具有电极的纳米通道形装置和相关方法与流程
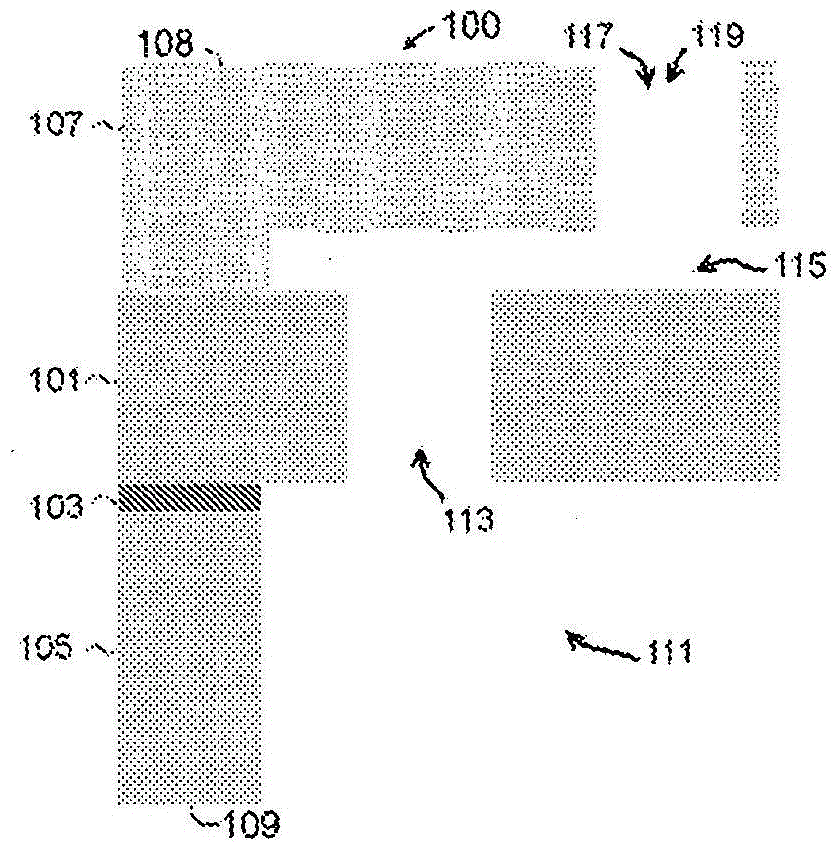
相关申请的交叉引用本申请要求2013年8月1日提交的美国临时专利申请序列号61/861,070的优先权,通过引用将其内容并入本文。背景信息本申请在来自nasa的拨款nnj06he06a和nnx08aw91g和来自国防部的dodw81xwh-09-1-0212的政府资助下完成。政府享有本发明的某些权利。在过去三十年中,在治疗剂(例如药物)递送
技术领域:
中已取得相当大的进步,在临床医学中产生了许多突破。设计能够以受控的方式递送治疗剂的治疗剂递送装置仍是挑战。可植入的药物递送装置的主要要求之一是治疗剂的受控释放,所述治疗剂范围从小药物分子到较大的生物分子。特别需要的是获得与零级动力学一致的持续被动药物释放曲线,由此在整个延长的递送期中血流中的药物浓度保持恒定。这些装置具有如下潜力:改善治疗效力,消除潜在的威胁生命的副作用,改善患者的顺应性,使健康护理人员的介入最小化,减少住院时间,以及减少管制药物向滥用的转变。纳米通道递送装置可以用于有效施用药物的药物递送产品。此外,纳米通道递送装置可以用于其中需要物质随时间的可控释放的其他应用。虽然已设计了一些可植入的被动药物递送装置以用于治疗剂的恒定施用,但是对于持续调节药物从这些装置释放的机制的缺乏是其使用的限制因素。主动控制从植入装置施用治疗剂的速率的能力将允许采用用于需要非恒定药物施用(例如时间治疗)的治疗的系统。在一些情况下,循环药物施用可增强治疗的疗效。事实上,时间生物学研究显示了疾病的病理生理学中的节律性。换言之,药物的疗效显示为强烈依赖于施用日程。从被动释放可植入系统中非恒定施用药物需要开发简单且安全的装置以用于药物释放的远程活化和减活以及精细调节。最近的研究已显示了重要的新型治疗范例的可能性,包括节律化递送和时间治疗,其中治疗施用的准确定时和位置对于效力和毒性具有显著影响。需要新型的药物递送结构从而不仅以精确的速率持续释放药物,而且还使其释放与昼夜周期同步。常规系统施用的化学疗法通常依赖于这样的给药方案:其中在2-3周恢复期之后将细胞毒性药物以最大可耐受剂量(mtd)一次递送或输注有限持续时间[31]。需要恢复期以使患者克服与该施用方式相关的副作用。新型递送方案,包括节律化递送[31,32]和时间治疗[33,34],在临床前试验中显示了通过调节递送的定时和/或频率,所需剂量可从mtd下调,且有害副作用随之减少,同时保持或甚至增加了疗效[31,33]。在节律性递送中,低于mtd的剂量以短时间增量施用,而不需要恢复期[31],然而在时间治疗中,递送与自然昼夜周期同步[34]。为了实现这些新型递送范例的全面潜力,已开发了大量的新型递送载体。在体内和体外均已显示了由基于硅的纳米流体膜被动控制的植入体能够进行持续的不依赖浓度的恒定和可再现的药物释放,即节律性递送的目标[35-37]。这样的被动释放膜是精确制造的,机械上牢固的,且能够在无需移动部件的情况下具有与其他递送形态相当的临床上合适的递送速率。然而,时间治疗的同步化还需要主动控制。到目前为止,文献中已出现了大量主动控制的药物递送结构。已在体内和体外由盖有薄金膜的具有贮器的硅微芯片实现了预编程的脉动递送,所述硅微芯片可被电破裂和单独寻址[38-39]。由于脉动递送受限于释放不连续量的药物,然而还研究了电动力学激励。通过在具有小至2nm[51]的临界尺寸的纳米通道中施加纵向和/或横向电场[40-50]已实现了纳米流体通道的直接电动力学控制。已研究了这样的装置作为预浓缩器[52-55]和分离器[56,57],以用于大量诊断技术,用于单一分子操作[58-61],和用于其在药物递送应用中的潜力[62]。基于电渗泵的递送装置使用了单独的电渗泵送室以施加压力至活塞或膈膜上从而使药物从第二含药室渗出[63,64]。这些装置,通常用玻璃料或轨迹蚀刻的聚合物膜实施[65],已显示为可切换的且能够进行持续释放。然而,这样的系统需要大的植入体积以容纳密封的泵送室,这对植入物的几何形状产生了另外的限制。用于样品加工[66]和药物递送[67]的电泳装置受益于整体流体净流动的缺少,所述整体流体净流动增大了环境压力梯度但在过去需要大功率。在电渗中,这样的压力梯度,对分子的缓释的潜在限制,必须包括复杂的植入物设计。已设计了用于治疗剂的恒定施用的一些可植入的被动药物递送装置[34]。然而,对于持续调节药物从这些装置释放的机制的缺乏是其使用的限制因素。主动控制从植入装置施用治疗剂的速率的能力将允许采用用于需要非恒定药物施用的治疗(例如时间治疗)的系统。在一些情况下,循环药物施用可增强治疗的疗效。事实上,时间生物学研究显示了疾病的病理生理学中的节律性[35]。换言之,药物的疗效显示为强烈依赖于施用日程。从被动释放可植入系统中非恒定施用药物需要开发简单且安全的装置以用于药物释放的远程活化和减活以及精细调节。微制作和纳米制作已能够产生具有足以研究和开发一系列的输送现象的尺寸的流体通道,所述输送现象包括零级被动扩散[1]、场效应流动控制[2,3]、表面控制的传导[2,4]、熵受限(trapping)[5]和浓差极化[2,7,8,9,10],这些是限于纳米级的流体所独有的。这些现象中的大部分在某种程度上与如下相关:如何聚集抗衡离子以及随后将其自身排布在溶液和纳米通道壁之间的界面处以筛选表面电荷,即称作电双层(edl)的结构[11]。已证实了控制分析物如何与edl相互作用对于应用例如药物递送[12]、分子筛[13,14]、控制燃料电池中的氢扩散[15]和生物样品的预浓缩[16]是有用的[12]。先前报道的基于硅的纳米通道膜是机械上牢固的,具有数十万个小至3nm的纳米制作的单分散的平行狭缝纳米通道[13,14]。已在体外和体内将该膜结构纳入到用于以临床相关的释放速率在延长的持续时间内的药剂的零级释放的可植入药物贮器中。纳米通道单分散性极其重要,因为零级被动释放依赖于有效纳米通道高度(物理高度加上壁和带电荷的分析物之间的任何静电相互作用)和扩散的分析物大小之间的确定关系[17,19]。然而,理想的是能够对用于这样的应用例如时间治疗的这些被动扩散装置进行释放速率调节[20]。时间治疗代表这样的给药方案:其中药剂的递送与天然昼夜周期同步,且已显示其对于一系列病理状况提高了疗效[21,22]。在这种背景下,我们先前已证实了利用电泳作为时间控制的主要模式的电动力学释放调节[17]。这些电泳控制的原型,源自较早期的较慢释放的100nm的纳米通道膜,其具有嵌入的铂电极,显示了在所施加的2v电势下释放速率的10x增加。还已经开发了基于电渗泵[23]、原电池、磁共振[24]和离子电渗法[25]等的其他替代性的可植入和经皮电动力学和电磁学装置。浓差极化通过在施加电场时纳米通道的入口和出口处的带电荷的分析物的累积和消耗来定义[8,9,10,26和27]。这些累积和消耗区域来自静电相互作用,在将相反极性的抗衡离子累积到纳米通道壁上的固有表面电荷时,该静电相互作用排斥具有相同极性的共生离子,同时仍保持整体溶液中的电荷中性。在消耗区域中,debye长度增加,导致对于共生离子移动通过纳米通道的进一步限制[26,28]。在分子检测之前,该现象已用于分析物的预浓缩[29]。还已证实其对于通过调节成分的等电点周围的ph来分离复杂混合物是有用的,甚至当debye长度相对短时处于中等离子强度[28]。该公开内容报道了改进快速释放纳米通道递送系统膜以能够电动力学调节药物释放。特别选择了树状富勒烯1(df-1)作为分析物,因为其具有高价态电荷(在ph7.4时为10.4,从而在较好地刺激体内环境的高离子强度溶液中使分析物对于所施加的电势的应答最大化)并且其可能用作自由基清除剂和mri造影剂的载体分子[30]。然而,在这些较小的纳米通道装置的情况下,与所施加的电压的极性无关,1.5v的施加导致了释放速率的下降,而不是利用先前产生的100nm膜所见的增加。根据铂电极之间的分隔距离(对于电子束沉积的薄膜为~1mm),该向下的释放速率调节可为多至两个数量级。这样的结果与电泳和电渗均不一致。该密封的基于uv的测试设备进一步排除了电渗,因为在这样的封闭系统中会预期相对于整体流体流动而快速建立相关压力梯度。因此在浓差极化中所见的debye长度调节会对利用改变离子强度以调节df-1释放的较早期实验的类似物所观察到的释放动力学作出可能的解释[20]。与利用被动释放膜获得的零级动力学相似,对于在浓差极化中所观察到的debye长度调节,还必须维持分析物大小和纳米通道高度之间的确切关系,以如本文所证实的那样显著影响分析物释放(尤其是存在于体内的高离子强度环境,预期该环境下debye长度大约为数纳米或更小)。概述在下文中,术语“结合”定义为连接,但是不必是直接的,而且也不必是机械上的。当在权利要求书和/或说明书中与术语“包括有”结合使用时,单词“一个”或“一种”的使用可以是指“一个”,但是它也与“一个或多个”或“至少一个”的含义一致。术语“约”一般是指所述值加或减5%。权利要求书中术语“或”的使用是用于表示“和/或”,除非另有说明是指仅替代物或替代物是互相排斥的,但是本公开内容支持指的是仅替代物和“和/或”的定义。术语“包括有”(comprise)(及其任何形式,例如“包括有”(comprises)和“包括有”(comprising)),“具有”(及其任何形式,例如“具有”(has)和“具有”(having)),“包括”(include)(及其任何形式,例如“包括”(includes)和“包括”(including))和“包含”(contain)(及其任何形式,例如“包含”(contains)和“包含”(containing))是开放式的连系动词。因此,“包括有”、“具有”、“包括”或“包含”一个或多个步骤或元件的方法或装置具有该一个或多个步骤或元件,但不限于仅具有该一个或多个元件。相似地,“包括有”、“具有”、“包括”或“包含”一个或多个特征的方法的步骤或装置的元件具有该一个或多个特征,但不限于仅具有该一个或多个特征。此外,以某方式配置的装置或结构以至少该方式来进行配置,但还可以以未列出的方式来进行配置。术语“入口微通道”定义为微通道,在进入纳米通道递送装置的纳米通道之前分子移动经过该微通道。术语“出口微通道”定义为微通道,在即将离开纳米通道递送装置之前分子移动经过该微通道。术语“纳米通道”定义为具有小于200nm的至少一个尺寸(例如,高度、宽度、直径等)的横截面的通道。术语“大通道”定义为具有大于约10μm的最大尺寸(例如,高度、宽度、直径等)的横截面的通道。本发明的其他目的、特征和优点将从以下详细描述中变得清楚。然而,虽然指出了本发明的特定实施方案,但是应理解详细描述和特定的实施例仅以说明的方式给出,因为对于本领域技术人员而言,本发明的精神和范围中的各种改变和变化都将从该详细描述中变得清楚。本公开内容的示例性实施方案包括主动控制的纳米流体膜,其利用电泳来控制药物释放的幅度、持续时间和定时。在示例性的实施方案中,使用高精度硅制作技术制备的膜具有在入口和出口处具有集成的铂电极,其允许采用低的施加电压(2vdc或以下)的分析物递送的放大和反转。用异硫氰酸荧光素缀合的牛血清白蛋白的溶液使用荧光光谱法和显微镜法证实了装置操作,并且对于长期分子释放和释放可逆性对其进行了表征。通过理论与实验分析的结合,确定了电泳和电渗的相对贡献。在2vdc下膜的临床相关的电泳释放速率超过被动释放接近一个数量级,这证实了实现治疗范例目标的潜力。示例性的实施方案包括在入口和出口处具有集成的铂电极的可植入的且机械上牢固的纳米流体膜。该电动力学驱动的纳米流体膜(纳米通道递送系统(nds))利用高度精确的硅纳米制作技术来制造。这样的设计使得在低的施加电压时以最小的电解使分析物的电泳输送通过其纳米流体通道。我们在30天周期中应用了荧光光谱法以测量与异硫氰酸荧光素(fitc-bsa)缀合的牛血清白蛋白的膜释放特性,和应用荧光显微镜法来显现微通道和纳米通道中的fitc-bsa的动力学。电泳输送是完全可逆的,且与浓度驱动的输送驱使的临床相关的释放速率[35]相比较,能够提高释放速率9倍以上。一些实施方案包括纳米通道递送装置,其包括:入口微通道;纳米通道;出口微通道;第一电极;和第二电极,其中所述入口微通道和所述出口微通道与所述纳米通道直接流体连通,和其中所述第一电极与所述纳米通道递送装置的第一表面直接结合。在特定的实施方案中,所述第二电极与所述纳米通道递送装置的第二表面直接结合。在一些实施方案中,所述第一电极包括在所述第一表面上沉积的导电材料,所述第二电极包括在所述第二表面上沉积的导电材料。在特别的实施方案中,所述第二表面是所述入口微通道、出口微通道或所述纳米通道的表面。在一些实施方案中,所述第一表面是所述入口微通道、出口微通道或所述纳米通道的表面。在特定的实施方案中,所述纳米通道递送装置经配置以通过在第一电极和第二电极之间施加电压来控制通过所述纳米通道的分子的扩散速率。在一些实施方案中,纳米通道与所述纳米通道递送装置的主平面平行取向。在特别的实施方案中,由入口微通道经纳米通道至出口微通道的流动路径需要在方向上有最多两个变化。在一些实施方案中,入口微通道具有长度、宽度和深度;出口微通道具有长度、宽度和深度;纳米通道具有长度、宽度和深度;纳米通道长度与入口微通道长度的比为0.01-50.0;和纳米通道长度与出口微通道长度的比为0.01-10.0。在特别的实施方案中,入口微通道通过单一纳米通道与所述出口微通道直接流体连通。一些实施方案包括纳米通道递送装置,其包括:入口微通道;纳米通道;出口微通道;第一电极;第二电极;和从所述入口微通道到所述出口微通道的流体流动路径,其中所述流体流动路径需要在方向上有最多两个变化,和所述第一电极与所述纳米通道递送装置的第一表面直接结合。在特别的实施方案中,所述第二电极与所述纳米通道递送装置的第二表面直接结合。在一些实施方案中,所述第一电极包括在所述第一表面上沉积的导电材料,所述第二电极包括在所述第二表面上沉积的导电材料。在特定的实施方案中,所述第二表面是所述入口微通道、出口微通道或所述纳米通道的表面。在一些实施方案中,所述第一表面是所述入口微通道、出口微通道或所述纳米通道的表面。在特别的实施方案中,所述纳米通道递送装置经配置以通过施加电压至第一电极和第二电极来控制通过所述纳米通道的分子的扩散速率。在一些实施方案中,纳米通道与所述纳米通道递送装置的主平面平行取向。在特定的实施方案中,所述入口微通道和所述出口微通道与所述纳米通道直接流体连通。一些实施方案包括纳米通道递送,包括:基本平坦的本体,所述本体包含第一表面和与所述第一表面相对的第二表面;位于所述基本平坦的本体内的纳米通道;第一电极;第二电极;与所述纳米通道流体连通的入口微通道;与所述纳米通道流体连通的出口微通道,其中所述入口微通道从所述纳米通道延伸至所述第一表面,和其中所述出口微通道从所述纳米通道延伸至第二表面;和从所述入口微通道到所述出口微通道的流体流动路径,其中所述流体流动路径需要在方向上有最多两个变化,和其中所述第一电极与所述纳米通道递送装置的第一表面直接结合。在特别的实施方案中,所述第二电极与所述纳米通道递送装置的第二表面直接结合。在一些实施方案中,所述第一电极包括在所述第一表面上沉积的导电材料和所述第二电极包括在所述第二表面上沉积的导电材料。在特别的实施方案中,所述第二表面是所述入口微通道、出口微通道或所述纳米通道的表面。在一些实施方案中,所述第一表面是所述入口微通道、出口微通道或所述纳米通道的表面。在特定的实施方案中,所述纳米通道递送装置经配置以通过施加电压至第一电极和第二电极来控制通过所述纳米通道的分子的扩散速率。在一些实施方案中,所述第一电极与和流体流动路径的第一端邻近的第一表面直接结合,和其中所述第二电极与和流体流动路径的第二端邻近的第二表面直接结合。特别的实施方案包括纳米通道递送装置,包括:多个入口微通道;第一电极;第二电极;多个纳米通道;和多个出口微通道,其中各入口微通道通过单一纳米通道与出口微通道直接流体连通,和其中所述第一电极与所述纳米通道递送装置的第一表面直接结合。在一些实施方案中,所述第二电极与所述纳米通道递送装置的第二表面直接结合。在一些实施方案中,所述第二表面是所述多个入口微通道的第一入口微通道、所述多个出口微通道的第一出口微通道或所述多个纳米通道的第一纳米通道的表面。在特别的实施方案中,所述第一表面是所述第一入口微通道、所述第一出口微通道或所述第一纳米通道的表面。在一些实施方案中,所述纳米通道递送装置经配置以通过施加电压至第一电极和第二电极来控制通过所述第一纳米通道的分子的扩散速率。在特定的实施方案中,所述纳米通道递送装置包括所述多个纳米通道的第二纳米通道,和其中施加电压至第一和第二电极不会控制分子通过所述第二纳米通道的扩散速率。在一些实施方案中,所述第一电极包括在所述第一表面上沉积的导电材料和所述第二电极包括在所述第二表面上沉积的导电材料。在特别的实施方案中,所述纳米通道与所述纳米通道递送装置的主平面平行取向。在一些实施方案中,所述多个入口微通道的入口微通道和所述多个出口微通道的出口微通道与所述多个纳米通道的共同纳米通道直接流体连通。在特定的实施方案中,各个入口和出口微通道布置为垂直于所述纳米通道递送装置的主平面;所述多个入口微通道形成第一阵列;所述多个出口微通道形成第二阵列;和所述第一阵列和所述第二阵列重叠,使得在沿垂直于所述主平面所取得的截面观察时各个入口微通道分布于各个出口微通道之间。一些实施方案包括纳米通道递送装置,包含:基本平坦的本体,所述本体包括长度、宽度和厚度,其中长度和宽度都大于厚度;在所述基本平坦的本体的第一侧上的入口表面,其中入口表面受到所述基本平坦的本体的长度和宽度的限制;在所述基本平坦的本体的第二侧上的出口表面,其中所述出口表面受到所述基本平坦的本体的长度和宽度的限制,并且其中入口表面基本上平行于所述出口表面;第一电极;第二电极;置于基本平坦的本体中的纳米通道,其中所述纳米通道包括入口端和出口端;与所述纳米通道流体连通的入口微通道;与所述纳米通道流体连通的出口微通道,其中所述入口微通道和纳米通道经配置使得第一线性轴可以在所述纳米通道的入口表面和入口端之间延伸,和其中所述第一电极与所述纳米通道递送装置的第一表面直接结合。在一些实施方案中,所述第二电极与所述纳米通道递送装置的第二表面直接结合。在特定的实施方案中,所述第一电极包括在所述第一表面上沉积的导电材料,和所述第二电极包括在所述第二表面上沉积的导电材料。在一些实施方案中,所述第二表面是所述入口微通道、所述出口微通道或所述纳米通道的表面。在特别的实施方案中,所述第一表面是所述入口微通道、所述出口微通道或所述纳米通道的表面。在一些实施方案中,所述纳米通道递送装置经配置以通过施加电压至第一电极和第二电极之间来控制通过所述纳米通道的分子的扩散速率。在特定的实施方案中,所述入口微通道和纳米通道经配置使得第二线性轴可以在所述纳米通道的出口表面和出口端之间延伸。在一些实施方案中,所述入口微通道的主轴垂直于与所述基本平坦的本体平行的平面。特别的实施方案还包括入口表面和入口微通道之间的入口大通道,其中入口大通道包含整体垂直于入口表面的边界壁。在一些实施方案中,入口大通道是由深度反应性离子蚀刻形成的。在特定的实施方案中,出口微通道的主轴垂直于与所述基本平坦的本体平行的平面。一些实施方案包括设备,所述设备包括:插入到封壳中的如本文中公开的第一纳米通道递送装置。在特别的实施方案中,所述第一纳米通道递送装置垂直于所述封壳的主轴安装。在一些实施方案中,封壳形状是盘形或卵形,具有小于封壳的任何直径测量的厚度。在特定的实施方案中,所述第一纳米通道递送装置安装于所述封壳的所述主平面中。在一些实施方案中,所述封壳包含隔件。在一些实施方案中,所述隔件包含自密封物质。在特别的实施方案中,所述隔件包含硅橡胶。在一些实施方案中,配置所述隔件以接收治疗剂的注射。特定的实施方案还包括覆盖所述隔件的盖体。在一些实施方案中,所述盖体包含经配置以引导针朝向隔件的孔口。在特别的实施方案中,所述封壳包含在第一纳米通道装置上方延伸的覆盖物。在一些实施方案中,所述覆盖物包含一个或多个孔口。在特定的实施方案中,所述一个或多个孔口的尺寸使得在使用期间它们不限制治疗剂从所述封壳中扩散。在一些实施方案中,配置所述覆盖物以保护所述第一纳米通道递送装置免受机械损害。在特别的实施方案中,配置所述覆盖物以保护所述第一纳米通道递送装置免于在将所述封壳植入到活体内后生物组织结构的侵入。在一些实施方案中,所述封壳包含第一内贮器。在特定的实施方案中,所述第一纳米通道递送装置与所述第一内贮器流体连通。在一些实施方案中,所述封壳包含与第二纳米通道递送装置流体连通的第二内贮器。在特别的实施方案中,所述第一和第二内贮器互相不流体连通。在一些实施方案中,所述第一和第二内贮器通过壁隔离。在特定的实施方案中,所述第一内贮器包含第一治疗剂,所述第二内贮器包含第二治疗剂。在一些实施方案中,配置所述第一纳米通道递送,以使第一治疗剂以第一扩散速率扩散,并且其中配置所述第二纳米通道递送装置以使第二治疗剂以第二扩散速率扩散。在特别的实施方案中,可以通过用较大的可移去组件替换封壳的第一可移去组件来改变第一内贮器的容积。在一些实施方案中,所述第一内贮器包含与治疗物质相容的涂层。在特定的实施方案中,所述封壳包含外涂层,其经配置以预防有害的组织包裹。在一些实施方案中,所述封壳包含柱形形状。在特别的实施方案中,所述封壳包含盘形形状。在一些实施方案中,所述封壳包含矩形表面和弓形表面。在特定的实施方案中,所述封壳包含均匀的横截面。在一些实施方案中,所述封壳包括一种或多种下列材料:不锈钢、钛、聚醚醚酮、聚砜、环氧树脂、硅橡胶、聚醚酮酮和热塑性聚氨酯。在特别的实施方案中,所述封壳包含锚定零件。在一些实施方案中,配置所述锚定部件以接受缝合。在特定的实施方案中,所述封壳包含色码,以指示封壳或纳米通道递送装置的特性。在一些实施方案中,所述色码指示包含在封壳内的治疗剂的特性。在特别的实施方案中,所述封壳包含在第一纳米通道递送装置上方延伸的半透明或透明的覆盖物。一些实施方案包括制作纳米通道递送装置的方法,其中所述方法包括:提供第一衬底;在所述第一衬底中形成多个纳米通道;在所述第一衬底的纳米通道中形成多个入口微通道;提供与入口微通道直接结合的第一电极;提供第二衬底;在所述第二衬底中形成多个出口微通道;提供与出口微通道直接结合的第二电极;和将所述第二衬底与所述第一衬底结合,其中各入口微通道与纳米通道直接流体连通。在特别的实施方案中,所述第一衬底包含绝缘体上硅晶片。在一些实施方案中,各纳米通道的高度是约2-250纳米。在特定的实施方案中,各纳米通道的高度是约100和500纳米。在一些实施方案中,所述第二衬底包含在硅上的氧化铟锡膜的牺牲释放层。特别的实施方案还包括在所述第二衬底中形成多个入口微通道前,在所述第二衬底上沉积玻璃膜。在一些实施方案中,所述第二衬底包含玻璃晶片;和所述玻璃晶片与第一衬底接合,且在形成多个出口微通道前将所述玻璃晶片研磨以减小厚度。特定的实施方案包括一种制作纳米通道递送装置的方法,其中所述方法包括:提供第一衬底;将第一电极与所述第一衬底直接结合;在所述第一衬底中形成多个纳米通道;将第一牺牲材料填充到多个入口微通道中;在所述第一衬底上形成多个纳米通道;将第二牺牲材料填充到多个纳米通道中形成覆盖多个纳米通道的盖层;在所述盖层中形成多个出口微通道;将第二电极与所述盖层直接结合;从多个纳米通道中去除第二牺牲材料;和从所述多个入口微通道中去除第一牺牲材料。在一些实施方案中,将入口微通道布置成垂直于所述第一衬底的主平面。在特别的实施方案中,将出口微通道布置成垂直于所述第一衬底的主平面。在一些实施方案中,入口微通道与纳米通道直接流体连通。在特定的实施方案中,出口微通道与纳米通道直接流体连通。在一些实施方案中,所述第一衬底包含绝缘体上硅晶片,其包含内氧化物层。在特别的实施方案中,用光刻工艺将入口和出口微通道图案化。在一些实施方案中,形成多个入口微通道包括从所述第一衬底蚀刻材料,其中所述蚀刻结束于内氧化物层处。在特定的实施方案中,形成多个入口大通道包括从所述第一衬底背侧蚀刻材料,和其中所述蚀刻结束于内氧化物层处。在一些实施方案中,在蚀刻材料以形成入口微通道和入口大通道后去除所述内氧化物层打开了入口微通道和入口大通道之间的路径。在一些实施方案中,每个纳米通道为约1-10纳米深。在特定的实施方案中,每个纳米通道为约10-20纳米深。在一些实施方案中,每个纳米通道为约20-30纳米深。在特别的实施方案中,每个纳米通道为约30-40纳米深。在一些实施方案中,每个纳米通道为约40-250纳米深。在特定的实施方案中,可以随后通过选择性蚀刻来去除第一牺牲材料。在一些实施方案中,所述第一牺牲材料选自:钨、铜、掺杂的玻璃和未掺杂的玻璃。在特定的实施方案中,将所述第一牺牲材料填充到多个入口微通道中,使得所述第二牺牲材料延伸高于入口微通道的顶部,并通过化学-机械平面化技术(cmp)得到平面化。在一些实施方案中,所述第二牺牲材料是钨、氮化钛、铝、钛钨或镍。在特别的实施方案中,可以随后通过选择性蚀刻来去除所述第二牺牲材料。在一些实施方案中,所述盖层选自氮化硅、氧化硅、碳氮化硅、碳化硅和硅。在一些实施方案中,所述盖层包含含有拉伸应力和压缩应力的材料的多重沉积物,使得盖层净应力为拉伸性的。在特定的实施方案中,所述盖层为约0.5-1.0微米厚。在一些实施方案中,所述盖层为约1.0-2.0微米厚。在特别的实施方案中,所述盖层为约2.0-4.0微米厚。在一些实施方案中,所述盖层为约4.0-10.0微米厚。在特别的实施方案中,所述盖层大于10.0微米厚。特定的实施方案包括制作纳米通道递送装置的方法,其中所述方法包括:提供第一衬底;在所述第一衬底的第一侧上形成多个纳米通道;将牺牲材料填充到多个纳米通道中;将初始盖层与所述第一衬底的第一侧结合;在所述盖层中形成多个入口微通道;制备具有接合层的第二衬底;将第一电极与所述第一衬底直接结合;将第二电极与所述第二衬底直接结合;将所述第二衬底与所述第一衬底的第二侧结合;去除所述第二衬底的第一部分;给所述第二衬底提供附加的盖层;在所述第二衬底中形成多个出口微通道;和去除所述牺牲材料以打开多个纳米通道。在一些实施方案中,所述第二衬底包含释放层,其中可以选择性地去除所述释放层以使第二衬底从第一衬底分离。在特别的实施方案中,出口微通道与所述纳米通道直接流体连通。在一些实施方案中,第一衬底包括绝缘体上硅晶片,其包括内层,该内层可以是例如介电层或金属层。在特定的实施方案中,形成多个入口微通道包括从所述盖层蚀刻材料,其中所述蚀刻结束于内层处。在一些实施方案中,形成多个入口大通道,包括从所述第一衬底背侧蚀刻材料,所述蚀刻结束于内层处。在特别的实施方案中,在蚀刻材料以形成入口微通道和入口大通道后去除所述内层打开了入口微通道和入口大通道之间的路径。在一些实施方案中,各纳米通道为约1-10纳米深。在特定的实施方案中,各纳米通道为约10-20纳米深。在一些实施方案中,各纳米通道为约20-30纳米深。在特别的实施方案中,各纳米通道为约30-40纳米深。在一些实施方案中,各纳米通道为约40-200纳米深。在特定的实施方案中,可以随后通过选择性蚀刻来去除所述牺牲材料。在一些实施方案中,所述牺牲材料是钨、氮化钛、铝、钛钨或镍。在特别的实施方案中,所述初始盖层是通过等离子体增强的化学气相沉积法沉积的氮化硅。在一些实施方案中,所述初始盖层为约0.01-0.5微米厚。在特定的实施方案中,所述初始盖层为约0.5-1.0微米厚。在一些实施方案中,所述初始盖层为约1.0-2.0微米厚。在特别的实施方案中,所述初始盖层为约2.0-4.0微米厚。在一些实施方案中,所述初始盖层为约4.0-10.0微米厚。在特定的实施方案中,所述初始盖层大于10.0微米厚。在特别的实施方案中,所述初始盖层选自氮化硅、氧化硅、碳氮化硅、碳化硅和硅。在一些实施方案中,所述初始盖层包含含有拉伸应力和压缩应力的材料的多重沉积物,使得盖层净应力为拉伸性的。在特定的实施方案中,所述接合层选自:苯并环丁烯、氧化硅、铜、掺杂的玻璃、金及金的合金。在一些实施方案中,将所述第二衬底与所述第一衬底结合的方法选自:阳极接合、融合接合和热压接合。特别的实施方案包括纳米通道递送装置,其包括:多个入口微通道,其中各入口微通道具有长度、宽度和深度,且其中入口微通道的长度大于入口微通道的宽度和深度;第一电极;第二电极;多个出口微通道,其中各出口微通道具有长度、宽度和深度;与多个入口微通道和出口微通道流体连通的多个纳米通道,其中多个入口微通道经配置使得入口微通道的宽度和深度限定出平行于所述纳米通道递送装置的主平面的第一平面;和其中多个出口微通道经配置使得所述出口微通道的宽度和深度限定出平行于所述纳米通道递送装置的主平面的第二表面,其中所述第一电极与所述纳米通道递送装置的第一表面直接结合。在一些实施方案中,所述第二电极与所述纳米通道递送装置的第二表面直接结合。在特定的实施方案中,所述第一电极包括在所述第一表面上沉积的导电材料,和所述第二电极包括在所述第二表面上沉积的导电材料。在一些实施方案中,所述第二表面是所述入口微通道、所述出口微通道或所述纳米通道的表面。在特别的实施方案中,所述第一表面是所述入口微通道、所述出口微通道或所述纳米通道的表面。在一些实施方案中,纳米通道递送装置经配置以通过施加电压至第一电极和第二电极来控制通过所述纳米通道的分子的扩散速率。一些实施方案包括纳米通道递送装置,其包括:入口通道;出口通道;和与所述入口通道和所述出口通道流体连通的纳米通道,其中所述纳米通道包括第一表面和第二表面,其中所述第一表面与所述第二表面基本上平行;和其中所述第一表面和所述第二表面是导电的。在特别的实施方案中,纳米通道递送装置经配置以通过施加电压至第一表面和第二表面来控制通过所述纳米通道的分子的扩散速率。在一些实施方案中,所述第一表面与所述第二表面电结合。在特定的实施方案中,所述纳米通道的所述第一表面和第二表面由小于500nm的距离间隔。在一些实施方案中,所述纳米通道的所述第一表面和第二表面由小于100nm的距离间隔。在特别的实施方案中,所述纳米通道的所述第一表面和第二表面由小于50nm的距离间隔。在一些实施方案中,所述纳米通道的所述第一表面和第二表面形成第一电极;所述纳米通道递送装置包括第二电极;和所述入口通道和所述出口通道中的至少一个在所述纳米通道和所述第二电极之间。在一些实施方案中,所述纳米通道的第一表面和第二表面经由第三表面电结合,所述第三表面基本上垂直于所述纳米通道的所述第一和第二表面而延伸。在特别的实施方案中,在操作过程中,可将电压施加至所述第一和第二表面,且可通过改变电压来控制流体移动经过所述纳米通道。在一些实施方案中,所述纳米通道与所述第一微通道和第二微通道直接流体连通。一些实施方案包括纳米通道递送,包括:第一外表面和第二外表面;第一电极和第二电极;纳米通道;与所述纳米通道流体连通的第一微通道;和与所述纳米通道流体连通的第二微通道,其中所述第一微通道从所述纳米通道延伸至所述第一外表面;其中所述第二微通道从所述纳米通道延伸至所述第二外表面;和其中所述第一和第二电极从所述纳米通道延伸至所述第一外表面。在特别的实施方案中,所述第一电极与所述纳米通道的第一表面直接结合。在一些实施方案中,所述第二电极与所述纳米通道的第二表面直接结合。在特定的实施方案中,所述第一电极与和所述第一微通道接近的所述纳米通道的第一端直接结合。在一些实施方案中,所述第二电极与和所述第二微通道接近的所述纳米通道的第二端直接结合。在特别的实施方案中,纳米通道递送装置经配置以通过施加电压至第一电极和第二电极来控制通过所述纳米通道的分子的扩散速率。在一些实施方案中,所述纳米通道递送装置包括基本平坦的本体和其中所述第一外表面和所述第二外表面是基本上平行的。在特别的实施方案中,所述纳米通道与所述第一和第二微通道直接流体连通。附图简述图1示出了制造不具有电极的纳米通道装置的示例性方法。图2-7示出了在纳米通道装置中提供电极的示例性方法。图8示出了具有电极的纳米通道装置的图像和示意图。图9示出了装置和微制作工艺流程的示例性实施方案的结构。图10示出了纳米通道装置、用该装置测试的df-1分子和在测试过程中所用的控制系统的示意图。图11示出了在纳米通道装置的测试过程中df-1的累积释放的图。图12示出了纳米通道装置的被动和主动释放模式的图。图13示出了纳米通道装置测试设备的一个实施方案的示意图。图14示出了通过纳米通道装置的fitc-bsa的累积释放。图15示出了正向偏置和反向偏置下fitc-bsa的电动力学输送的荧光图像,表明了电动力学流动的可逆性。图16示出了用于模拟纳米通道流体系统的电学性质的电阻网络。图17示出了电渗和电泳之间的平衡的示意图。图18示出了纳米通道膜的模型。图19示出了不同的纳米通道布置的模型。图20示出了不同的电极布置的示意图。图21示出了测试的特定纳米通道装置构造的实验结果的比较。图22示出了不同的构造和纳米通道深度之间的平均时间常数的比较。图23示出了纳米通道装置测试装置。图24示出了用于图23的测试装置中的纳米通道装置电极上的接合点。图25示出了用于图23的纳米通道装置测试装置的组件。图26示出了电渗输送对截留于纳米通道装置的微通道内部的空气泡的运动的影响。图27示出了纳米通道装置中系列和非系列稀释的荧光相对于浓度的内推拟合曲线之间的比较。图28-33示出了显示随时间推移从纳米通道装置中释放的bsa-fitc的图。图34示出了显示随时间推移从纳米通道装置中释放的葡萄糖的图。图35-36示出了施加至纳米通道装置的dc电压随时间推移的电流曲线。图37示出了用于测试纳米通道装置的电路的示意图。图38-39示出了从图37的电路生成的电流相对于电压的图。图40示出了从图37的电路生成的随时间推移的电流的图。图41和42示出了经配置以测试纳米通道装置的flowfet装置的示意图。图43-45示出了经配置以与纳米通道装置一起使用的药物递送植入物。图46示出了涂覆电极的纳米通道装置和内部结构的示意图。图47示出了用于测试的原型纳米通道装置的示意图。图48-50示出了在图47的装置的测试过程中qdots、bsa和df1的主动释放的实验结果。说明性实施方案的详细描述纳米装置设计和制作本文所描述的整体微制作的纳米通道膜(也称作“nds”)的一些示例性实施方案由微机加工硅晶片和pyrex盖壳体电极组成。示例性的实施方案包括美国专利公开2007/0066138(“diffusiondeliverysystemsandmethodsoffabrication”)和美国专利公开2010/0152699(“nanochanneleddeviceandmethodoffabrication”)中描述的特征和制作技术,通过引用将其并入本文。除了前述设计和制作技术以外,还可根据美国专利公开2007/0066138(“diffusiondeliverysystemsandmethodsoffabrication”)和美国专利公开2010/0152699(“nanochanneleddeviceandmethodoffabrication”)中公开的方法和设备来设计和制作ndd装置,所述专利通过引用并入本文。可利用“基准”nds装置使用不同的方法来将导电性(例如,金属)涂层用作电极。基准nds装置的示例性实施方案包括平面本体(具有第一和第二相对表面),其包括多个纳米通道线。沿所述纳米通道线为交替的入口微通道和出口微通道结构。入口微通道延伸至装置的本体中以将纳米通道与第一相对表面结合,任选地与有较大的大通道相交。出口微通道结合具有纳米通道的第二相对表面。可将多个纳米通道线与各个大通道结合,且所述装置可包含多个大通道。此外,可通过以下描述的方法在较大的绝缘体上硅(soi)硅晶片上同时制作多个nds装置。通过切开该较大晶片可获得单独的nds装置。现参见图1,显示了nds装置100的横截面,其具有位于平面本体中的纳米通道线,所述平面本体具有相对的表面108和109。在该实施方案中,包括与微通道和大通道结合的纳米通道线,以下描述了制造不具有电极的基准nds构造的一种示例性方法。在该实施方案中,在将入口微通道113图案化和进行蚀刻之前,可将底层和硬掩模层(未示出)沉积在soi晶片上(101,103,105)。在清洁步骤后,可沉积衬层(未示出),然后可沉积插头(plug)并进行化学-机械抛光。在另一清洁步骤之后,可沉积将形成纳米通道115的层。然后可沉积覆盖层(未示出)并将纳米通道115图案化和进行蚀刻。在另一清洁步骤之后,可沉积介电层或介电层堆叠体107,将出口图案化,并蚀刻第一出口微通道117。在另一清洁步骤之后,可沉积一个或多个保护性衬层并蚀刻第二出口微通道119。清洁之后,可沉积正面保护层(未示出),且将设备反转(从图1中所示的位置)。然后可沉积硬掩模层,并将大通道111图案化和进行蚀刻以在埋入的氧化物层103上停止。然后将埋入的氧化物层103与残余硬掩模一起蚀刻。清洁晶片以去除入口和衬牺牲层。可沉积保护性衬层(未显示)。可替代地,可在晶片水平上去除其他牺牲材料,且然后可将晶片分为单独的纳米通道装置。在另一方法中,可形成不具有保护层的出口117、入口113、纳米通道115和大通道111,且可在去除全部牺牲材料后施加保护层。现参见图2,在另一个实施方案中,可纳入另外的步骤以在纳米通道装置中提供电极。例如,可在将入口113图案化和进行蚀刻之前将底层和硬掩模层(未示出)沉积在晶片上。在清洁步骤之后,可沉积衬层(未示出)且然后可沉积插头并进行化学-机械抛光。在另一清洁步骤之后,可沉积将形成纳米通道115的层。然后可沉积覆盖层(未示出)并将纳米通道115图案化和进行蚀刻。在另一清洁步骤之后,可沉积介电层107。然而该实施方案不同于以上图1的讨论中描述的实施方案。在该实施方案中,在沉积了介电层107之后,还沉积了传导层121(其可用作电极)。如本文所用的,术语“电极”包括任何导电材料,包括例如金属。在所示的实施方案中,传导层121直接结合到纳米通道装置的表面。在示例性的实施方案中,可将金属层(例如,电极)与纳米通道装置的表面直接结合,所述纳米通道装置的表面包括例如所述装置的外表面的表面、入口或出口微通道表面的表面或纳米通道表面的表面。在沉积介电层107和传导层121之后,可将出口图案化并蚀刻第一出口117。接下来的几个步骤与图1中所显示和描述的实施方案中所用的步骤相同。例如,在另一清洁步骤之后,可沉积保护性衬层并蚀刻第二出口119。在清洁之后,可沉积正面保护层(未示出),且将设备反转(从图2中所示的位置)。然后可沉积硬掩模层并将大通道111图案化和进行蚀刻以在埋入的氧化物层103上停止。然后蚀刻该埋入的氧化物层103,清洁晶片并沉积衬层(未示出)。不同于先前的实施方案,然后可沉积非保形传导层123以作为第二电极。在一些实施方案中,可将另外的任选的层122和124分别施加至层121和123,以保护层121和123和/或将其与外界环境隔开。理解到本文所描述的其他实施方案可包括与层122和124相似的层以保护传导层和/或将其与外界环境隔开。再次地,可将晶片分为单独的纳米通道装置并去除牺牲层。现参见图3,在又一个实施方案中,可纳入另外的步骤以提供具有与图3所示的不同构造的电极。在该实施方案中,沉积另外的材料以提供电极,所述电极与先前描述的实施方案相比较距纳米通道115甚至更近。第一几个步骤整体上与图2中描述的那些相同。例如,可在将入口113图案化和进行蚀刻之前在晶片上沉积底层和硬掩模层(未示出)。在清洁步骤之后,可沉积衬层(未示出),且然后可沉积插头并进行化学-机械抛光。在另一清洁步骤之后,可沉积将形成纳米通道115的层。然后可沉积覆盖层(未示出)并将纳米通道115图案化和进行蚀刻。在另一清洁步骤之后,可沉积介电层107。在沉积介电层107之后,还沉积了传导层121(其可作为电极的部分)。在沉积介电层107和传导层121之后,可将出口图案化并蚀刻第一出口117。在清洁步骤之后,可沉积金属衬层131,其延伸到第一出口117中。金属衬层131以这样的方式和位置沉积:使得其与先前沉积的金属衬层121电结合。因此,金属衬层121和131可用作连续的单元并作为单一电极发挥作用。为了保护该电极侧壁层不受进一步的加工所引起的损坏,可将牺牲层沉积到该传导层131的顶部,随后可将其选择性去除。接下来的几个步骤与图2中所显示和描述的实施方案中所用的那些相同。例如,然后可蚀刻第二出口119,并且在清洁之后,可沉积正面保护层(未示出)。然后可将设备从图3中所示的位置反转。然后可沉积硬掩模层,并将大通道111图案化和进行蚀刻以在埋入的氧化物层103上停止。然后可将该埋入的氧化物层103与残余硬掩模一起蚀刻。清洁晶片以去除入口和衬牺牲层。沉积保护衬层(未示出)。然后可沉积保形金属层133以用作第二电极。与图2中所沉积的非保形金属层不同,保形金属层133延伸到大通道111中。这再次将第二电极置于与纳米通道115非常邻近。在一些特定的实施方案中,金属层131和133在纳米通道115的约30微米之内。在特别的实施方案中,金属层131和133在纳米通道115的10微米、15微米、20微米、21微米、22微米、23微米、24微米、25微米、26微米、27微米、28微米、29微米、30微米、31微米、32微米、33微米、34微米、35微米、36微米、37微米、38微米、39微米、40微米、45微米或50微米之内。在沉积金属层131和133之后,可随后将晶片分成单独的纳米通道装置并去除牺牲层。或者,可在晶片水平下去除其他牺牲材料,然后将晶片分成单独的纳米通道装置。现参见图4,在又一个实施方案中,可纳入另外的步骤以提供具有与图2和图3中所示不同构造的电极。在该实施方案中,沉积另外的材料以提供比先前描述的实施方案与纳米通道115甚至更接近的电极。在起始步骤中,可在将入口113图案化和进行蚀刻之前将底层和硬掩模层(未示出)沉积在晶片上。在清洁步骤之后,可将金属衬层135沉积在入口113上。此外,可沉积随后可易于在工艺中去除的任何牺牲衬层以保护电极材料(如果需要)。其后是可沉积和进行化学-机械抛光的插头。其余加工步骤等同于以上图3的描述中提供的那些。例如,在另一清洁步骤之后,可沉积将形成纳米通道115的层。然后可沉积覆盖层(未示出)并将纳米通道115图案化并进行蚀刻。在另一清洁步骤之后,可沉积介电层107。在沉积了一个或多个介电层107之后,还沉积了传导层121(其可用作电极的部分)。在沉积介电层107和传导层121之后,可将出口图案化并蚀刻第一出口117。在清洁步骤之后,可沉积金属衬层131,其延伸至第一出口117中。金属衬层131以这样的方式和位置沉积:使得其与先前沉积的金属衬层121电结合。因此,传导衬层121和131可用作连续的单元并作为单一电极发挥作用。为了保护该电极侧壁层不受进一步的工艺所引起的损害,可在该传导层131的顶部沉积牺牲层,随后可选择性地将其去除。然后可蚀刻第二出口119,并且在清洁之后,可沉积正面保护层(未示出)。然后可将该设备从图4中所示的位置反转。然后可沉积硬掩模层,并且将大通道111图案化和进行蚀刻以在埋入的氧化物层103上停止。然后将该埋入的氧化物层103与残余硬掩模一起蚀刻并去除入口牺牲材料。然后可沉积保形金属层133以用作第二电极的部分。保形金属层133延伸到大通道111中,并与金属衬层135电结合。因此,金属衬层133和135可用作连续的单元并作为单一电极发挥作用。图4中所示的构造将第二电极非常邻近(例如,邻接)纳米通道115。在一些特定的实施方案中,金属层131和135在纳米通道115的约15微米内。在特别的实施方案中,传导层131在纳米通道115的0.0、0.1、0.2、0.3、0.4、0.5、0.6、0.7、0.8、0.9、1、2、3、4、5、6、7、8、9或10微米内,且传导层135在纳米通道115的0.0、0.1、0.2、0.3、0.4、0.5、0.6、0.7、0.8、0.9、1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30、31、32、33、34、35、36、37、38、39、40、45或50微米内。在一些实施方案中,金属层135完全延伸至入口113中使得其直接邻接纳米通道115(例如,在纳米通道115的1微米内)。在沉积金属层131和133之后,可将晶片分为单独的纳米通道装置并去除牺牲层。可替代地,首先在晶片水平上去除牺牲材料,然后将晶片分为单独的装置。现参见图5,另一个实施方案包括允许在操作过程中用可用作电极的材料涂覆纳米通道115的顶表面和底表面的加工步骤。在所示的实施方案中,在沉积硬掩模层(未示出)之前可起初将金属底层137沉积在晶片上。然后可将金属入口113图案化和进行蚀刻,随后进行清洁步骤。可将金属衬层135沉积在入口113上,然后可沉积插头并进行化学-机械抛光。在示例性的实施方案中,可以以这样的方式和位置沉积金属衬层135使得其与金属底层137电结合。在另一清洁步骤之后,可沉积将形成纳米通道115的层。然后可沉积覆盖层139并将纳米通道115图案化和进行蚀刻。其余的加工步骤大体上等同于以上图4的描述中所提供的那些。例如,在另一清洁步骤之后,可沉积介电层107。在沉积了介电层107之后,还沉积了传导层121(其可用作电极的部分)。在沉积介电层107和传导层121之后,可将出口图案化并蚀刻第一出口117。在清洁步骤之后,可沉积传导衬层131,其延伸至第一出口117中。传导衬层131以这样的方式和位置沉积:使得其与先前沉积的传导层121和传导底层139电结合。因此,传导层121、131和139可用作连续的单元并作为单一电极发挥作用。然后可沉积任何另外的牺牲层以保护电极侧壁。然后可蚀刻第二出口119,并且在清洁之后可沉积正面的保护层(未示出)。然后可将设备从图5中所示的位置反转。然后可沉积硬掩模层,并且将大通道111图案化和进行蚀刻以在埋入的氧化物层103上停止。然后将该埋入的氧化物层103与残余硬掩模一起蚀刻并去除入口牺牲材料。然后可沉积保形金属层133以用作第二电极的部分。保形金属层133延伸到大通道111中,并与金属衬层135电结合,金属衬层135又与金属底层137电结合。因此,金属衬层133、135和137可用作连续的单元并作为单一电极发挥作用。在沉积传导层131之后,可将晶片分为单独的纳米通道装置,并去除牺牲层。可替代地,首先在晶片水平上去除牺牲材料,然后将晶片分为单独的装置。图5中所示的构造将第一和第二电极的金属层137和139置于纳米通道115的相对表面上。在一些特定的实施方案中,金属层137和139在彼此约250纳米(nm)内。在特别的实施方案中,金属层137和139在彼此500、400、300、200、100、90、80、70、60、50、40、30、20或1、2、3、4、5、6、7、8、9或10nm内。直接在纳米通道115的表面上的金属层137和139的邻近和位置在具有电极的纳米通道递送系统(ndse)的操作过程中提供了大量的优点。例如,可利用比采用将电极置于更远离纳米通道的构造会需要的更小的电压影响或控制通过纳米通道115的分子的移动。图5中显示和描述的构造还可在操作过程中提供分子通过纳米通道115的移动的更精确的控制。例如,在操作过程中,使用者可将正电压和负电压施加到纳米通道115中彼此相对的整个表面。这可将通过纳米通道115的分子暴露于来自电极的电能(相较于仅在入口和出口微通道上具有电极的构造,其具有比纳米通道大得多的横截面积)。通过入口和出口微通道的中间部分的分子将不会与非常邻近电极的分子一样受电能影响。通过将电极定位在纳米通道表面上(比位于微通道表面或大通道表面上的电极,其相互之间更靠近),通过纳米通道的每个分子将相对地紧密邻近电极。该构造将允许来自电极的电能影响分子移动至更高的程度,并将进而对分子通过纳米通道115提供更大的控制(例如,经过纳米通道115的分子的扩散速率)。现参见图6,另一个实施方案包括与图1中显示和描述的“基准”nds设计中所用的那些相似的加工步骤。在该实施方案中,可在晶片上将入口113图案化和进行蚀刻。在清洁步骤之后,可沉积衬层(未示出),然后可沉积插头并进行化学机械抛光。在另一清洁步骤之后,可沉积将形成纳米通道115的层。然后可沉积覆盖层(未示出),并将纳米通道115图案化和进行蚀刻。在另一清洁步骤之后,可沉积介电层107,将出口图案化,和蚀刻第一出口117和第二出口119。在清洁之后,可沉积正面保护层(未示出),且将设备反转(从图6中所示的位置)。然后可沉积硬掩模层,并将大通道111图案化和进行蚀刻至在埋入的氧化物层103上停止。然后可将该嵌入的氧化物层103与残余硬掩模一起蚀刻并去除入口牺牲材料。然后可将保形金属层133沉积在入口113的一侧上的暴露表面上,如图6中所示。然后可将晶片分为单独的纳米通道装置,并去除牺牲层。在将晶片分为单独的纳米通道装置之后,可沉积保形金属层141至纳米通道115和出口117、119上暴露的表面。可替代地,首先在晶片水平上去除牺牲材料。然后将晶片分为单独的装置,且可将保形金属层141沉积至纳米通道115和出口117、119上暴露的表面。与图5中所示的先前的实施方案相似地,该实施方案包括纳米通道115的任一侧上的金属层。然而,与先前的实施方案不同地,纳米通道115的任一侧上的金属层141是电结合的,且因此将具有相同的电极性(例如,如相对于地测量的正电压或负电压)。可与控制阀相似地有效使用纳米通道115,在纳米通道115的每侧上具有电结合的金属层141。例如,将相同类型的电荷施加到金属层141可排斥纳米通道115中的流体分子远离纳米通道的相对表面上的金属层141。这可有效地使通过纳米通道115的路径变窄,所述纳米通道能够使分子通过且限制了流体分子的运动。现参见图7,该实施方案包括与先前描述的那些显著不同的方案。该实施方案还提供了与先前实施方案相比的几个结构差别。例如,在提供在纳米通道的相对侧上的表面上具有相反极性的电极的同时,其还允许从纳米通道的同“侧”对电极进行外部电连接(如以下更详细地解释的)。图7中的结构可通过多种方法来实现,并且提供了三个实例。通过线和接触通孔来连接将外部与纳米通道表面上的电极连接的接合垫。在一个实施方案中,可初始将介电层151沉积在晶片上。然后可金属底层图案化和进行蚀刻。在清洁步骤之后,可填充金属底层,然后进行金属底层的化学-机械抛光/平面化(cmp)。接着,可清洁晶片以完成金属底层152。可将入口113图案化和进行蚀刻,然后清洁入口113并给其加衬。然后可沉积插头(未示出)并对其进行cmp工艺。可沉积介电层153,然后将纳米通道115的空间图案化和进行蚀刻。在清洁步骤之后,可沉积纳米通道层154(例如,在与纳米通道115相同的平面上的层)并对其进行cmp工艺。然后可沉积介电层155并将覆盖层空间图案化和进行蚀刻。在清洁步骤之后,可沉积覆盖金属层156,对其进行cmp工艺,然后清洁。可替代地,可沉积覆盖金属层、将其图案化、进行蚀刻并清洁。然后可沉积介电层107,之后沉积介电覆盖层157。可将线159和接合垫160图案化和进行蚀刻,然后进行清洁步骤。然后可将接触通孔161图案化并进行蚀刻至金属覆盖层156或金属底层152。在另一清洁步骤之后,可将互连金属沉积在线159、接合垫160和通孔161中。然后可沉积钝化层162,将接合垫163图案化并进行蚀刻直至线159。在另一清洁步骤之后,可在钝化层162中将出口图案化。然后可在钝化层162、介电覆盖层157、介电层107和金属覆盖层156中蚀刻出口117,止于纳米通道层154处。在另一清洁步骤之后,可沉积保护层,并将设备反转(从图7中所示的位置)。然后可沉积硬掩模层,并将大通道111图案化和进行蚀刻至在埋入的氧化物层103上停止。然后蚀刻该埋入的氧化物层103,清洁晶片并沉积衬层(未示出)。然后可将晶片分成单独的纳米通道装置并去除牺牲层。在第二实施方案中,可初始将介电层151沉积在晶片上。然后将电介质中待稍后用底层电极填充的沟槽图案化和进行蚀刻。在清洁步骤之后,沉积金属底层以填充沟槽,然后进行过载金属的化学-机械抛光/平面化(cmp)至止于介电层151。可清洁晶片以完成底层金属电极152。可在层152中将入口113图案化和进行蚀刻,并进入到硅中,止于埋入的氧化物,然后清洁入口113并给其加衬。然后可沉积插头(未示出)并对其进行cmp工艺。可沉积牺牲纳米通道层154,且可将任选的牺牲介电覆盖层和纳米通道线图案化和进行蚀刻,并去除牺牲材料。然后可沉积介电层155,然后将电介质中待稍后用覆盖层电极填充的沟槽图案化和进行蚀刻。在清洁步骤之后,可沉积覆盖金属层156以填充沟槽,然后进行过载金属的cmp工艺至止于电介质155,然后清洁以完成覆盖层金属电极156。然后可沉积厚的介电层或堆叠体107,之后沉积介电覆盖层157。可在介电层157中将线159和接合垫160图案化和进行蚀刻,之后进行清洁步骤。然后可将接触通孔161图案化并向下蚀刻至金属覆盖层156或金属底层152。在另一清洁步骤之后,可在线159、接合垫160中沉积互连的金属并填充通孔161。再次使用cmp工艺以去除多余的过载。任选地,随后可沉积钝化层162并将接合垫163图案化、进行蚀刻和清洁。这导致了接合垫和金属底层或覆盖层之间通过线和接触通孔的电接触。在另一清洁步骤之后,可在钝化层162上将出口图案化。然后可在钝化层162、介电覆盖层157、介电层107中蚀刻出口117,并止于金属覆盖层156中。在清洁和施加保护衬层之后,进行第二蚀刻,止于纳米通道层154或金属底层152处。在另一清洁步骤之后,可沉积保护层,且将设备反转(从图7中所示的位置)。然后可沉积硬掩模层,并将大通道111图案化和进行蚀刻至停止于埋入的氧化物层103上。然后蚀刻该埋入的氧化物层103和残余硬掩模层,清洁晶片并沉积衬层(未示出)。然后可将晶片分成单独的纳米通道装置并可去除牺牲层,或者去除牺牲层然后分成单独的纳米通道装置。可替代的实施方案以沉积金属底层152开始,将其图案化,对其进行蚀刻和清洁。然后将介电层151施加在该表面上,并利用cmp工艺,去除金属层152的顶部上的电介质。可沉积牺牲纳米通道层154,且可将任选的牺牲介电覆盖层和纳米通道线图案化和进行蚀刻,并可去除牺牲覆盖层。然后沉积金属覆盖层并将其图案化、进行蚀刻和清洁以形成覆盖层156。然后视需要沉积和平面化厚的介电层107。然后可将接触通孔161图案化并向下蚀刻至金属覆盖层156或金属底层152。然后在接触所述通孔的表面上将金属线和接合垫图案化。这导致了接合垫和金属底层或覆盖层之间通过线和接触通孔的电接触。任选地,随后可沉积钝化层162并通过图案化、蚀刻和清洁打开接合垫163。可在钝化层162上将出口图案化。然后可在钝化层162、介电覆盖层157、介电层107中蚀刻出口117,并止于金属覆盖层156中。在清洁并施加保护衬层之后,进行第二蚀刻,止于纳米通道层154或金属底层152处。在另一清洁步骤之后,可沉积保护层,且将设备反转(从图7中所示的位置)。然后可沉积硬掩模层,并将大通道111图案化和进行蚀刻至停止于埋入的氧化物层103上。然后蚀刻该埋入的氧化物层103和残余硬掩模层,清洁晶片并沉积衬层(未示出)。然后可将晶片分成单独的纳米通道装置并去除牺牲层,或者去除牺牲层然后分成单独的纳米通道装置。其他实施方案的讨论利用如美国专利8,480,637中所描述的高精度硅微制作技术产生了本章节中所讨论的nds膜。在该工作中,使用了呈现5.7nm的nds膜。另外,使用了微通道膜,其通过从结构中去除sin层来获得。所采用的所有膜呈现微通道,其具有用于将电极集成到nds上的1×3μm2的横截面(图8)。为了电子束沉积,使用了离子辅助的电子束蒸发器(chaindustries,inc.,fremont,ca,usa)。利用电极堆叠体涂覆nds膜的两侧。使用了两种不同的堆叠体组合:i)sic-ti(厚度)和pt(厚度),沉积角度为10°;ii)sin-ta(厚度)和pt(厚度),沉积角度为90°。涂覆工序按如下进行:首先,从出口侧去除钨牺牲层并将膜在热皮伦尼亚(piranha)浴(h2so4+h2o2)中清洁。将nds膜的硅侧朝下置于4”硅载体晶片上,该硅载体晶片安装在由doublesided可复用接头(3mcompany,st.paul,mn,usa)制成的专用u形膜支架上,其用于在离子辅助的电子束蒸发器内部产生成角度的沉积。将带(dupont,wilmington,de,usa)切割成小条并将其置于膜的非常边缘的上方,然后固定到可复用接头的顶部以将nds膜保持在合适位置,并避免由于芯片的侧面上可能的金属沉积导致的电短路。以10°来沉积具有纳米通道的膜的出口侧从而避免堵塞纳米通道。极限角度对于出口微通道的低纵横比(深度比宽度=1.5比5)是必要的。以90°沉积仅具有微通道的芯片。在下面题目为“df-1扩散”的章节中描述了将nds膜组装到扩散装置中的细节。如下图9中所示,在示例性的实施方案中,硅晶片呈现了交叉指型几何形状,由通过垂直的纳米通道的阵列相互连接的平行微通道组成。在一些实施方案中,通过将pyrex盖晶片阳极接合到锚点网格来获得微通道和纳米通道的顶表面。在示例性的实施方案中,纳米通道的纳米尺寸是深度。因为施加到嵌入膜电极的电势差,药物分子能通过钻通pyrex的入口进入膜,移动通过一组136个微通道,然后进入到120个纳米通道的网孔中(两组,每个微通道上60个),并最终通过另一组136个微通道到达湿蚀刻穿过硅晶片的出口。图9显示了装置和微制作工艺流程的示例性实施方案的结构。在该实施方案中,首先在热皮伦尼亚(piranha)溶液(3:1h2so4(96体积%):h2o2(30体积%))中清洁双侧抛光(dsp)的硅衬底,在120℃下持续10分钟。然后在950℃下使50nm的氧化硅垫层热生长,然后利用低压化学气相沉积(lpcvd)沉积150nm厚的低应力氮化物掩蔽层。该掩蔽层用作氧扩散的阻挡,防止硅表面的氧化。在该示例性实施方案中,利用evg620对准器(evgroup,inc.)在氮化物氧化掩蔽层的顶部通过标准光刻法限定出纳米沟槽,稍后利用he+sf6等离子体反应性离子蚀刻(rie)去除该掩蔽层。然后可利用1:10hf:h2o湿蚀刻溶液从氮化物掩蔽层中的开口去除防止因先前的rie的过度蚀刻而生长的氧化物垫层(参见下图9a)。然后可使牺牲氧化物膜热生长至需要的厚度(f)穿过氮化物掩蔽层(参见下图9b)。利用关系h=0.46·f可确定纳米沟槽的深度(h),该关系代表了生长的氧化物和消耗的硅之间的比率。在该示例性的实施方案中,随后可在稀释的hf溶液中剥离垫氧化物、氮化物和牺牲氧化物层。然后可通过由lpcvd将500nm厚的低温氧化物(lto)掩蔽膜沉积到dsp硅衬底上来制作微沟槽。在该实施方案中,可将入口和出口下方和上方的室以及微沟槽光刻限定在lto表面上。在该实施方案中利用he+chf3+cf4等离子体rie可在限定的区域中蚀刻lto掩模。对于20或30μm的深度,可使用感应耦合等离子体(icp)深度硅蚀刻方法(oxfordplasmalab100)。然而,对于2μm深度特征,可在80℃下利用45wt%koh溶液进行湿蚀刻(图9c)。在该示例性实施方案中可随后在1:10稀释的hf溶液中剥离lto掩模。在一个示例性的实施方案中,可利用150nm厚的lpcvd氮化物掩蔽膜的沉积来启动穿过dsp硅衬底的出口的制作。然后可进行背面光刻法从而限定出口的区域。在该实施方案中利用he+sf6等离子体rie将氮化物掩模蚀刻在限定的区域中。可在80℃下在45wt%koh溶液中进行贯穿晶片蚀刻(参见以下图9d),然后在稀释的hf溶液中去除氮化物掩模(参见以下图9e)。在该实施方案中,可将pyrex7740晶片用于顶部衬底制作,因为其具有与硅的优良的接合相容性。首先,可对用于容纳金属电极的沟槽进行光刻限定并利用he+chf3+cf4等离子体rie将其蚀刻到pyrex衬底中至0.5μm的厚度(参见以下图9f)。然后可在皮伦尼亚溶液中剥离掉光刻胶。接着,可进行第二次光刻步骤以将金属电极的位置限定在沟槽中。在该实施方案中,可使用电子束(e-束)金属蒸发器以层状沉积钛(ti0.05μm)/铂(pt0.15μm)电极到衬底表面上。然后,光刻胶的顶部上多余的金属可被“揭掉”而将金属电极留在0.5μm沟槽中(参见以下图9g)。然后可通过在低于pyrex转变温度的200℃下进行pecvd工艺用1μm厚的氧化物覆盖衬底,以覆盖其中新沉积的电极。然后可进行化学机械抛光/平面化(cmp)步骤以去除回到原始的pyrex表面的多余沉积的氧化物并且仅留下覆盖电极的薄层(参见以下图9h)。接着,使流体区域中的金属电极的区域以及装置接触垫上方的区域再次暴露以确保与溶液和装置两者良好的电接触,且同时防止流体泄露。在该实施方案中,可对这些窗口进行光刻限定并利用he+chf3+cf4等离子体rie蚀刻至0.6μm的深度,留下小的氧化物插头以防止流体泄露(参见以下图9i)。可将该内部窗口设计为具有与入口和出口下方和上方的微流体室的25μm重叠。该区域中金属的暴露对于通过装置的电动力学流动的建立是重要的。然后可通过超声钻孔将该出口机加工到衬底中(例如,利用来自bullenultrasonics,inc.的技术)。在一些实施方案中,可利用在evg520晶片接合器上进行的阳极接合技术将pyrex晶片和dsp硅衬底最终接合在一起以盖住和形成微通道和纳米通道(参见以下图9j)。在特定的实施方案中,接合条件为350℃下400伏,持续10分钟。在特别的实施方案中,可随后将光刻胶旋涂到接合的晶片上以在膜切片过程中保护纳米通道和微通道免受碎片。在一些实施方案中,可最终从接合的晶片切出3×5.5mm的ndd装置并利用酸喷射工具(spt)在臭氧中进行清洁。df-1扩散df-1(mw=2827da)呈现了2.5nm的流体力学直径,且在ph7.4下具有-10.4e的高负电荷(图10的第1部分)。其为高度水溶性的且其在λ=320nm处呈现吸收峰。借助uv-vis分光光度法在所施加的ac电场的影响下进行其通过nds膜的扩散的研究。通过利用在“关”(无源)和“开”(有源)状态之间的33250a功能/任意波形功能发生器(agilenttechnologies,santaclara,ca,usa)控制的i052c2ro和i051c2ro高速舌簧式继电器(americanrelays,inc.,santafe,ca,usa)调节施加到nds膜的dc电压来产生ac电场(图10的第4和5部分)。利用elenco9440试验板(digi-key,thiefriverfalls,mn,usa),100欧姆分流电阻器和3952xmolex插头(molex,lisle,il,usa)构建了电路。为了进行吸光率测量,使用了与常规48-管机器转盘集成的variancary50biouv-vis分光光度计(agilenttechnologies,santaclara,ca,usa)(由quantumnorthwest与agilenttechnologies和我们集团联合开发)。利用由通过nds膜隔开的两个容纳聚醚醚酮(peek)本体的源贮器和下沉贮器(sinkreservoir)组成的常规扩散装置进行扩散研究(图2b)。通过利用og116-31uv-固化环氧树脂(epoxytechnology,inc.,billerica,ma,usa)将peek本体之一附接到uv-大管(sigma-aldrich,st.luis,mo,usa)获得了下沉贮器。两个本体呈现了机加工的管路以允许连接电极与控制电路的线交叉(图10的第3部分)。两个硅橡胶o-环(applerubber,lancaster,ny,usa)将peek本体之间的膜密封。对于该实验使用两个复制品。利用传导h20e10z环氧树脂(epoxytechnology,billerica,ma,usa)将电引线附接至膜。将bare36awg红和黑线端部附接到膜并在150℃下固化10分钟。首先将膜浸入到异丙醇(ipa)中2小时以促进所有通道的润湿,然后利用millipore水冲洗。最后,在进行扩散测试之前将芯片浸入到所选择的缓冲溶液中过夜。在从(1.56至100μg/ml)的df-1浓度下获得了吸光度标准曲线(λ=320nm)。将芯片组装到常规的扩散装置中,用50mmnacl中的3mg/ml浓度的200μldf-1溶液装载源贮器,并用4.25ml的50mmnacl溶液装载下沉贮器。常规的转盘允许通过在300rpm下进行磁搅拌而使下沉溶液均匀化。测试在室温(23±0.1℃)下进行。将收集的数据关于t=0处的吸光度归一化并计算df-1的累积释放曲线。iv表征在一系列外部施加的增高的电压步长下进行了不同电极堆叠体组合的电学性质的表征。这包括电解诱导冒泡的定性观察。进行该表征以优化电极堆叠体和偏压条件两者以延长nds装置的工作寿命并利用由大量气体形成所标记的上边界确定nds装置的所施加的电压方面的有效工作范围。利用与先前实验中所用的相同程序将nds膜浸泡在ipa中,并在90℃下在烘箱中干燥约1小时。然后,将其破裂成两半,其中大致相等地沉积电极堆叠体(每侧正方形或矩形≥1cm)。36awg黑和红线从其绝缘体剥离,且将h20e10z环氧树脂(epoxytechnology,billerica,ma,usa)施加至红线的剥离端之一。将红线的环氧树脂涂覆端置于电极侧附着的芯片之一的角落上。一旦将线就位,将芯片置于150℃的等温烘箱上持续10分钟以固化环氧树脂。在另一个附着的芯片上用黑色线重复该工艺。将可复用的接头切成覆盖每个芯片的全部宽度和一半长度的尺寸。采用两个电极之间1mm的距离,将两半置于彼此面对的可复用接头上以构建夹层。利用millipore水制备了50mmnacl溶液。利用合适尺寸的可复用接头片将芯片夹层的端部附接至洁净室签棒的轴,且线/绝缘带端与该签棒接触。将铝箔作为镜子置于培养皿下(fisherscientificinc.,hampton,nh,usa)以允许更容易观察气泡形成。然后利用签棒将芯片夹层悬于培养皿上方。将50mmnacl溶液倾倒入培养皿中,达到芯片夹层上的绝缘底部的水平。利用e3643adc电源(agilenttechnologies,santaclara,ca,usa)在膜之间施加一系列增高的dc电压步长,并每秒利用万用表(agilenttechnologies,santaclara,ca,usa)记录产生的电流,直至对于该特别电压产生稳定的电流。所施加的dc电压范围从0.5v至2v,以0.25v为步长。在施加电压时,周期性检查芯片由施加电压导致的气泡生成。利用dc电源和34970adataacquisition/switchunit(agilenttechnologies,santaclara,ca,usa)测试芯片构造。表面电荷测量利用复制膜的表面化学和结构的晶片(尺寸33×37mm)进行膜的表面电荷测量。将晶片浸泡在ipa中,用millipore水冲洗,干燥并装入delsananocsubmicronparticlesize和zetapotentialanalyzer(beckmancoulter,inc.,ca,usa)的平坦表面池中,电极侧面向下。然后将其浸入到10mmnacl溶液中。在一系列短时(20-40分钟)和长时(5小时)运行中在20v下进行测量,暴露表面积为562mm2。结果图11和12描述了间歇地受先前所描述的电波形影响,df-1通过5nm(图11)和1μm(图12)膜的累积释放量对时间。这些膜中的5.7nm的纳米通道衬有氮化硅(5nm通道),其具有通过经验确定的约24mv的ζ电势。讨论通过将表面沉积的铂电极纳入我们先前报道的纳米通道膜上,我们利用机械上牢固的和临床相关的平台以暂时控制药物释放而不会显著增加制作或设计复杂性。此外,这些膜不需要分开的泵送区室或任何移动部件。可将这些先前描述的药物递送膜纳入高柔性封壳设计中,该设计可屏蔽见于体内环境中的较大的生物元件(包括细胞和大的细胞器结构)与调节电势的任何直接相互作用。考虑到在1-50μamp的邻近区域中需要一定电流来产生释放调节,这是重要的。递送构造的制作不足和设计复杂性导致了可被认为是更有限的范围的分析物,因为考虑到体内环境中存在高离子强度,递送的药剂必须是高度带电荷的,或需要高度带电荷的递送载体,例如本文所用的与df-1相似的胶束或富勒烯络合物。通过移动电极至更靠近纳米通道,可获得进一步增强和较低的泄露电流,非常现实的可能性是由于高柔性硅制作技术用于制作这些膜。该制作工艺允许制造高纳米通道密度,同时允许对其总数目和借助于所选分子的大小来优化的几何形状的精密控制。确定了优化的分子流体力学直径与纳米通道高度比率为约1:3以保证分子的释放。这导致了brownian运动的限制和关于浓度的质量通量高于阈值的饱和。可植入封壳的设计意在容纳和保护活性的nds膜,可高度适应于由患者病状所设置的要求。该类定制可通过贮器的容积和制造其的材料来控制。为了实现药物释放的显著降低,需要相对高的电压。使用图10中(第5部分)所示的正方形波形来降低平均电流。所施加的电势为1.5v,且所测量的功率消耗在1.5μw和75μw之间,对于电池而言是非常低的功率要求。图12描述了关于表面沉积的铂电极的电势ψ(mv)和归一化的数目浓度n。该图显示了对于5.7nm纳米通道的被动和主动释放模式之间的差异。电势的施加在装置中产生了浓差极化,因此调节了导致通道内部不同的电势的debye长度。由于较少的具有足够热能的粒子能够跨越能量位垒,所以一旦其因由于离子消耗导致的电荷分子的浓度变化而增高时,便导致释放速率的调节。如本文使用的术语“直接流体连通”应当解释为两个直接连接的本体之间的流体连通,例如使得流体可以从一个本体中出来并立即进入第二个本体,而不流经中间体。此外,本文使用的术语“入口”应当解释为纳米通道递送装置内的室或贮器,其初始保存经由该纳米通道递送装置递送的物质。类似地,“出口”应当解释为纳米通道递送装置内的室或贮器,其保存即将离开该纳米通道递送装置的物质。根据本公开内容,可在没有过多实验的情况下进行并完成本文所公开和要求保护的所有装置、系统和/或方法。尽管已经根据特别的实施方案描述了本发明的装置、系统和方法,但是本领域技术人员将清楚的是,可以对本文所述的装置、系统和/或方法的步骤或方法的步骤顺序进行改变,而不脱离本发明的概念、精神和范围。对于本领域技术人员清楚的是所有这样的类似变更和改变都视为在所附权利要求限定的本发明的精神、范围和概念范围内。通过将表面沉积的铂电极纳入我们先前报道的纳米通道膜上,可以利用机械上牢固的和临床相关的平台以暂时控制药物释放而不会显著增加制作或设计复杂性。此外,这些膜不需要分开的泵送区室或任何移动部件。可将这些先前描述的药物递送膜纳入高柔性封壳设计中,该设计可屏蔽见于体内环境中的较大生物元件(包括细胞和大的细胞器结构)与调节电势的任何直接相互作用。考虑到在1-50μamp的邻近区域中需要一定电流来产生释放调节,这是重要的。递送构造的制作不足和设计复杂性导致了可被认为是更有限的范围的分析物,因为考虑到体内环境中存在高离子强度,递送的药剂必须是高度带电荷的,或需要高度带电荷的递送载体,例如与本文所用的df-1相似的胶束或富勒烯络合物。通过移动电极至更靠近纳米通道,可获得进一步增强和较低的泄露电流,非常现实的可能性是由于高柔性硅制作技术用于制作这些膜。该制作工艺允许制造高纳米通道密度,同时允许对其总数目和借助于所选分子的大小来优化的几何形状的精密控制。确定了优化的分子流体力学直径与纳米通道高度比率为约1:3以保证分子的释放。这导致了brownian运动的限制和关于浓度的质量通量高于阈值的饱和。可植入封壳的设计意在容纳和保护活性的nds膜,可高度适应于由患者病状所设置的要求。该类定制可通过贮器的容积和制造其的材料来控制。实验设备和程序荧光显微成像在一些实施方案中,可使用测试设备来进行示例性的装置的微通道和纳米通道中分析物的电泳输送的荧光显微成像。在一些实施方案中,测试设备由双室系统组成,其中源和下沉溶液同轴地组装到玻璃盖玻片的顶部。图13(a部分)显示了测试设备的一个实施方案的示意图。在该实施方案中,用环氧树脂将有线的ndd胶合至聚乙烯环底部,且pyrex盖朝向柱体之外以构建源贮器。接着,通过利用光学uv-胶(68,norlandopticaladhesive)将该源贮器胶粘到玻璃盖玻片上。小心进行该程序以1)在盖玻片和膜出口之间构建平行的空间隙,和2)借助于倒置荧光显微镜(zeisslsm510)构建关于ndd的内部结构的成像的未堵塞的光学路径。最终,将第二个较大的环用环氧树脂胶合至盖玻片的同侧,其围绕源篮(basket),且因此构建了下沉贮器。利用磷酸缓冲盐溶液(pbs)缓冲液装载下沉贮器,且膜从出口润湿以防止气泡在膜和盖玻片之间的间隙中积聚。24小时之后,可通过光学显微术来验证膜的完全润湿。在该实施方案中,用pbs装填源贮器以构建流体在贮器之间和贯穿膜的连续性。用2%wtfitc-bsa(sigma-aldrich)的pbs溶液替换源贮器中的pbs。然后可将3vdc的电势差施加到在出口电极处具有正极性的ndd电极。然后可以以几秒的间隔通过荧光显微镜摄像机收集荧光图谱的快照。累积的主动释放为了进行累积主动释放测量,可将ndd布线和组装到由插入到细胞培养孔(well)的细胞培养篮(basket)组成的测试装置中(参见图13)。电接触在该实施方案中,可利用填充固体银的传导性环氧树脂(h20e)对ndd膜进行布线,a部分:b部分的比率为1:1。可通过将小滴的环氧树脂施加到电极的接触垫将线(36awg)与膜电极接合,从而传导性环氧树脂不会碰到膜的切片侧。在该实施方案中,可在120℃下的热板上将环氧树脂固化15分钟,然后在110℃下在烘箱中进行约1小时。插入组件可以在a部分:b部分比例为10:1下利用353nd环氧树脂胶将ndd组装到聚苯乙烯细胞培养插入物中(12孔形式,bectondikinson,usa)。然后可将环氧树脂混合物装载到具有191/2g针的注射器中。可将有线的ndd膜置于软硅橡胶膜上,所述硅橡胶膜良好贴附于孔足以防止环氧树脂的浮凸。可去除篮的底部的聚合物膜并将插入物以包围贴附的膜的方式置于硅橡胶上。然后可注射环氧树脂混合物以填充篮和硅橡胶表面上的膜之间的空间,同时防止环氧树脂沉积到膜的顶部上。然后,剥离底部硅橡胶膜之后,可将环氧树脂在热板上固化1分钟并在110℃下在烘箱中完全固化另外30分钟。一旦固化了环氧树脂,线和电极可以是完全隔绝的。润湿程序在该实施方案中,在先前利用一滴乙醇(防止在膜出口处形成气泡)润湿膜出口之后,可随后利用2ml的乙醇填充细胞培养孔,并将篮置于所填充的孔中。在几秒内,乙醇填充该膜直至膜入口打开而不截留微通道中的任何气泡,这可通过光学显微术来验证。然后可用乙醇填充篮,保证源贮器和下沉贮器之间的流体的连续性。然后可利用去离子水替换乙醇(每12小时交换2次),并最终在上游和下游贮器中用pbs来替换持续24小时。fitc-bsa释放测试在该实施方案中,在浓度为10mg/mlpbs(ph7.2,gibco)中制备了fitc-bsa(sigma-aldrich)溶液。可将叠氮化钠(0.05%wt)添加到溶液以防止细菌的生长。将1.6ml的pbs装载入到细胞培养孔中,同时将0.8ml的溶液装载入到篮插入物中。可用粘结剂teflon带(goregaskettape)盖住该篮。在该实施方案中,可将两个不锈钢(ss-316l)球添加到每个孔中以混合下沉溶液,并利用相同的teflon带气密性地盖住12孔板。可通过稳定的dc电源(例如,e3643a,agilenttechnologies)将1和2vdc施加到一组3个复制品的每个。在该实施方案中,未将电压施加到用作被动释放对照物的另外3个篮。最后,可将板储存在黑盒子中以防止样品的光漂白。可将盒子置于摇板(rockerplate)(vwr)上,导致不锈钢球恒定地混合并均匀化下沉溶液。以大约3天的时间间隔,可将200μl的下沉溶液装载到96孔板中。可使用荧光分光光度计(fluostaroptima,bmglabtech)来测量其荧光。然后可将样品与新鲜pbs一起重新添加到细胞培养孔以补偿从孔中的蒸发。在该实施方案中,最终通过荧光-浓度标准曲线获得样品的释放浓度。结果图14描绘了在33天周期内的11个不同的时间,fitc-bsa经过ndd膜的累积释放。fitc-bsa的被动释放显示了快的瞬态分布,其饱和至低得多的稳态速率。该瞬态可与跨越膜的长结构的浓度梯度的平衡的获得相关联。另外,这可能归因于与氧化硅壁上fitc-bsa的吸收有关的有效纳米通道横截面的逐渐减小。氧化硅衬底上的bsa吸收其实已被广泛引证38-42,并报道对从较小纳米通道(22nm)5的释放速率具有影响。在ndd电极上施加1或2vdc的电势时的释放速率为在获得浓度梯度平衡之后测量的被动释放速率的约3.9和9.8倍:13.5(1v)和33.9(2v)相对于3.5(被动释放)μg/天。图15描绘了正向和反向偏置下fitc-bsa的电动力学输送的荧光图像,表明了电动力学流动的可逆性。讨论电极在基于硅的纳米流体膜的入口和出口处的集成是该类有前景的牢固的药物递送构造的重要扩展。可使用电极以电动力学启动和调节在低施加电压下治疗剂的释放,而不需要通常施加至介入活塞或柔性膜的压力梯度33,34。在诱导电极表面的最小化的电解的电压下(需要最小化的电解以维持离子电流),短电极间隔产生了高电场。完全的倒流和低功率控制的释放可从膜获得,该膜的释放速率在2v下为34μg/天,这与其他优化的药物递送构造5相比相当,并达到了利用相似设计的系统中被动输送所未能达到的关注的治疗剂的范围44,45。分析了实验数据以理解电泳和电渗的电动力学现象之间的平衡。我们从每秒释放的平均药物量确定了系统的有效迁移率,在1v下为1.57×10-7mg/s,且在2v下为3.93×10-7mg/s。然后通过将平均释放速率除以出口微通道的累积横截面(1.064×103μm2)获得了平均通量,在1v下为0.0147mg/cm2,并且在2v下为0.0369mg/cm2。考虑到在稳态下入口和出口通量必须相等,这是可行的。通过将通量除以初始浓度10mg/ml46,计算了最大有效释放速度(veff),其在1v下等于14.7μm/s且在2v下等于36.9μm/s。最后,如下确定有效迁移率(μeff)47:其中ex为轴向上外部施加的电场。在该背景下,进行模拟以确定ex的合适值。图16的插图显示了用于模拟流体系统的电性质的电阻网络。因为用于皮下施用药剂的水溶液通常具有高离子强度以更密切地匹配体内生理环境,电双层(edl)的debye长度(λd)通常在纳米或亚纳米范围中,如通过以下所确定的48:其中εr是溶液的相对介电常数,εo是自由空间的介电常数,k是玻尔兹曼氏常数,t是以开尔文为单位的温度,na是阿伏加德罗常数,ci是第i个离子的摩尔浓度,zi是第i个离子的电荷价态,且e是基本电荷。将用于表征该电动力学递送系统的fitc-bsa装载在标准的pbs缓冲液(137mmnacl,2.7mmkc1,8mmna2hpo4,2mmkh2po449)中,产生所计算的0.75nm的λd。考虑到这样的薄edl,将电阻(r)作为贯穿所有网络分支的常规比例来对待。因此从以下获得每个区段的近似几何电阻:其中l是区段的长度,a是横截面面积。然后将跨越网络中的每个电阻器的电压降定义为可数值求解的等式系统(matlab,themathworks,inc.)。当变量的最大残余变得小于10-9时,则答案是收敛的。由于与微通道(590μm和9.16μm2)相比的纳米通道的短长度(5μm)及其显著的横截面(4.5μm2),其对于总电阻的整体贡献相对小。如图12所示,这导致了跨整个系统的拟线性电压降。因此可通过将所施加的电压除以725μm(从入口到出口的最长路径)来估计ex(假设在铂电极处为最小接触电阻)。所计算的有效迁移率在每个所施加的电压处应该是相等的。有效的迁移率在1v下为1.12×10-4cm2/v且在2v下为1.41×10-4cm2/v,其是合理一致的。与有效迁移率相关的电渗(μeof)和电泳(μef)迁移率为50:μeff=μeof+μep(4)在薄的λd近似值下,可如下计算电渗和电泳迁移率51:其中ζ是ζ电势,η是粘度(对于pbs缓冲液52,先前在室温下测定为0.93mpa·s),且r和q是fitc-bsa的流体力学半径和总电荷。考虑到二氧化硅表面和fitc-bsa具有净负电荷,预期这些计算的迁移率导致相反方向的速度,如图17中所示。然后利用grahame等式53获得了给通道加衬的二氧化硅表面的ζ电势:其中σ(ζ)是二氧化硅表面电荷,先前所估计为-1μc/cm210。从该等式中获得的ζ电势为-10.5mv。μeof为7.997×10-5cm2/v。该值代表甚至考虑到复杂构造的良好近似值,因为先前已证实电场和电渗速度两者具有符合复杂几何形状的能力。在该实施方案中,还利用6nm的fitc-bsa流体力学半径计算μep55。该分子上电荷的选择是更复杂的。表明bsa的最大理论电荷为-11的滴定值是可用的;然而,溶剂的屏蔽对有效电荷起重要作用,所述有效电荷在电场存在下产生电动势56。在文献中已报道了估计的比率7:10fitc/bsa,且对于各fitc理论电荷为-1。因此,在ph6.8中分子上的总电荷可能理论地接近-18(7*-1+-11)56,55。然而,这样的电荷导致的有效速度超过了我们凭经验测定的值。bsa的有效电荷在ph6.8中还可以低至-8.456,并且,与表面的ζ电势相似地,其可受离子强度的进一步影响。-13和-15之间的电荷范围足以导致-1.980×10-4和-2.28×10-4cm2/vs之间的电泳迁移率(符号基于导电惯例),其与计算的电渗迁移率和等式4结合得出了与我们实验数据非常一致的有效迁移率。还在纯pbs中测量了跨膜电流,其在1vdc和2vdc下分别确定为17和52μa。这分别等同于17和104μw的消耗功率,并且与其他已报道的递送构造33相比非常有利。虽然这些计算包括大量近似值,但是它们表明了理论和实验分析之间的良好一致,使得可获得合理设计的高度可控的药物递送构造。并且,它们阐明了电泳和电渗对电动力学输送的相对贡献。在体内存在的高离子强度环境中,这样的考虑对于决定递送何种药物和将需要多大功率是重要的。未来的分析会包括在药物递送环境中对系统参数进行彻底探测,以及研究能够进一步利用这些现象的新型递送结构体,其可产生更宽范围的递送速率和甚至更低的操作功率。利用荧光光谱学和显微术用fitc-bsa对本文所呈现的示例性实施方案的累积的长期分子释放和释放可逆性进行了表征。此外,确定了电泳和电渗的相对贡献,并观察了临床相关的释放速率。该构造能够最终允许对药物释放进行连续的调节,这是包括时间疗法的许多新兴的治疗方案的要求。可利用嵌入有动力的可植入封壳中的预编程的控制单元来获得该持续的节律性递送。另外,这样的系统可包括递送方案的激活或调节的远程外部控制的遥测硬件。最后,植入物可纳入传感器,其能够转换环境、物理和生物变化,具有调节治疗剂的施用的逻辑单元,能够实现人工腺体。除了以上的设计和制作技术以外,还可根据以下中公开的方法和设备设计和制作ndd装置:美国专利公开2007/0066138("diffusiondeliverysystemsandmethodsoffabrication")和美国专利公开2010/0152699("nanochanneleddeviceandmethodoffabrication"),通过引用将其并入本文。具有电极的纳米通道递送系统1示例性实施方案的制作方法具有电极的纳米通道递送系统(ndse)的示例性实施方案由两个主要的组件组成:硅衬底和具有集成电极的pyrex盖体。1.1硅衬底制作在该实施方案中,利用双侧抛光的p型4英寸晶片开始硅衬底的加工。硅衬底的第一主要工艺是限定纳米通道。为了制作纳米通道,在晶片上沉积垫氧化物层并随后沉积氮化物层。然后,施加标准光刻法来产生纳米通道图案。之后,使用cf4反应性离子蚀刻(rie)来去除这些通道图案上的氮化物。控制蚀刻时间使得保留二氧化硅的薄层以防止反应性离子侵蚀下面的硅。使用稀释的hf溶液来清洁通道图案中的氧化物,然后,将硅晶片放入热氧化物炉中,并生长所需量的氧化硅,氧化物的厚度限定了纳米通道的高度。通过利用椭圆偏振测量术测量了氧化物膜的厚度。然后用hf溶液去除衬底上的所有电介质。对于第二主要步骤,沉积新的氮化硅层以限定微通道。在纳米通道的顶部将微通道图案化。然后,重复rie。之后,进行koh蚀刻以获得3微米深的微通道。第三主要步骤是背面开口。将新的氮化硅层沉积到硅衬底上。通过两侧对准在晶片的背部使出口图案化。使用cf4rie来打开出口图案。然后进行穿过晶片的koh蚀刻(约8小时)以蚀刻穿过硅晶片。利用hf溶液去除介电材料之后,沉积15nm厚的氧化硅层作为用于容易线接合的隔绝层。利用afm测量了真实的纳米通道高度,对于每个衬底测量了5个选择的点。1.2pyrex玻璃制作这里总结了制作具有电极的pyrex玻璃盖体的示例性方法作为实例。在该工艺中有三个主要步骤。第一步骤是得到电极。在4英寸的pyrex晶片上蒸发钛、金和钛的夹层。然后,使用标准的光刻法来将电极图案化。施加多步骤湿蚀刻来蚀刻ti-au-ti夹层,然后清洁掉残余的光刻胶。这里,电极的高度高于pyrex表面150nm。然后,进行剥离工艺以将电极的厚度与蒸发的pyrex玻璃膜相匹配。为如此,进行光刻法以覆盖电极,然后利用cha蒸发150nm玻璃膜。接着,在丙酮中对晶片进行超声处理以去除光刻胶并清洁电极顶部沉积的pyrex。之后,蒸发100nm厚pyrex膜以覆盖电极。然后施加cmp步骤以抛光表面从而达到所需的粗糙度,并优化cmp工艺。在cmp工艺过程中,需要控制许多参数例如压力、速度、时间和垫。在合适控制压力、速度、时间以及其他参数的情况下尝试了许多不同的制作技术。最终将表面粗糙度降低至~1nm。1.3层接合在pyrex晶片中钻孔,并利用520进行阳极接合。对不同的纳米通道高度调节接合条件。在接合步骤之后,对晶片进行清洁、干燥,实现了ndde装置。1.4膜清洁从制作组接收了5个ndde晶片。在任何类型的测试之前,必须清洁所有的膜以去除任何制作残余物。首先,从所有膜上去除有机硅层。然后,将膜分为10个一组,并放入塑料微型管中。用乙醇填充微型管并将所述微型管置于板式振动器(plateshaker)上持续30分钟。在该时间过后,去除乙醇并在培养皿上分离膜。最终,将培养皿置于115℃下的真空烘箱中过夜以让膜完全干燥。2.新型ndde膜构造描述对于新型的ndde膜有11个构造。每个构造的不同之处在于电极和结构支撑柱的数目,微通道和纳米通道结构的类型,以及通道内电极的存在。图18显示了膜的简单布置。新型ndde和先前的构造之间的主要区别在于用于主动释放的电极的添加。在这些实施方案中,存在三种类型的纳米通道:笔直的、垂直的和倾斜的(titled)纳米通道。在具有笔直纳米通道的构造中,流体从入口直接穿越到出口。在垂直的通道中,入口和出口不直接连接。微通道和纳米通道是交叉指型的。在倾斜的纳米通道中为相同的情形,但通道处于一定角度。图19显示了这些差异。在这些实施方案中,存在具有笔直纳米通道的三种构造,构造#1,5和10。构造#1和#5每个具有两个电极,但不同之处在于纳米通道长度。构造#10具有第三电极,其横越通道的中部。三种构造具有倾斜的纳米通道(#7,#8和#9),五种构造具有垂直的纳米通道(#2,#3,#4,#6和#11)。构造#2,3和4中的纳米通道比构造#6-9和#11中的纳米通道短。结果,在构造#2,3和4中存在更多微尺寸和纳米尺寸的通道,6个构造具有4个电极(#3-4,#6,#8-9和#11)。将它们布置为沿膜的较长尺寸运行的两对相对的电极。构造#9和#11仅在微通道的一侧具有长的纳米通道。信息总结于以下表1中。在进行气体测试之后获取这些图像。3膜表征3.1afm测量通过原子力显微镜法(afm)(digitalinstrumentdimension3000,columbus,ohio)进行纳米通道深度的测量。在位于晶片的不同区域中的晶片的4-5个不同的点上进行测量。对于每个ndde衬底观察到小于2.62%的纳米通道尺寸的变化。3.2光学显微镜法在进行气体测试之前,在乙醇中洗涤所有的膜,同时在板式振动器上进行摇动。在烘箱中完全干燥之后,通过利用竖式光学显微镜对其进行选择。为了选择用于进一步电渗测试的装置的目的,抛弃呈现可见缺陷(微尺寸)的所有膜。所筛选的膜为20nm(不具有电极),50nm(具有或不具有电极),100nm(具有电极),和150nm(具有电极)。将不具有电极的膜命名为“wo”,而具有电极的那些膜命名为“w”。通常,对于不同的纳米通道深度,构造#3,6,7,8,9,10和11比构造#1,2,4和5具有更大量的存活膜。首先,检查膜的可见的或主要的缺陷例如过度蚀刻,破裂边缘和丢失的玻璃。约62-75%的膜通过了该检查,最常见的缺陷是硅的过度蚀刻(尤其是纳米通道区域中),在一些膜中,在通道中存在大的针孔。在构造和尺寸中随机分配了这三个主要的缺陷。第二检查检测通道区域中的尘埃粒子,在锚点和纳米通道之间的具有不良对比度(contrast)的膜,和玻璃的附着问题。通常,构造#2和#3具有对比度的问题。难以验证通道是否存在。对于所有纳米通道深度,构造#4表现出电极的过度沉积。对于深度100nm和150nm,对于随后的测试没有膜是可行的。在筛选过程结束时,对于进一步的测试,68%的20nmwo,67%的50nmwo,63%的50nmw,74%的100nmw和71%的150nmw是存活的。还存在一些缺陷,其是相同纳米通道深度尺寸的膜所特有的。入口将指玻璃侧上的开口,并且出口将指硅侧上的开口。3.3电极电阻测试通过在两个电极之间和在一个电极和硅表面之间测量电阻来进行电极电阻测试,以确定在它们之间是否存在电连接。所测试的膜是50nm,100nm和150nm深度。所测量的构造是#1,2,3,4(适用时),5,6,7,8和10。每个电极的命名呈现于以下显示的图20中。对于仅具有两个电极的膜,获取了两次电阻测量。首先,测量跨越电极b和c的电阻。然后,跨越电极b和膜的硅表面进行电阻的测量。对于具有三个电极的膜,跨越a-c,a-d和电极a与膜的硅表面测量电阻。最终,对于具有四个电极的膜,跨越a-b,a-c,b-d,a-d和电极a与膜的硅表面测量了电阻。测量了234个膜。每个膜在电极之间具有大于1mω的电阻值。所有膜具有在电极a与膜的硅表面之间大于60mω的值(超过万用表的极限)。3.4气体测试系统设计了常规气体测试系统以提供用于示例性实施方案的测试设备,所述系统包括:高纯度氮,n2,槽(研究纯度级别99.9999%,matheson),双级压力调节器(3120-580,matheson),膜支架,夹持系统,压力传感器(fullscale200psi,准确度0.05%,px01c1-200g5t,omegadyneinc.),气体过滤器(mathesontrigas200nm)和阀门管道和连接的系统。设计膜支架以确保容纳一个ndde的有效密封和硅橡胶常规密封(由applerubber,lancaster,ny,usa成型)。其由不锈钢ss316机加工过的杆和机械夹具制成。夹持系统使用了两个相对的螺纹杆,它们通过中间的螺母而相互抵住。3.4.1测量边界条件测量了零电压v0,而系统与气体槽隔离,且系统内部的压力为0psi。在200psi的满刻度压力(pfs)下测量满刻度电压(vfs)。在大约45psi的初始压力下开始先前在较老的膜版本上的气体压力测试。这对应于1.125v的初始电压。以下的表2中所列的边界条件用于生成等式1(与电压比压力有关的线性等式,其中vl是瞬时电压)。3.4.2气体测试程序首先,将膜置于柱形的硅橡胶密封的沟内。然后将密封置于螺纹杆中。接着,用六角螺母将杆锁到位。间隔距离为37.00±.05mm。固定膜之后,简单地打开真空阀以从系统中去除任何空气(绝对压力15.6kpa)。一旦关闭真空阀,就允许经过滤的n2气体进入系统中直至手持万用表读数1.134+.005v(对应于约0.31mpa的压力)。在系统已饱和有n2气体之后,关闭所有阀门,在t0=0秒时将系统与气体槽隔离。测量了由于贯穿膜的气体流动所致的系统压力下降,并利用数字万用表(34410a型,agilenttechnologies,santaclara,ca,usa)在0.1hz下收集数据,持续600秒。3.4.3气体测试系统性能测试3.4.3.1泄露分析进行一系列测试以确保装置所固有的任何泄露不会影响系统的可重复性和可再现性。首先,通过用整体硅橡胶盘替代硅密封和膜来进行气体测试。通过利用整体硅芯片(呈现与ndde相同的尺寸,但不具有入口和出口,并且不具有通道)进行第二测试,以分析常规硅密封的效力。两个测试均重复三次。关于数据的均值计算泄露测试的复制品的标准偏差stdeva。百分比压力下降与初始ap=3.110spa有关并在测试结束时进行测量(逝去时间为600秒)。还测量了压力传感器的噪音。观察了满刻度(fs)值的噪音<0.004%,其以压力为单位,对应于51pa的压力测量中的绝对噪音(0.51巴10-3)。噪音的幅值与因泄露导致的压力下降相当。因此,在两种情况下,认为泄露都是可以忽略的。3.4.3.2系统可重复性还通过利用相同的ndd装置进行10次气体测试来验证系统的可再现性。在2个膜上进行测试,所述2个膜呈现了“高”和“低”的n2流量(分别为ndse150nm构造#2,ndde150nm构造#5)。图21显示了ndde150nm构造#2的实验结果的比较。发现数据的标准偏差在压力传感器的可重复性极限内(±0.05%fs=6.8-102pa)。计算数据群的偏离以估计数据分布的正态性。偏离的值表明数据呈现从gaussian分布大的小偏离。3.4.4实验数据处理通过等式1将从压力传感器收集的电压数据转化为对应的压力数据。通过单一指数衰减性良好的描述了压力下降,与一维瞬态问题的答案相一致。因此,将收集的压力数据关于指数函数(等式2)进行拟合,其中d是时间常数。p(t)=k*e-dt(2)在600s的时间范围中进行了对每个曲线重新取样的插值,且该插值起始于k=0.31mpa的相对压力。对于每个所测试的ndde构造计算了实验数据和插入的拟合曲线之间的最大标准偏差,s,和相关系数,r2。从标准化的数据中,每个膜可用d来表征,其与膜的气体流量成比例。对于每个不同的膜构造计算平均时间常数davg,而不排除任何孤立的膜。在排除孤立的数据之后还计算了基于大部分聚集在一起的膜分布的选择的平均时间常数(ds)。两个平均值之间的差异可表示数据的一致性。在t=600秒处计算相对于起始压力(0.31pa)的压力数据的百分比标准偏差。另外,计算了数据分布的偏差。这些数值与所测试的装置的数目和抛弃的膜的数目一起列于表10中。3.4.5结果和讨论气体测试的第一目标是分析膜结构以提供对制作过程的反馈。第二目标是选择对于进一步的电渗测试最能存活的膜。通常,对于每个构造所测试的装置的数目不足以提供完全的统计学分析。然而,列于表x中的值定性地表明选择最能存活的膜的范围。对于100和150nm的纳米通道深度的构造4对于该测试是不可用的。气体流动对于纳米通道深度和膜构造中的差异是敏感的。图22显示了不同构造和纳米通道深度之间的平均时间常数ds的比较。构造#2和3显示了对于每个纳米通道深度的最大ds值。这归因于较短的纳米通道长度和较大的交叉指型微通道数目。构造#3相对于构造#2的较低ds值是由入口和出口和通道区域之间的额外的微尺寸间隔所致。由于增加的纳米通道长度,构造#6,7和8包含较少量的微通道。这导致了这些构造相对于构造#2和3的ds值的显著降低。仅包含笔直的纳米通道的三个膜构造#1,5和10表现出非常低的气体流量。这与相对于其他构造显著较小的横截面通道面积是一致的(约2-3个数量级)。对于这些膜,气体测试所检测的压力范围与压力传感器的可重复性范围的数量级相同(+0.05%fs=6.9毫巴)。因此,关于这些构造的结果对于选择的目的而言是不可靠的。对于该原因,需要通过显著降低系统贮器的容积来改变气体测试系统。4ndde电渗测试4.1测试插入物组件为了进行电渗(eo)测试,将ndde布线并装配到测试装置中。测试装置由细胞培养篮和细胞培养孔组成。图23显示了一个测试装置的图片。通过利用环氧树脂胶将膜组装到细胞培养篮中。该篮用作溶液贮器,被置于包含下沉溶液的孔中。4.1.1线接合通过h20e胶将导线接合到电极来组装ndde膜。h20e是双组分的填充100%固体银的传导性环氧树脂。为制备环氧树脂,以重量计1:1(a部分:b部分)的混合比进行混合。切割5厘米的线并将端部剥离。向该线添加一小滴环氧树脂混合物并且将其与电极接合。将该线置于电极的中部,但是使硅表面氧化以获得电绝缘。图24显示了ndde电极上的接合点。验证膜:没有传导性环氧树脂接触膜的切片侧。将膜在120゜c的热板上固化15分钟。最后,在110℃下的烘箱中烘烤至少1小时。4.1.2篮制备通过利用353nd环氧树脂胶将ndde组装到篮中。353nd是双组分高温环氧树脂。为了制备环氧树脂,以重量计10:1(a部分:b部分)的混合比进行混合。利用191/2g针将环氧树脂混合物装载到注射器中。使针的尖端变平以防止对膜的任何损害,并优化到细胞培养篮中的环氧树脂注射。对于细胞培养用途,篮是可商购的。它们在篮的底部呈现了聚合物膜。从篮中去除膜,并将插入物置于围绕膜的硅橡胶正方形上。将环氧树脂混合物注射到围绕膜的篮中,填充硅橡胶表面(在膜的顶部不添加环氧树脂)。利用环氧树脂胶小心涂覆线以构建用于流体的密封和电绝缘。保持培养插入物向下,持续1分钟或直至加热固化环氧树脂至琥珀色。在固化程序过程中去除任何空气泡。将玻璃板留在热平板上大于5分钟以完全固化环氧树脂。在环氧树脂硬化之后,从硅正方形去除篮,如果环氧树脂未填充电极空间,则在空的空间中施加环氧树脂滴,并在110℃下将篮在烘箱中烘烤另外30分钟。最后,从硅中去除掩蔽带。图25显示了组装的篮。4.2润湿程序在运行电渗测试之前,用在以下实验过程中所用的溶剂填充膜(膜润湿)。通过利用不同的程序进行润湿。4.2.1pbs润湿用800μl的pbs填充篮并将其置于12孔板中。留下pbs以通过毛细填充来润湿膜。每个膜的出口朝空气开放。膜留置润湿。24小时之后,在光学显微镜下检查膜。发现润湿不完全。尤其是对于所有ndde装置的具有支撑柱的出口区域是完全干燥的。发现pbs不能润湿膜,出口微通道与连接出口的大开口连接。另外在微通道纳米通道区域中发现一些气泡。4.2.2pbs+真空为了协助利用pbs进行ndde润湿,将真空施加于膜的出口侧。在20inhg下保持真空12小时。通过光学显微术观察润湿区域的量。真空的施加有助于增加pbs润湿的量。对呈现柱的出口区域的大部件进行填充。然而,许多ndde仍在通道区域和出口区展现出大量的未填充区。总之,真空的应用不足以促进协助利用pbs进行的ndde润湿。4.2.3pbs+真空+电渗在ndde膜的电极之间施加1伏的电势差以分析电渗流是否可用于润湿程序。观察了被截留在微通道内部的空气泡的运动,表明周围微纳米通道区中对应的pbs运动。图26显示了在60分钟的时间间隔内电渗输送对气泡运动的影响。将电压保持恒定12小时。在该程序结束时,膜润湿仍不完全。总之,虽然作为所施加电场的结果观察到了流体的运动,但是完全ndde润湿将是耗时的。4.2.4乙醇、去离子水和pbs证明了利用pbs进行的直接润湿是无效率的。为此,对其他ndde膜采用了不同的润湿程序。用2ml的乙醇填充细胞培养孔。通过先用一滴乙醇润湿膜出口来将篮置于填充的孔中。该步骤避免了膜出口中的空气泡。乙醇在几秒内填充了膜,到达膜入口开口。在通过光学显微镜验证完全润湿之后,用乙醇填充篮,保证了篮贮器和孔流体之间的流体的连续性,通过利用光学显微镜,验证了在膜中没有截留气泡。用去离子水替换篮和孔中的乙醇,每12小时替换去离子水2次。需要24小时以允许乙醇扩散出膜并被去离子水替换。然后用pbs替换水,留置于上游和下游贮器中24小时。对于避免来自与乙醇直接接触的pbs的盐沉淀,去离子水中间步骤是必要的。总之,乙醇润湿程序保证了即时和完全的ndde膜润湿。4.2.5bsa-fitc标准曲线该实验的目的是从系列稀释和非系列稀释中确定缀合的bsa-fitc(sigmaaldrich)的标准曲线。制备了浓度为10mg/ml的bsa-fitc的初始溶液。将181.48mg的bsa-fitc添加到18.148ml的缓冲溶液(pbs(gibco)ph7.2氯化钙+0.05%wt叠氮化钠)。通过10mg/mlbsa-fitc溶液的1:10稀释制备了1mg/mlbsa-fitc的起始溶液。通过系列稀释(迭代稀释)和非系列稀释(来自1mg/mlbsa-fitc起始溶液)制备了标准曲线。表3显示了用于非系列稀释的13种溶液。其包括稀释比率,bsa-fitc溶液的量和缓冲溶液以及最终浓度。表3.用1mg/mlbsa-fitc溶液开始的非系列稀释表表4显示了制备系列稀释所采用的三个稀释序列表5通过列出bsa-fitc溶液的量、缓冲溶液的量、取自先前稀释的溶液的量、溶液的总量和其浓度描述了三个序列。表4.系列稀释的序列表5.利用1mg/mlbsa-fitc溶液开始的系列稀释使用fluostaroptima分光光度计(bmglabtech)来测量样品的荧光。以下表6显示了用于进行测量的分光光度计的设置。制备和测量了系列和非系列的标准曲线样品的三个复制品。向每个中添加100μl。表6.分光光度计的说明发射滤波器485激发滤波器520增益735光底部微板greiner96f底定位延迟0.2s将收集的荧光数据拟合为指数函数f=k(1-e-dc)(1)计算反函数f1以将样品的荧光强度与样品浓度相关联。f1的表达式为计算了标准曲线的三个复制品的平均荧光值。对于系列和非系列标准曲线计算了平均值的指数拟合。对于系列和非系列稀释两者计算了关于三个复制品的平均值的拟合曲线的相关系数r和百分比标准偏差。通常,三个复制品标准曲线的荧光结果显示了小于5%的标准偏差。仅在系列稀释的小浓度时观察到例外,其中多次稀释可引入较大误差。对于高于5μg/tnl的浓度,系列和非系列标准的拟合曲线关于实验数据表现出小于10%的百分比偏差。然而,非系列稀释的样品的测量显示了较大的散射。对于低浓度,系列稀释比非系列稀释更准确,因为初始添加的bsa-fitc溶液的量小于总溶液。对于小于5μg/ml的浓度,也观察到了较大的变化。发现了用系列稀释和非系列稀释制备的标准曲线之间的整体一致性。表7和图27示出了系列稀释和非系列稀释的插值拟合曲线之间的比较。选择拟合曲线的平均作为bsa-fitc样品的最终标准偏差曲线。计算了相对于两个插值拟合曲线的平均值的插值拟合曲线的百分比标准偏差。表7.系列和非系列的插值曲线之间的比较总之,浓度和荧光之间的关系式表示为:其中导入了所计算的系数d和k。4.34.4电渗测试微-纳米通道中的电渗(eo)输送强烈地受如下参数影响:与膜结构和表面性质相关的参数,以及与溶液化学相关的参数,特别地,强烈影响输送的参数是:-所施加的电压(dc或ac)-ac电压的形状、比率和频率-通道尺寸-电极位置,称作通道位置-缓冲溶液的ph以下段落中所描述的测试的目的是对以上参数的影响进行定量以对电渗输送的控制提供深入了解。4.4.1测试1.bsa-fitc的电渗输送4.4.1.1该测试的目的是获得与通道尺寸、所施加的dc电压和电极构造对eo输送的影响相关的初步结果。为了进行该测试,选择了14个膜:6ndse50#3,2ndse50#4,3ndse100#3,3ndse150#3/通过先前描述的光学显微术和气体测试选择了膜。以下表8总结了测试装置,展示了测试配置。表8测试设置。用于各测试配置的膜的数目。该表列出了用于电压影响测试、电极配置测试和纳米通道尺寸效应测试的膜的数目。如以上表8中所示,通过比较被动bsa-fitc扩散(2个复制品)与主动输送0.9vdc(2个复制品)和1.8vdc(2个复制品)评估了所施加的电压对eo输送的影响。为了评估电极配置的影响,选择了构造#3的2个膜和呈现构造#4的2个膜。ndde#3呈现了与ndde#4相同的通道结构。然而,虽然ndde#3的电极置于入口和出口区,但是ndde#4电极覆盖了交叉指型微通道的整个顶表面。为此,选择了2个ndde50#4与ndde50#3相比较。通过关于50、100和150nm的纳米通道尺寸测试ndde#3,评估了纳米通道尺寸效应。4.4.1.2结果和讨论测试1施加的dc电压。在图28中显示了bsa-fitc贯穿ndde100#3膜的主动释放的结果(12天中)。该图显示了施加1.8伏和0.9伏的累积释放曲线与累积被动释放的比较。发现了主动释放速率和被动释放速率之间的显著差异。尤其是在施加0.9伏和1.8伏下分别测量了约300%和400%的释放量的增加。对于每个测试配置,bsa-fitc的释放速率随时间推移而增大。在施加1.8伏的情况下,释放速率的增大显著。事实上,在测试的最初9天,施加1.8伏的释放速率结果比施加0.9伏的小。结果显示了施加电压对膜的释放速率的明显影响。然而,由于小的复制品数目和大的数据标准偏差(所测量数据的约30%),仅可将结果作定性考虑。通道尺寸的影响。在图29中显示了bsa-fitc贯穿ndde50#3,100#3和150#3膜的主动累积释放(12天中)的比较。结果显示了,施加相同的电压(1.8伏),在增大的纳米通道尺寸下累积释放增加。尤其是,如果与ndde50#3的释放结果相比较,对于呈现100nm和150nm的纳米通道的膜,测量了约400%和600%的增大。如先前所描述的,对于每个测试配置,bsa-fitc的释放速率随时间推移而增大。在实验测试的第6天之后,测量了显著的增大。结果显示了纳米通道尺寸对膜的释放速率的明显影响。然而,由于小的复制品数目和大的数据标准偏差(所测量数据的约30%),仅可将结果作定性考虑。电极配置的作用。通过测试ndde50#3(入口和出口区中的电极)和ndde50#4(微通道中的电极)的1.8伏主动释放,分析了电极配置对bsa-fitc的释放速率的影响。在图30中显示了结果(在12天周期内)。如图30所示,未测量到累积结果中的显著差异。释放似乎未受不同的电极配置的影响。由于小的复制品数目和大的数据标准偏差(所测量数据的约40%),仅可将结果作定性考虑。4.4.2bsa-fitc的电渗输送4.4.2.1该测试的目的是利用不同的ndde构造重复测试1(如上),分析通道尺寸、所施加的dc电压和电极配置对eo输送的影响,为了进行该测试,选择了17个膜:9ndse150#7,2ndse150#8,3ndse100#7,3ndse50#7。通过如先前所描述的光学显微术和气体测试来选择膜。以下表9总结了测试装置,展现了测试配置。表9.测试装置。用于各测试配置的膜的数目。该表列出了用于电压作用测试、电极配置测试和纳米通道尺寸效应测试的膜的数目。如该表中所示,通过比较被动bsa-fitc扩散(3个复制品)与主动输送0.9vdc(3个复制品)和1.8vdc(3个复制品)评估了所施加的电压对eo输送的作用。为评估电极配置3的作用,选择了构造#4的3个膜和呈现构造#8的2个膜。ndde#7呈现了与ndde#8相同的通道结构。然而,虽然ndde#7的电极置于入口和出口区域中,但是ndde#8电极覆盖了交叉指型微通道的整个顶表面。为此选择2个ndde150#8与ndde150#7相比较。通过测试纳米通道尺寸50,100和150nm的ndde#7评估了纳米通道尺寸效应。4.4.2.2测试2.结果和讨论所施加的dc电压。在图31中显示了bsa-fitc贯穿ndde150#7膜的主动释放的结果(9天内)。该图显示了施加1.8伏和0.9伏的累积释放曲线与累积被动释放的比较。发现了主动释放速率和被动释放速率之间的显著差异。尤其是在实验分析9天之后,对于0.9伏和1.8伏的主动释放的释放量为被动释放量的约45倍。并且,在该情况中,对于每个测试配置,bsa-fitc的释放速率随时间推移而增大。对于施加1.8伏的情况,释放速率的增加再次是更显著的。然而,对于施加0.9伏的总体释放量的结果大于通过施加1.8伏的释放量。相似的结果见于测试1。虽然施加电压的作用明显,但是由于小的复制品数目和大的数据标准偏差(所测量数据的约35%),仅可将结果作定性考虑。通道尺寸效应。在图32中显示了bsa-fitc贯穿ndde50#7;100#7,150#7膜的主动累积释放(12天中)的比较。该结果显示了施加相同的电压(1.8伏),在增大的纳米通道尺寸下累积释放增加。在该情况中,通过比较,ndde100#7和ndde150#7的释放数据结果比ndde50#7的结果大两个数量级。如先前所描述的,对于每个测试配置,bsa-fitc的释放速率随时间推移而增大,且在从检测开始约6天时发生显著的增加。该结果显示了纳米通道尺寸对膜的释放速率的比例作用。然而,由于小的复制品数目和大的数据标准偏差(所测量数据的约35%),仅可将结果作定性考虑。电极配置的作用。通过测试ndde150#7(入口和出口区中的电极)和ndde150#8(微通道中的电极)的1.8伏主动释放,分析了电极配置对bsa-fitc的释放速率的作用。在图33中显示了结果(在9天的周期中)。未测量到累积结果中的显著差异。释放似乎未受不同电极配置影响。由于小的复制品数目和大的数据标准偏差(所测量数据的约40%),仅可将结果作定性考虑。4.4.2测试3葡萄糖的电渗输送4.4.2.1测试3.目的该测试的目标是为了通过应用名义上不带电荷的分子例如葡萄糖来显示eo输送。为了进行该测试,选择了6个ndde50#1膜。为了将主动释放与被动释放相比较,从不呈现电极的不同晶片选择了3个ndde#150。通过光学显微术来选择所有膜。对于构造#1的气体测试显示了气体流动太小以至于不能可靠地测量。将每个膜固定在用于其他eo测试的篮装置中。对于接收施加电压的膜,每个篮装有.8ml的4m葡萄糖溶液。其各自的孔装有1.6ml的去离子水+.05%wt叠氮化钠。对于被动释放的膜,每个篮装有1ml的4m葡萄糖溶液。其各自的孔装有2ml的去离子水+.05%wt叠氮化钠。每隔几天,用血糖仪对来自每个孔的两份1μl样品进行读数。每天加回读数之间通过的5μl的去离子水以补偿(accoutfor)溶液的蒸发。4.4.2.2测试3.结果和讨论图34显示了在29天中的实验结果。重要的是注意,对于被动释放膜,在第54天对第一可测量浓度进行读数。为此,数据点对应于通过假设膜的随时间推移的线性累积释放从第54天外推的数据。此外,用于被动释放实验的膜来自不同于主动释放实验中所用的那些的晶片。其来自在玻璃上不具有电极的晶片。该数据显示了改变施加电压不会显著影响经过膜的葡萄糖的释放速率。复制品数目小,但主动释放量和被动释放量之间的差异非常大。因此,该数据定性地明确表明了eo增大了葡萄糖经过膜的输送速率。为了测试以上比较的被动释放数据的有效性,通过考虑作为浓度的函数的扩散d计算了葡萄糖经过的理论释放。该假设有助于生成等式6。是质量流量。n是纳米通道的数目。w,h和l(μm)是单一纳米通道的宽度、高度和长度。扩散常数df是浓度系数b是cin(μm)和cout(μm)分别是2.75e6和0。基于第54天的实验数据,葡萄糖被动释放的量为430μg。在第54天释放的葡萄糖的理论量是632.778μg。计算比实验结果大出约30%,验证了被动释放的数量级是合理的。4.4.3测试4.v-i特性曲线通过施加电场产生的电渗流在于离子沿通道的定向运动。通过向电极施加电势差,产生了离子流。电流给出了对应于所施加电压的电渗输送的幅度的指示。获得与特定膜构造相关的v-i曲线允许设计对主动输送的控制。v-i曲线的研究允许确定重要的参数例如电极随时间推移的稳定性。4.4.3.1测试4.材料和方法以与用于nddeeo测试的篮相同的方式准备了用于每个纳米通道尺寸和每个构造的一个膜(如4.1.2段落中所描述)。在用乙醇和去离子水润湿膜(参见4.2.4段落)之后,用0.8ml的pbs填充篮并将其置于12孔板中。用1.6ml的pbs填充孔。然后此时将一个膜与用于在膜的电极处施加恒定dc电压的电源(agilente3643a)连接。所连接的电极位于与通道区最接近处。数字万用表(agilent34410a)与膜串联连接以测量通过eo现象产生的离子流所产生的电流。遵循步长曲线,将增大的dc电压施加到膜。步长包括0.4v,0.8v,1.2v,1.6v和1.9v。在将dc电压施加到膜之后,在将电压水平增高至接下来的步长值之前,使电流平衡。以1分钟间隔收集电流数据。最后对于每个ndde配置绘制作为所施加电压的函数的平衡点。4.4.3.2测试4.结果和讨论作为实验测量的结果,收集了随时间推移的电流曲线。图35显示了关于一个膜ndde150#2收集的数据。该图显示了在增大的施加dc电压的步长值处的电流曲线。尤其显示了步长0.4v,0.8v,1.2v和1.6v的序列。每次电压水平在较高步长下增加时,在电流曲线中观察到峰。然后在较低值下电流逐渐平衡。在降低的施加电压下观察到了相似的结果。在该情况中,通过逐步下降电压水平,在电流曲线中观察到负峰值。图36显示了用ndde150#2膜收集的数据。在该情况中,电压从0.8v降低到0.4v导致了电流降低达到负值(或电流的暂时反向)。图35和36中显示的行为是在电容器和电阻器之间呈现并联的典型电路。以下图37显示了该电路的示意图。在示意图中,电容器代表了电极及其积聚电荷的能力。rl代表电极和接线的电阻,r代表流体的电阻。通过瞬时施加恒定电势e,电容器的节点处的电压vc等于零。因此,在时间t=0处,仅通过电极的电阻rl确定了电流i。接着为瞬态,其中vc逐渐增高,确定了电流i朝向较低稳态水平的指数下降瞬态在电流中呈现尖峰,且每次电压瞬时改变时发生指数平衡。在增大的电压下出现电流的正峰(如图35中所示),在减小的电压下出现电流的负峰(如图36中所示)。本文推导了描述电路情况的微分方程并计算其解。i=ic+ir(8)推导方程式(13),通过施加恒定电势e,因此,其中:积分方程式(15)的特定解是其通过强加边界条件来计算:和ic(t=∞)=0(17)然后简单地获得电流i(t)vc=e-rsi(19)通过将(17)和(19)导入到方程式(18),获得了电流的表达式在t=0处获得了电流的峰值稳态下的电流是对于对应于电容器的充电的增大电压的情况计算解析解。对于降低电压的情况相似地推导解。简化的模型给出了实验分析过程中测量的电流的正峰值和负峰值的解释。该模型还描述了在电压变化下电流峰的绝对值的增大。该模型未考虑流体电阻r的变化。通过对施加的电压绘制电流平衡点,获得了膜的v-1曲线。图38显示了对于一个膜ndde150#1的v-1曲线。电流随施加的电压呈指数增大。对于其他测试的构造观察到相似的结果(ndse150#2,#6,#7,#8,#9)。在图38中,显示了所测试的膜的v-1曲线。全部构造的测量的电流处于μa的数量级。在这方面,ndde150#1表现出一个例外:在0-30na的范围中表现出电流值。不同的膜的电流值的差异与通道的结构和电极的配置紧密相关。呈现运行于每个微通道顶部上的交叉指型电极(构造#8和#9)如果与在微通道外部呈现电极的构造相比较显示了较高的电流。换言之,电流取决于由电势差产生的电场。其还取决于可用于离子运动的通道的数目和尺寸。在这方面,膜ndde#1合理地显示了电流的最小值,其纳米通道的数目为全部其他构造中最小的。v-1曲线协助理解从一个电极朝向另一个电极的离子运动的强度。然而,v-1特性曲线和分子的主动释放之间无直接关联。明确的实例由ndde构造150#7和#8所代表。在对于150#7和#8所测量的v-1曲线之间观察到显著的差异。然而,主动释放结果(测试2)显示了,在第一分析中在2个不同的构造的情况中存在可以忽略的差异。额外的实验结果对于ndde150#2膜测量了增大和减小电压的v-1曲线。图39显示了代表所测量的数据的图形。在下降的电压下测量了较大的电流值。在测量增大的步长之后马上进行了减小的步长下的测量。因此,电极处的电荷积累可导致反向电流的暂时增大,因此导致迟滞效应。在增大的电压步长下测量电流时,在一些情况下观察到电流的突然的不规则峰。发现该效应发生在1.6伏或1.9伏。图40显示了对于ndde150#2膜观察到的不规则峰的实例。该峰达到两倍于预期的电流值。该效应可通过电极表面层的快速劣化而导致。除了先前的设计和制作技术之外,还可根据以下中所公开的方法和设备来设计和制作ndd装置:美国专利公开2007/0066138(“diffusiondeliverysystemsandmethodsoffabrication”)和美国专利公开2010/0152699(“nanochanneleddeviceandmethodoffabrication”),通过引用将其并入本文。流场效应晶体管(flowfet)实施方案本公开内容的额外的示例性实施方案提供了利用集成的顶部和底部电极对经过纳米通道的治疗组分(例如药物)的电动力学输送或被动扩散的控制。示例性的实施方案可称作双栅极流场效应晶体管(flowfet)。已证明了生物源的分子从纳米流体膜的被动的(cd.fine等人,labchip,2010.10.3074-3083)和主动的(fine等人“alow-voltageelectrokineticnanochanneldrugdeliverysystem”labchip,接收)零级释放。在被动释放的情况下,释放是零级和连续的,具有的速率由膜构造从机械上确定。在主动释放的情况下,释放速率由纳米通道的入口和出口处的电极所施加的电场来确定,其诱导了电渗、电泳或二者。开发了能够影响分析物经过微通道和纳米通道的主动和被动输送的几个原型,所述影响通过调节一个或多个纳米通道表面的表面电荷进而导致ζ电势的改变和通过延长延伸至纳米流体通道中的双电层来实现(r.b.m.schasfoort等人,science,1999,286,942-945,r.karnik等人,nanoletters,2005,5,943-948,r.karnik等人,applphys.lett.,2006,88,123114,和c.s.lee,w.c.blanchard,和c.t.wu,analyticalchemistry,1990,62,1550-1552)。该调节导致了带电荷的分析物的可用横截面的挤压以在纳米通道中扩散或漂移经过,或影响所诱导的电渗流,在一些情况下使其反向。使用具有集成金属栅极或半导体栅极(相对于毛细管)的微制作和纳米制作的装置的实施方式,称作流场效应晶体管(flowfet)。目前可用的flowfet的主要缺点是栅极仅影响纳米通道的表面之一。这是个问题,因为二氧化硅(通常涂覆硅纳米通道表面的材料)的中性ph的固有ζ电势非常高。此外,大部分生理学溶液的离子强度也非常高,导致非常有效的电荷遮蔽和非常薄的debye长度。这样,调节纳米通道的顶部和底部(这两侧占总纳米通道周长的99%以上)二者上表面电荷的能力将是对现有设计的巨大改进,且能够使得该技术拓展到高度充电的生理环境中的药物递送。在该结构中需要源电极和漏电极以保证对于两个栅极电极以相同的方向(增高的或降低的)调节表面电荷以使得作用最大化。这样的装置在电学领域中具有关于鳍式场效应晶体管(finfet)的扩展。一个finfet具有在由电介质和栅极电极缠绕的薄台面的顶部和底部上构建的源电极和漏电极。该几何形状允许通道完全耗尽固有电荷,大大改进了晶体管的转换速度。除了dcζ电势调节以控制扩散、漂移或dc电渗以外,该实施方案所能够进行的第二操作模式是ac偶联的行波电渗(a.ramos等人,proc.spie,5839.305-313和a.ramos等人j.appl.phys.,2005,97.084906-084906-8)。该现象发生于将一系列的相移ac电波形施加于紧密隔开的电极阵列时。如果在准确的频率下适当的相移,可在通道的仅一个表面上具有电极的微通道中获得整体的单向流。因此,整体的电场仅在短距离内下降,但通过粘性耦合,使通道的整个横截面上的溶液移出。可在纳米尺度下实施这样的系统。这可通过串联放置大量前述的flowfet双栅极并施加合适的ac电势波形而在纳米流体膜中得以实施。该装置的示例性的实施方案可见于图41和42中。可与此设计一起使用的纳米通道膜描述于该章节末的制作程序中,或描述于p.fine等人.labchip,2010.10.3074-3083中(阳极接合的纳米通道膜和利用牺牲层技术制作的纳米通道膜)。用于激发行波电渗的双栅极flowfet或串联的flowfet的制作取决于纳入其的纳米流体膜的制作方法。对于阳极接合膜,以高剂量将底栅极和/或源电极/漏电极离子植入到高电阻率绝缘体上硅(soi)衬底,所述衬底覆盖有光刻图案化的离子植入物掩蔽层(优选具有超过1000欧姆*cm的固有电阻率或利用隔离氧化和化学机械抛光(cmp))。顶栅极和/或源电极/漏电极如下制备:将沟槽蚀刻到顶部pyrex层中并随后用ti/au或ti/pt(但并不限于这些金属)进行填充至高于沟槽的水平。然后对电极进行化学机械抛光回到原始的pyrex表面。然后如p.fine等人.labchip,2010.10.3074-3083中所描述的完成膜的制作,不同之处是必须对准阳极接头使得pyrex中的电极将与soi衬底中的纳米通道特征合适地对准。对于利用牺牲金属制造的纳米通道,首先将底栅极和/或源电极/漏电极沉积在soi衬底上,然后图案化,蚀刻,并且最后用电介质或其他适当的纳米通道围绕材料进行覆盖。然后对电介质进行化学机械抛光回到电极表面,然后进行牺牲金属的沉积、图案化和蚀刻,一旦在后续步骤中去除所述牺牲金属,其限定出纳米通道。然后该牺牲金属被涂覆电介质,并被化学机械抛光回到牺牲金属表面,然后进行顶栅极和/或源电极/漏电极的沉积、图案化和蚀刻。然后将整个堆叠体嵌入适当量的电介质或其他材料中。在每个cmp步骤中,可保留薄的电介质以分隔金属层(如果需要)。然后根据p.fine等人.labchip,2010,10,3074-3083继续该工艺。因为纳米通道非常薄,所以电极宽度和间距可能也必须非常小(亚微米)。目前的微制作和纳米制作光刻能力可产生小于25nm的特征。还可以使用纳米压印光刻产生小至20nm的电极。纳米装置设计和制作(第二原型)用于本研究的整体微制作的纳米通道递送装置(ndd)膜由纳米微机加工的硅晶片和pyrex盖壳体电极组成。硅晶片呈现了由通过垂直的纳米通道阵列相互连接的平行微通道组成的交叉指型几何形状。微通道和纳米通道的顶表面通过将pyrex盖晶片阳极接合到锚点的网点而获得。纳米通道的纳米尺寸是深度。作为施加到嵌入的膜电极的电势差的结果,药物分子通过钻通pyrex的入口进入膜,移动通过一组136个微通道,然后进入到120个纳米通道的网孔中(两组,每个微通道上60个),并最终通过另一组136个微通道到达湿蚀刻穿过硅晶片的出口。示例性的实施方案提供了在低功率下对于被动扩散和电动力学驱动的膜两者在体内调节药物释放的能力,包括完全关闭药物释放(例如,静电阀门)。双栅极结构的优点包括允许在比采用现有设计可能的(即在flowfet表面之一上)多的纳米通道表面上调节表面电荷。这在高离子强度环境,大部分生理环境的情况,或高固有表面电荷材料如二氧化硅中是重要的。除了先前的设计和制作技术以外,还可根据以下中公开的方法和设备来设计和制作ndd装置:美国专利公开2007/0066138(“diffusiondeliverysystemsandmethodsoffabrication”)和美国专利公开2010/0152699(“nanochanneleddeviceandmethodoffabrication”),通过引用将其并入本文。本公开内容的示例性实施方案还提供了电动力学纳米通道植入物的被动或主动流体和压力补偿。药物递送植入物通常由刚性封壳构成以防止内部和外部施加的应力使药物贮器破裂,导致灾难性的植入物失效。从这些刚性体通过纳米通道膜进行的电动力学药物递送可产生流体流动,且因此可产生压力,必须对其进行补偿以防止可能过早终止药物释放的负压的建立。本公开内容的示例性实施方案提供了补偿该电动力学产生的流体流量的方法和结构,该方法和结构利用了第二纳米通道膜被动地通过压力诱导的流动或主动地通过电动力学诱导的流动。实施方案的示例性说明可见于图43、44和45中。可与此设计一起使用的纳米通道膜描述于该章节末的制作程序中,或描述于p.fine等人.labchip,2010.10.3074-3083中。该植入物构造由策略性放置在植入体的相对端处的纳米通道膜组成。一个纳米通道膜(递送膜)具有集成的电极以产生用于递送药物的向外电动力学流动。第二纳米通道膜(补偿膜)可以是被动的或主动的,并具有向内的流动,其由递送膜诱导的负压或由主动的电动力学诱导的与递送膜相似的流动来启动。需要递送膜和补偿膜之间的总纳米通道横截面的仔细平衡来保证合适的补偿,且所述平衡取决于该补偿是主动的还是被动的。这样,如果有必要还可使用多个补偿膜。虽然基于正递送的药物来常规选择递送膜,但是可基于许多标准来选择补偿膜,所述标准包括对于药物在贮器中时其所需免疫应答的保护水平。在流体被吸入到补偿膜中的情况下,这种考虑是重要的。如果有效负载对免疫标志物或蛋白质例如胰蛋白酶高度敏感,可使用较大量的较小横截面的纳米通道。通过利用生理流体补偿流体流动,还可能补偿渗透压。纳米装置设计和制作(第二实施方案)用于该研究的整体微加作的纳米通道膜(nds)由微机加工的硅晶片和pyrex盖壳体电极组成。硅晶片呈现了交叉指型几何形状,其由通过垂直的纳米通道的阵列相互连接的平行微通道组成。通过将pyrex盖晶片阳极接合到锚点网格获得了微通道和纳米通道的顶表面。纳米通道的纳米尺寸是深度。因为施加到嵌入的膜电极的电势差,药物分子能通过钻通pyrex的入口进入膜,移动通过一组136个微通道,然后进入到120个纳米通道的网孔(两组,每个微通道上60个)中,并最终通过另一组136个微通道到达湿蚀刻穿过硅晶片的出口。示例性的实施方案能够通过适当地补偿任何诱导的流体流动或压力进行电动力学药物递送。电动力学纳米通道药物递送是高度可控的过程,且其代表了允许药物释放的模拟和数字调节的少数药物递送技术之一。先前的方法包括利用可变形的膜围绕纳米通道膜或纳米多孔玻璃料。这样的实施方式需要分开的泵送室,其具有封闭的泵送贮器,并根据施加到膜或活塞的压力将药物推出,或需要被设计到封壳中的可变形内衬。除了先前的设计和制作技术以外,还可根据以下中所公开的方法和设备设计和制作ndd装置:美国专利公开2007/0066138(“diffusiondeliverysystemsandmethodsoffabrication”)和美国专利公开2010/0152699(“nanochanneleddeviceandmethodoffabrication”),通过引用将其并入本文。本公开内容的示例性实施方案还包括用于主动控制引导的(led)药物递送系统的可植入的涂覆电极的膜,包括用于以下中所公开的制作的结构和方法:美国专利公开2007/0066138(“diffusiondeliverysystemsandmethodsoffabrication”)和美国专利公开2010/0152699(“nanochanneleddeviceandmethodoffabrication”),通过引用将其并入本文。特定的示例性实施方案包括邻近微通道和纳米通道的涂覆金属电极(铂,金,银,多晶硅等)的可植入的牢固纳米流体膜。利用高度精确和准确的硅纳米制作技术制造了膜。这样的设计能够在药物溶液流经纳米通道时在直接施加到药物溶液的低电压下使药物电动力学(电泳,电渗)输送经过纳米流体通道。与浓度驱动的输送相比较,该实施方式允许显著增加并精细调节药物的释放速率,能够实现临床相关的释放速率。开发了装置的示例性实施方案来以主动控制的动态方式控制药物从可植入的药物贮器中释放。利用紧密集成到纳米通道的电极来施加电场,允许在低功率下进行药物释放的动态调节。被动纳米通道膜可维持恒定的药物零级释放持续延长的时间,但不能被转换或具有能够动态控制的输出,即不能调节绝对释放,也不能开启和关闭释放。需要电极接近以允许足够的电场强度,同时保持在导致显著电解所需的电势以下。所发明的装置允许调节、增强和精细调节从纳米通道膜的释放速率,所述纳米通道膜已能够释放治疗相关范围内的药物。示例性的实施方案包括优点:例如动态释放控制,低功率和植入物形状柔性和简单的实施方式。示例性的实施方案包括用于主动和电动力学控制药物和粒子从如本公开内容其他地方所描述的可植入装置中释放的系统。示例性的实施方案还涉及利用金属电极涂覆被动纳米通道膜的入口和出口面,所述金属电极可用于跨纳米流体膜施加电场。通过直接真空蒸发被动膜的硅表面和pyrex表面上的钛层(10nm)和金层(100nm)制作了原型。图46显示了涂覆电极的膜及其内部结构的示意图。通过本发明的说明,利用20nm纳米通道原型进行了实验。在由两个并排放置并将膜夹在其间的uv-管组成的装置中测量了牛血清白蛋白(bsa)、13nmodots和枝状富勒烯(dfl)的释放的主动控制。在杯的侧壁上钻的2个孔的对应者中对膜进行环氧树脂胶合。以下图47显示了装置的示例性实施方案的示意图。odots、bsa和dfl的主动释放的实验结果显示于图48-50中。该图显示了通过施加跨膜的电势,可以显著增强分子和粒子的释放。示例性的实施方案包括大量的优点,包括药物和粒子从植入物释放的主动电动力学控制。此外,对释放的控制可以是预编程的,或远程控制的。并且,由于纳米通道中电渗的效率,能量消耗降低,且该特征可作为纳米通道膜的制作步骤来实施。还可将涂覆电极的膜集成到可植入的装置中。系统可能够进行长期时间治疗,且可根据需要来调节装置对药物和颗粒的电泳或电渗输送。可用一系列的生物相容性材料来制作电极,包括金属(例如,钛、铂、金)、聚合物和陶瓷。电极可用ito(透明的)来制作以允许光学品质控制。此外,该技术可用于根据分子/颗粒(particular)电荷和尺寸对分子和粒子进行的电动力学分离。参考文献通过引用将以下参考文献的内容并入本文:参考文献(通过引用将每个并入本文)[1]f.martin,r.walczak,a.boiarski,m.cohen,t.westa,c.cosentinoandm.ferrari,j.controlledrelease,2005,102,123-133.[2]r.karnik,k.castelinoanda.majumdar,appl.phys.lett.,2006,88,123114.[3]w.sparreboom,a.vandenbergandj.c.t.eijkel,nat.nanotech.,2009,4,713-720.[4]r.b.schochandp.renaud,appl.phys.lett.,2005,86,253111.[5]j.hanandh.g.craighead,j.vac.sci.technol.a,1999,17,2142.[6]d.kim,j.d.posnerandj.g.santiago,sensorsandactuatorsa:physical,2008,141,201-212.[7]m.z.bazant,k.thorntonanda.ajdari,phys.rev.e,2004,70,021506.[8]s.j.kim,y.c.wang,j.h.lee,h.jangandj.han,phys.rev.lett.,2007,99,044501.[9]a.mani,t.a.zangleandj.g.santiago,langmuir,2009,25,3898-3908.[10]t.a.zangle,a.maniandj.g.santiago,langmuir,2009,25,3909-3916.[11]q.pu,j.yun,h.temkinands.liu,nanolett.,2004,4,1099-1103.[12]d.fine,a.grattoni,e.zabre,f.hussein,m.ferrariandx.liu,labchip,2011,11,2526-2534.[13]j.fu,r.b.schoch,a.l.stevens,s.r.tannenbaumandj.han,nat.nanotech.,20072,121-128.[14]w.chu,t.huen,j.tuandm.ferrari,proceedingsofspie,1997,2978,111.[15]s.oleksandrov,j.w.lee,j.h.jang,s.haamandc.h.chung,j.ofnanosc.andnanotech.,2009,9,1551-1554.[16]s.m.kim,m.a.burnsande.f.hasselbrink,anal.chem.,2006,78,4779-4785.[17]d.fine,a.grattoni,s.hosali,a.ziemys,e.derosa,j.gill,r.medema,l.hudson,m.kojic,m.milosevic,l.brousseauiii,r.goodall,m.ferrariandx.liu,labchip,2010,10,3074-3083.[18]a.grattoni,h.shen,d.fine,a.ziemys,j.s.gill,l.hudson,s.hosali,r.goodall,x.liuandm.ferrari,pharm.res.,2011,28,292-300.[19]a.grattoni,d.fine,e.zabre,a.ziemys,j.gill,y.mackeyev,m.a.cheney,d.c.danila,s.hosali,l.j.wilson,f.hussainandm.ferrari,acsnano,2011,5,9382-9391.[20]r.farra,n.f.jr.sheppard,l.mccabe,r.m.neer,j.m.anderson,j.t.jr.santini,m.j.cimaandr.langer,sci.transl.med.,2012,4,121-122.[21]m.c.mormontandf.levi,cancer,2003,97,155-169.[22]m.h.smolenskyandn.a.peppas,advancedrugdeliveryreviews,2007,59,828-851.[23]c.h.chenandj.g.santiago,j.ofmicroelectromechanicalsystems,2002,11,672-683.[24]t.hoare,j.santamaria,g.f.goya,s.irusta,d.lin,s.lau,r.padera,r.langerandd.s.kohane,nanolett.,2009,9,3651-3657.[25]m.r.prausnitz,c.s.lee,c.h.liu,j.c.pang,t.p.singh,r.langer,j.c.weaver,j.controlledrelease,1996,38,205-217.[26]r.b.schoch,j.hanandp.renaud,rev.mod.phys.,2008,80,839-883.[27]x.liu,f.pu,y.fan,x.deng,d.liands.li,am.j.physiol.heartcirc.physiol,2009,297,h163-h170.[28]a.plecis,r.b.schochandp.renaud,nanolett.,2005,5,1147-1155.[29]y.c.wang,a.l.stevensandj.han,anal.chem.,2005,77,4293-4299.[30]r.bakry,r.m.vallant,m.najam-ul-haq,m.rainer,z.szabo,c.w.huckandg.k.bonn,int.j.nanomedicine,2007,2,639-649.[31]1.r.s.kerbelandb.a.kamen,natrevcancer,2004,4,423-436.[32]e.pasquier,m.kavallaris,andn.andre,natrevclinoncol,2010,7,455-465.[33]m.h.smolenskyandn.a.peppas,advanceddrugdeliveryreviews,2007,59,828-851.[34]m.mormontandf.levi,cancer,2003,97,155-169.[35]d.fine,a.grattoni,s.hosali,a.ziemys,e.derosa,j.gill,r.medema,l.hudson,m.kojic,m.milosevic,l.brousseauiii,r.goodall,m.ferrari,andx.liu,labchip,2010,10,3074-3083.[36]a.grattoni,h.shen,d.fine,a.ziemys,j.s.gill,l.hudson,s.hosali,r.goodall,x.liu,andm.ferrari,pharm.res,2010.[37]r.walczak,a.boiarski,m.cohen,t.west,k.melnik,j.shapiro,s.sharma,andm.ferrari,nanobiotechnology,2005,1,35-42.[38]j.t.santini,m.j.cima,andr.langer,nature,1999,397,335-338.[39]j.h.prescott,s.lipka,s.baldwin,n.f.sheppard,j.m.maloney,j.coppeta,b.yomtov,m.a.staples,andj.t.santini,nat.biotechnol.,2006,24,437-438.[40]r.karnik,r.fan,m.yue,d.li,p.yang,anda.majumdar,nanoletters,2005,5,943-948.[41]t.kuo,l.a.sloan,j.v.sweedler,andp.w.bohn,langmuir,2001,17,6298-6303.[42]s.pennathurandj.g.santiago,analyticalchemistry,2005,77,6782-6789.[43]s.pennathurandj.g.santiago,analyticalchemistry,2005,77,6772-6781.[44]a.plecis,r.b.schoch,andp.renaud,nanolett,2005,5,1147-1155.[45]r.b.m.schasfoort,s.schlautmann,j.hendrikse,anda.vandenberg,science,1999,286,942-945.[46]r.karnik,k.castelino,anda.majumdar,appl.phys.lett.,2006,88,123114.[47]p.vonguggenberg,inelectricalinsulationanddielectricphenomena,1993.annualreport.,conferenceon,1993,pp.122-127.[48]s.a.miller,v.y.young,andc.r.martin,journaloftheamericanchemicalsociety,2001,123,12335-12342.[49]c.r.martin,m.nishizawa,k.jirage,m.kang,ands.b.lee,advmater,2001,13,1351-1362.[50]l.sunandr.m.crooks,journaloftheamericanchemicalsociety,2000,122,12340-12345.[51]c.duananda.majumdar,natnano,2010,5,848-852.[52]s.j.kim,y.wang,j.h.lee,h.jang,andj.han,physrevlett,2007,99,044501-044501.[53]t.a.zangle,a.mani,andj.g.santiago,chemsocrev,2010,39,1014-1035.[54]a.plecis,c.nanteuil,a.haghiri-gosnet,andy.chen,analyticalchemistry,2008,80,9542-9550.[55]j.h.leeandj.han,microfluidnanofluidics,2010,9,973-979.[56]j.fu,r.b.schoch,a.l.stevens,s.r.tannenbaum,andj.han,natnano,2007,2,121-128.[57]j.han,j.fu,andr.b.schoch,labchip,2008,8,23-33.[58]j.hanandh.g.craighead,science,2000,288,1026-1029.[59]b.r.cipriany,r.zhao,p.j.murphy,s.l.levy,c.p.tan,h.g.craighead,andp.d.soloway,analyticalchemistry,2010,82,2480-2487.[60]m.j.levene,j.korlach,s.w.turner,m.foquet,h.g.craighead,andw.w.webb,science,2003,299,682-686.[61]s.pennathur,f.baldessari,j.g.santiago,m.g.kattah,j.b.steinman,andp.j.utz,anal.chem,2007,79,8316-8322.[62]l.chen,j.choo,andb.yan,expertopindrugdeliv,2007,4,119-129.[63]s.litster,m.e.suss,andj.g.santiago,sensoractuata-phys,2010,163,311-314.[64]s.bhavaraju,j.gordon,anda.joshi,drugdeliverytechnology,2010,10,24-31.[65]p.apel,radiat.meas.,2001,34,559-566.[66]y.peng,a.pallandre,n.t.tran,andm.taverna,electrophoresis,2008,29,157-178.[67]a.d'emanueleandj.n.staniforth,pharmres,1991,8,913-918.[68]c.a.haynesandw.norde,j.colloidinterf.scl.,1995,169,313-328.[69]o.svenssonandt.arnebrant,jcolloidinterfacesci,2010,344,44-47.[70]k.c.kwok,k.m.yeung,andn.h.cheung,langmuir,2007,23,1948-1952.[71]y.i.tarasevich,theor.exp.chem.,2001,37,98-102.[72]t.maruyama,s.katoh,m.nakajima,andh.nabetani,biotechnol.bioeng,2001,75,233-238.[73]w.r.whitney,jphyschem,1903,7,190-193.[74]p.m.sinha,g.valco,s.sharma,x.liu,andm.ferrari,nanotechnology,2004,15,s585-s589.[75]g.b.lesinski,s.sharma,k.a.varker,p.sinha,m.ferrari,andw.e.carson,biomed.microdevices,2005,7,71-79.[76]d.marro,y.n.kalia,m.b.delgado-charro,andr.h.guy,pharmres,2001,18,1701-1708.[77]m.h.oddyandj.g.santiago,jcolloidinterfacesci,2004,269,192-204.[78]w.sparreboom,a.vandenberg,andj.c.eijkel,newjphys,12,1-23.[79]ambion,inc.,2008.[80]j.h.knoxandk.a.mccormack,chromatographica,1994,38,207-214.[81]g.j.sommer,s.m.kim,r.j.littrell,ande.f.hasselbrink,labchip,2007,7,898-907.[82]a.seiyama,y.suzuki,andn.maeda,colloidpolymsci,1993,271,63-69.[83]f.vanderheyden,d.stein,andc.dekker,phys.rev.lett.,2005,95.[84]g.o.f.parikesit,a.p.markesteijn,v.g.kutchoukov,o.piciu,a.bossche,j.westerweel,y.garini,andi.t.young,labchip,2005,5,1067.[85]p.ramasamy,r.elmaghrabi,g.halada,andm.rafailovich,mater.res.soc.symp.proc,1061,1-6.[86]u.andu.scheler,chem.phys.lett,2007,435,342-345.santen,r.j.,yue,w.,naftolin,f.,mor,g.,berstein,l.thepotentialofaromataseinhibitorsinbreastcancerprevention.endocrine-relatedcancer.6,235-243(1999).goss,p.e.,strasser,k.aromataseinhibitorsinthetreatmentandpreventionofbreastcancer.j.clin.oncol.19,881-894(2001).chlebowski,r.t.reducingtheriskofbreastcancer.n.engl.j.med.,343,191-198(2000).dowsett,m.,jones,a.,johnston,s.r.,jacobs,s.,trunet,p.,smith,i.e.invivomeasurementofaromataseinhibitionbyletrozole(cgs20267)inpostmenopausalpatientswithbreastcancer.clin.cancerres.1,1511-1515(1995).brueggemeier,r.w.,hackett,j.c.,diaz-cruz,e.s.aromataseinhibitorsinthetreatmentofbreastcancer.endocrinereviews26,331-345(2005).coates,a.s.,keshaviah,a.,thürlimann,b.,etal.fiveyearsofletrozolecomparedwithtamoxifenasinitialadjuvanttherapyforpostmenopausalwomenwithendocrine-responsiveearlybreastcancer:updateofstudybig1-98.j.clin.oncol.25,486-492(2007).goss,p.e.,ingle,j.n.,martino,s.,etal.arandomizedtrialofletrozoleinpostmenopausalwomenafterfiveyearsoftamoxifentherapyforearly-stagebreastcancer.n.engl.j.med.349,1793-1802(2003).garreau,j.r.,delamelena,t.,walts,d.,karamlou,k.,johnson,ν.sideeffectsofaromataseinhibitorsversustamoxifen:thepatients'perspective.am.j.surg.192,496-8(2006).luthra,r.,kirma,ν.,jones,j.,tekmal,r.r.useofletrozoleasachemopreventiveagentinaromataseoverexpressingtransgenicmice.thejournalofsteroidbiochemistryandmolecularbiology.86,461-467(2003).harper-wynne,c.,ross,g.,sacks,ν.,salter,j.,nasiri,n.,iqbal,j.,a'hern,r.,dowsett,m.effectsofthearomataseinhibitorletrozoleonnormalbreastepithelialcellproliferationandmetabolicindicesinpostmenopausalwomen:apilotstudyforbreastcancerprevention.cancerepidemiol.biomarkersprev.11,614-21(2002).当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1