白色固体,用四氢呋喃洗涤几次,真空烘箱中烘干,得到中间体i(季铵盐醇)保存于瓶中待用,该中间体的产率为93%,核磁共振数据为:1h nmr(600mhz,d2o)9.01(d,2h),8.22(d,2h),4.61(s,2h),4.38(s,3h)。
[0199]
取中间体i(季铵盐醇)2.04g和n,n-二异丙基乙胺2.6g溶于20ml二氯甲烷中,加入滴液漏斗中;称量2.0g三聚氯氰(cc)溶于30ml二氯甲烷置于圆底烧瓶中,在室温搅拌15min;将滴液漏斗中的溶液滴入圆底烧瓶,滴速为1h,滴加完后反应30min,tlc监控反应;反应结束后,真空减压除去溶剂,石油醚洗涤多次得到产物,所述产物的产率88%,核磁共振数据为:1h nmr(600mhz,chloroform-d)9.01(d,2h),8.22(d,2h),4.83(s,2h),4.38(s,3h)。
[0200]
实施例16
[0201]
在圆底烧瓶中加入化合物a 1.40g和化合物b 4.52g,置于65℃恒温油浴中反应1h;量取20ml异丙醇作为溶剂加入圆底烧瓶中,接着继续反应24h;反应结束后,60℃条件下减压旋干溶剂,得白色固体,用四氢呋喃洗涤几次,真空烘箱中烘干,得到中间体i(季铵盐醇)保存于瓶中待用,该中间体的产率为91%,核磁共振数据为:1h nmr(600mhz,d2o)9.01(d,2h),8.22(d,2h),4.81(d,2h),4.61(d,2h),4.38(s,3h),1.26-1.95(m,32)。
[0202]
取中间体i(季铵盐醇)6.38g和n,n-二异丙基乙胺2.6g溶于20ml二氯甲烷中,加入滴液漏斗中;称量2.0g三聚氯氰(cc)溶于30ml二氯甲烷置于圆底烧瓶中,在室温搅拌15min;将滴液漏斗中的溶液滴入圆底烧瓶,滴速为2h,滴加完后反应1h,tlc监控反应;反应结束后,真空减压除去溶剂,石油醚洗涤得到产物,所述产物的产率83%,核磁共振数据为:1h nmr(600mhz,chloroform-d)4.01(t,4h),2.60(t,2h),2.26(t,2h),1.26-1.30(m,40h),0.88(s,6h)。
[0203]
下文实施例17至19的制备过程分三步进行,反应路线如下:
[0204]
a+b
→
中间体i
[0205]
中间体i+m
→
中间体ii
[0206]
中间体ii+d
→
e。
[0207]
实施例17至19中化合物a、b、中间体i、m、中间体ii、d和e的结构式分别如表3所示。
[0208]
表3
[0209][0210]
[0211]
每个实施例的反应条件如下。
[0212]
实施例17
[0213]
称取1.69g的化合物a和2.2g化合物b加入250ml圆底烧瓶里,室温下搅拌2h,加入50ml丙酮并在此温度下继续搅拌反应12h,得中间体中间体i。然后在搅拌下加入化合物c1.37 g和n,n-二异丙基乙胺1.3g,超纯氮气保护下继续反应12h,得到含羟基的双季铵盐(中间体ii),将其加入滴液漏斗中。称量1.85g化合物d溶于100ml四氢呋喃并加入到圆底烧瓶中,在0℃下搅拌5min;将滴液漏斗中的溶液滴入到圆底烧瓶中,控制滴加速度为30min,滴加完后接着反应30min,tlc监控反应;反应结束后,旋干得到白色固体,丙酮多次洗涤纯化,得到产物,该产物的产率为78%。该产物的核磁共振数据为1h nmr(600mhz,chloroform-d):3.42(t,2h),3.34(t,4h),3.30(s,12h),3.22(t,4h),2.80(t,4h),2.57(t,2h),1.71(m,4h),1.26-1.29(m,36h),0.88(t,6h)。
[0214]
实施例18
[0215]
称取6.08g的化合物a和1.88g化合物b加入250ml圆底烧瓶里,室温下搅拌2h,加入100ml丙酮并在此温度下继续搅拌反应12h,得中间体中间体i。然后在搅拌下加入化合物c1.37 g和n,n-二异丙基乙胺1.3g,超纯氮气保护下继续反应12h,得到含羟基的双季铵盐(中间体ii),将其加入滴液漏斗中。称量1.85g化合物d溶于100ml四氢呋喃并加入到圆底烧瓶中,在0℃下搅拌5min;将滴液漏斗中的溶液滴入到圆底烧瓶中,控制滴加速度为30min,滴加完后接着反应30min,tlc监控反应;反应结束后,旋干得到白色固体,丙酮多次洗涤纯化,得到产物,该产物的产率为85%。该产物的核磁共振数据为1h nmr(600mhz,chloroform-d):3.42(t,2h),3.30(s,18h),3.22(t,4h),2.46(t,4h),2.57(t,2h),1.71(m,4h),1.29-136(m,60h)。
[0216]
实施例19
[0217]
称取1.31g的化合物a和4.42g化合物b加入250ml圆底烧瓶里,室温下搅拌2h,加入50ml丙酮并在此温度下继续搅拌反应12h,得中间体中间体i。然后在搅拌下加入化合物c 0.88g和n,n-二异丙基乙胺1.3g,超纯氮气保护下继续反应12h,得到含羟基的双季铵盐(中间体ii),将其加入滴液漏斗中。称量1.85g化合物d溶于100ml四氢呋喃并加入到圆底烧瓶中,在0℃下搅拌5min;将滴液漏斗中的溶液滴入到圆底烧瓶中,控制滴加速度为30min,滴加完后接着反应30min,tlc监控反应;反应结束后,旋干得到白色固体,丙酮多次洗涤纯化,得到产物,该产物的产率为80%。该产物的核磁共振数据为1h nmr(600mhz,chloroform-d):4.31(s,4h),3.42(t,2h),3.30(s,12h),3.22(t,4h),2.53(t,2h),1.71(m,4h),1.26-1.29(m,28h),0.88(t,6h)。
[0218]
下文实施例20至21的制备过程分两步进行,反应路线如下:
[0219]
a+b
→
中间体i
[0220]
中间体i+d
→
e。
[0221]
实施例20至21中化合物a、b、中间体i、d和e的结构式如表4所示。
[0222]
表4
[0223]
[0224][0225]
每个实施例的反应条件如下。
[0226]
实施例20
[0227]
取2.62g化合物a溶于20ml甲苯中并转移到100ml的三口圆底烧饼中,加入1.5g的化合物b在高纯氮气和50℃温度和持续搅拌下反应36h,反应混合物经过滤和石油醚反复洗涤得到中间产物i。
[0228]
称量中间产物i 4.02g和n,n-二异丙基乙胺2.6g溶于30ml四氢呋钠,加入滴液漏斗中,然后在圆底烧瓶中加入1.85g化合物d(溶于20ml四氢呋喃),在0℃下搅拌5min;将滴液漏斗中的溶液滴入到圆底烧瓶中,控制滴加速度为30min,滴加完后接着反应30min。tlc监控反应;反应结束后,用二氯甲烷萃取三次,取有机相用无水硫酸钠干燥,旋干得到白色固体,丙酮多次洗涤纯化,得到产物反应性的季鏻盐,即化合物e,该产物的总产率为55%。该产物的核磁共振数据为:1h nmr(600mhz,chloroform-d)1.5(m,2h),2.46(t,2h),3.49(t,2h),7.33-7.36(m,15h)。
[0229]
实施例21
[0230]
取2.62g化合物a溶于20ml甲苯中并转移到100ml的三口圆底烧饼中,加入2.4g的化合物b在高纯氮气和50℃温度和持续搅拌下反应36h,反应混合物经过滤和石油醚反复洗涤得到中间产物i。
[0231]
称量中间产物i 6.22g和n,n-二异丙基乙胺2.6g溶于30ml四氢呋钠,加入滴液漏斗中,然后在圆底烧瓶中加入1.85g化合物d(溶于20ml四氢呋喃),在0℃下搅拌5min;将滴液漏斗中的溶液滴入到圆底烧瓶中,控制滴加速度为30min,滴加完后接着反应30min。tlc监控反应;反应结束后,用二氯甲烷萃取三次,取有机相用无水硫酸钠干燥,旋干得到白色固体,丙酮多次洗涤纯化,得到产物反应性的季鏻盐,即化合物e,该产物的总产率为52%。该产物的核磁共振数据为:1h nmr(600mhz,chloroform-d)1.22-1.39(m,10h),1.49(m,2h),2.43(t,2h),3.45(t,3h),7.33-7.36(m,15h)。
[0232]
本发明所有实施例最终制备的产物均可用于制作抗菌整理液,并且不同使用条件
的抗菌效果有区别。
[0233]
比较实施例1至21所得化合物e的抗菌效果,具体如下表5。
[0234]
表5
[0235][0236]
备注:1)表5抗菌数据数据,实施例1-9采用浸渍-辊轧-烘干工艺对棉织品进行整理,其中浴液比为20:1,轧余率为90%,烘干工艺为85℃焙烘30min;实施例10-18采用浸渍-高速甩干-烘干法进行整理,其中浴液比为20:1,余率为90%,烘干工艺为85℃焙烘20min;实施例19-21为喷涂-甩干-焙烘工艺进行整理,浴液比为30:1,甩干后余液率为85%,烘干烘干工艺为105℃焙烘5min。
[0237]
2)表5数据的测试方法:fz/t73023-2006抗菌针织品d.8震荡法。此外,采用晕圈法(fz/t73023-2006抗菌针织品-附录e-晕圈法)测试得上述21个实施例和两性离子的对比实验的抑菌圈结果均为0mm,产品均完全不溶出。对比实验所用抗菌整理剂为两性离子,其分子结构为整理工艺和浓度均与实施例1-9一致。
[0238]
3)表5中整理液的组成为:化合物e1.0wt%,碳酸氢钠1.0wt%,余量为水。
[0239]
此外,本发明还提供了部分抗菌结果示例。
[0240]
(1)实施例2所得化合物e浸渍织物和未浸渍织物的抗菌效果对比如图1,实验过程
是:采用振荡法(gb/t 20944.3-2007)对整理纺织品的抗菌性能进行测试。受试细菌有革兰氏阴性的大肠杆菌(e.coli,atcc 25922)和革兰氏阳性的金黄色葡萄球菌(s.aureus,atcc 6538),具体实验步骤如下:1)接种菌落,37℃下,培养14h;用已灭菌的pbs缓冲液(ph=7.2)梯度稀释菌液至10-5
,得到稀释菌液。2)取相同尺寸的空白纺织品和整理后纺织品(已灭菌)于不同50ml试管,各加入5ml稀释菌液浸没织物,密封后放至摇床,37℃,190r/min,充分振荡24h。3)培养24h后,取出试管,用pbs液进行10倍梯度稀释,取100μl至营养琼脂培养基中,均匀涂抹至干,放入培养箱中,37℃下,培养16h后,取出,计算平皿中菌落个数。按公式计算:
[0241]
抗菌率(%)=[(n
1-n0)/n0]
×
100%。
[0242]
注:n1整理后棉布菌落数;n0空白棉布菌落数。
[0243]
并利用抑菌圈法测试整理纺织品的溶出性。
[0244]
结果显示整理棉织物的对e.coli和s.aureus的抗菌率分别大于99.5%、99.9%,且属于非溶出性抗菌织物。
[0245]
(2)实施例11所得化合物e浸渍织物和未浸渍织物的抗菌效果对比如图2,实验过程是:采用采用活细胞计数法对整理纺织品的抗菌性能进行测试,按照国家标准方法(gb/t 24253-2009)对房屋尘螨的防螨活性进行评价。结果显示对革兰氏阴性菌(大肠杆菌)、革兰氏阳性菌(金黄色葡萄球菌)和真菌(黑曲霉菌)的抑菌率均达99.99%以上,对房屋尘螨有较好的杀螨活性。
[0246]
(3)实施例19所得化合物e不同浓度下对大肠杆菌和金黄色葡萄球菌的抑制效果如图3至5。实验过程是:使用酶标法测定抗菌药物的mic值,将稀释后不同浓度的抗菌药物溶液分别加到灭菌的100孔板中,如:第1至11孔加10ul药液,浓度分别为100μg/ml、90μg/ml、80μg/ml、70μg/ml、60μg/ml、50μg/ml、40μg/ml、30μg/ml、20μg/ml、10μg/ml、5μg/ml,第12孔不加药做对照组;经lb肉汤稀释后的菌悬液(0.5麦氏比浊标准稀释1000倍,菌液浓度为105cfu/ml),每孔加入90μl,此时,第1孔至第11孔药物浓度分别为10μg/ml、9μg/ml、8μg/ml、7μg/ml、6μg/ml、5μg/ml、4μg/ml、3μg/ml、2μg/ml、1μg/ml、0.5μg/ml,在酶标仪(全自动生长曲线仪)中测定24h的od600值的变化曲线。分别配置2.0wt%、1.0wt%、0.5wt%、0.1wt%、0.05wt%抗菌整理液,按照所述的整理工艺整理,并采用振荡法(gb/t 20944.3-2007)对整理纺织品的抗菌性能进行测试。结果显示:对大肠杆菌的最低抑菌浓度为4μg/ml,对金黄色葡萄球菌的最低抑菌浓度为1μg/ml。抗菌整理液浓度》0.1%时整理的纺织品对金黄色葡萄球菌(s.aureus)和大肠杆菌(e.coli)的抗菌率》99.9%。
[0247]
(4)比较实施例1和两性离子对大肠杆菌和金黄色葡萄球菌的抑制效果,结果如图6所示。实验过程是:配置相同浓度均为1.0wt%的季铵盐(实施例1)和两性离子抗菌整理液对纺织品进行抗菌整理,采用振荡法(gb/t 20944.3-2007)对整理纺织品的抗菌性能进行测试。结果显示:相同浓度下,季铵盐整理液整理的纺织品对金黄色葡萄球菌(s.aureus)和大肠杆菌(e.coli)的抗菌活性均大于两性离子整理液整理的纺织品。
[0248]
(5)实施例1所得化合物e抗菌整理棉布(化合e在整理液中的浓度为1.0wt%)的红外光谱分析结果如图7,实验过程是:采用红外光谱反射法(atr)对比研究了整理前后纺织品的结构,扫描范围(400~4000cm-1
),扫描次数(32次),分辨率(4cm-1
)。结果显示:相比于未整理的纺织品,经化合物e整理的纺织品存在三嗪环和季铵基团,表明化合物e接枝在纺
织品表面。
[0249]
(6)实施例1所得化合物e抗菌整理棉布(化合e在整理液中的浓度为1.0wt%)的x射线光电子能谱分析结果如图8,实验过程是:采用x射线光电子能谱(xps,ulvac-phi 1800)分析产物的元素组成及确定氮(n)元素的化学价态。以al靶(1486.6ev)为辐射源,其测试仰角为75
°
。
[0250]
结果显示:经化合物e整理的棉纺织品在扫描光谱中多了n信号峰,n1s可以拟合分成三个峰,结合能为398.5ev、399.9ev、402ev,分别属于c=n、c-n、n
+
,这三种类型的峰与化合物e上n的类型相吻合,表明化合物e以化学键合的方式接枝在纺织品表面。
[0251]
最后应说明的是:以上各实施例仅用以说明本发明的技术方案,而非对其限制;尽管参照前述各实施例对本发明进行了详细的说明,本领域的普通技术人员应当理解:其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分或者全部技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本发明各实施例技术方案的范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1