用于遗尿的药物制剂及其使用方法与流程
本申请为申请日为2013年6月28日,申请号为201380037454x,发明名称为“用于遗尿的药物制剂及其使用方法”的发明专利申请(国际申请号:pct/us2013/048627)的分案申请。
本申请要求享有在2013年3月13日提交的序号为13/800,761的美国专利申请,在2013年3月20日提交的序号为13/847,940的美国专利申请和在2012年7月27日提交的序号为13/560,665的美国专利申请的优先权。
本申请主要涉及用于抑制膀胱的平滑肌的方法和组合物,具体地,涉及用于治疗遗尿的方法和组合物。
背景技术:
夜间遗尿症(通常称为遗尿)是最常见的儿童泌尿道疾病和最常见的小儿健康问题之一。大约13%的6岁儿童尿床,同时大约5%的10岁儿童也尿床。在朋友家或在宿营地对于尿床小孩儿能够感觉到尴尬和内疚并且焦虑地度过夜晚。
而在多数案例中遗尿只是发育延迟并将随时间自行消除,其可持续到少年并对家庭极有压力。小比例(5%至10%)的遗尿案例是真正由特定医学状况引起的。遗尿也与家族史的情况相关。据此,需要治疗遗尿的组合物和方法。
技术实现要素:
本申请的一个方面涉及一种治疗受试者遗尿的方法,其包括向有此需要的受试者施用一种药物组合物,所述药物组合物包含:活性成分,所述活性成分以每剂1-1000mg的量包含一种或多种镇痛剂,其中,所述一种或多种镇痛剂选自阿司匹林、布洛芬、萘普生、萘普生钠、吲哚美辛、萘丁美酮和对乙酰氨基酚。所述药物组合物可以被配制成立即释放、延迟释放、延长释放或其结合。
本申请的另一个方面涉及一种治疗受试者遗尿的方法,其包括:向有此需要的受试者施用一种药物组合物,所述药物组合物包含:第一活性成分,所述活性成分包含选自镇痛剂、抗毒蕈碱剂、抗利尿剂、解痉剂、pde5抑制剂的一种或多种试剂;和第二活性成分,所述第二活性成分包含选自镇痛剂、抗毒蕈碱剂、抗利尿剂、解痉剂和pde5抑制剂的一种或多种试剂,其中,所述第一活性成分被配制成立即释放,以及其中,所述第二活性成分被配置成延长释放。
本申请的另一个方面涉及一种药物组合物,其包含:一种或多种镇痛剂、去氨加压素和药学可接受的载体。
附图说明
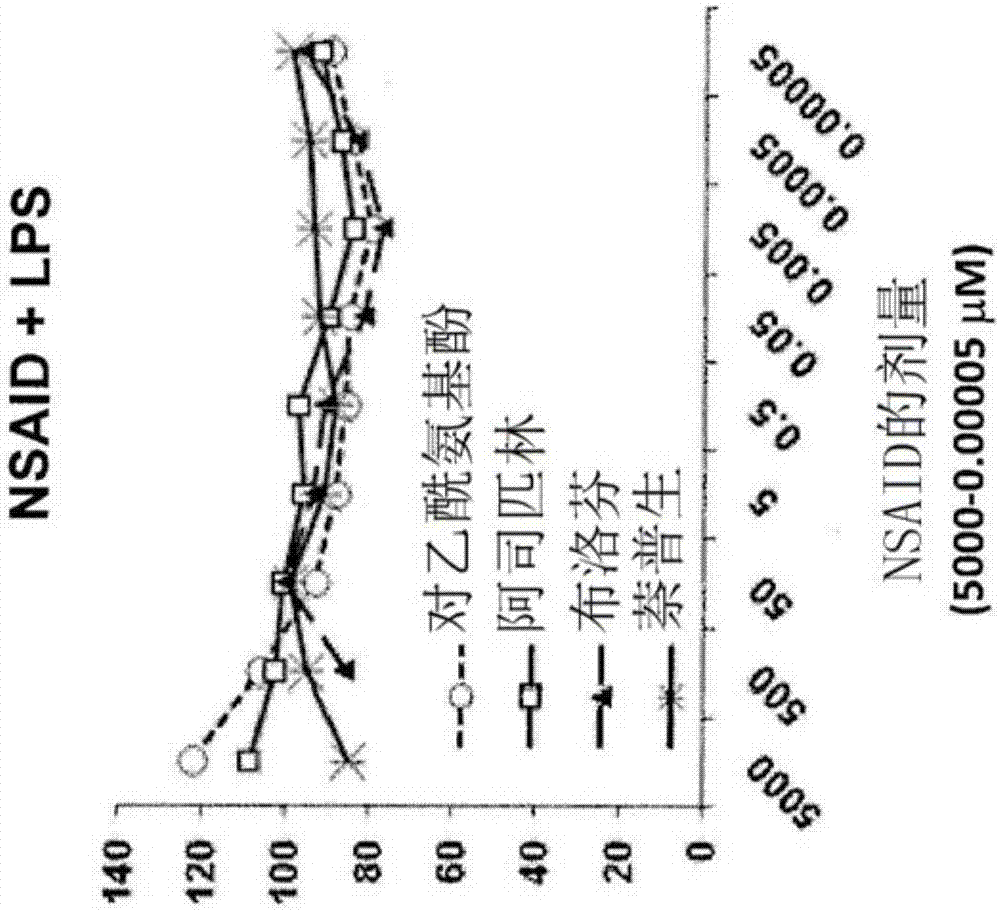
图1a和图1b是显示在缺少lps下(图1a)或存在lps下(图1b)镇痛剂调节raw264巨噬细胞的辅刺激分子的表达的图。细胞在镇痛剂单独存在或与鼠伤寒沙门氏菌(salmonellatyphimurium)lps(0.05μg/ml)共同存在下培养24小时。结果是cd40+cd80+细胞的平均相对百分比。
具体实施方式
提出以下的详细说明以使本领域的技术人员实施和使用本发明。为了解释的目的,以下阐述具体术语以充分理解本发明。然而,对于本领域的技术人员来说,显然这些具体的细节并不需要在本发明中实施。提供具体应用的表述仅用作典型的实施例。本发明并不意欲限于所示的实施方式,而是希望包括与在此公开的原理和特征相一致的可能的最宽范围。
在此使用的术语“遗尿”是指儿童在通常发生膀胱控制的年龄之后在睡觉时无意识的排尿。
在此使用的术语“有效量”是指达到选定的结果所需的量。
在此使用的术语“镇痛剂”是指用于缓解疼痛且包含抗炎化合物的试剂、化合物或药物。示例性的镇痛和/或抗炎试剂、化合物或药物包括但不限于非甾体抗炎药(nsaids)、水杨酸盐、阿司匹林、水杨酸、水杨酸甲酯、二氟尼柳、双水杨酯、奥沙拉秦、柳氮磺吡啶、对氨基苯酚的衍生物、乙酰苯胺、对乙酰氨基酚、非那西丁、灭酸酯、甲灭酸、甲氯灭酸酯、甲氯灭酸钠、杂芳基乙酸衍生物、托美丁、酮咯酸、双氯芬酸、丙酸衍生物、布洛芬、萘普生钠、萘普生、非诺洛芬、酮洛芬、氟比洛芬、奥沙普秦;烯醇酸、昔康(oxicam)衍生物、吡罗昔康、美洛昔康、替诺昔康、安吡昔康、屈噁昔康、匹伏昔康(pivoxicam)、吡唑酮衍生物、保泰松、羟基保泰松、安替比林、氨基比林、安乃近、考昔类药物、塞来考昔、罗非考昔、萘丁美酮、阿扎丙宗、吲哚美辛、舒林酸、依托度酸、异丁基苯基丙酸、鲁米考昔(lumiracoxib)、艾托考昔、帕瑞考昔、伐地考昔、替拉考昔(tiracoxib)、依托度酸、达布非酮、右酮洛芬、醋氯芬酸、利克飞龙(licofelone)、溴芬酸、氯索洛芬、普拉洛芬、吡罗昔康、尼美舒利、西唑来汀、3-甲酰基氨基-7-甲基磺酰基氨基-6-苯氧基-4h-1-苯并吡喃-4-酮、美洛昔康、氯诺昔康、右旋吲哚布芬、莫苯唑酸、呱氨托美丁(amtolmetin)、普拉洛芬、托芬那酸、氟比洛芬、舒洛芬、奥沙普秦、扎托洛芬、阿明洛芬、噻洛芬酸,及其药用盐、其水合物和其溶剂合物。
在此使用的术语“考昔(coxib)”是指含有能够抑制cox-2酶的活性或表达或者能够抑制或缓解严重的炎症反应的严重程度(包括疼痛和肿胀)的化合物的组合物。
在此使用的术语“衍生物”是指化学改性的化合物,其中所述改性是被普通的熟练化学工作者认为是惯用手段,例如,酸的酯或酰胺,或者保护基(例如对于醇或硫醇的苄基或对于胺的叔丁氧羰基)。
在此使用的术语“类似物”是指包括特定的化合物或其类的化学修饰形式且保持所述化合物或所述那类化合物的药学的和/或药理学的活性特征的化合物。
在此使用的术语“药学上可接受的盐”是指所公开的化合物的衍生物,其中,母体化合物通过形成其酸式盐或碱式盐而被改性。药学上可接受的盐的实例包括,但不限于,碱性残基(例如胺)的矿物盐或有机酸盐;酸性残基(例如羧酸)的碱盐或有机盐;等等。所述药学上可接受的盐包括(例如,由非毒性的无机酸或有机酸)形成的母体化合物的常规的非毒性盐或者季铵盐。例如,这样常规的非毒性盐包括:由无机酸得到的盐,如盐酸、氢溴酸、硫酸、氨基磺酸、磷酸、硝酸等等;以及由有机酸制备的盐,如乙酸、丙酸、琥珀酸、羟乙酸、硬脂酸、乳酸、苹果酸、酒石酸、柠檬酸、抗坏血酸、双羟萘酸、马来酸、羟基马来酸、苯乙酸、谷氨酸、苯甲酸、水杨酸、对氨基苯磺酸、2-乙酰氧基苯甲酸、反丁烯二酸、甲苯磺酸、甲磺酸、乙烷二磺酸、草酸、羟乙磺酸等。
在此使用的短语“药学上可接受的”是同化合物、材料、组合物和/或剂型一起使用,其在合理的医学判断范围内,适合与人类和动物的组织接触使用,具有合理的利益/风险比而无过高的毒性、刺激性、过敏反应、或其它问题或并发症。
在此使用的术语“延长释放”,又称持续释放(sustained-release、sr)、持续作用(sustained-action、sa)、限时释放(time-release、tr)、控制释放(controlled-release、cr)、改良释放(modifiedrelease、mr)或缓释(continuous-release、cr),指一种在药物片剂或胶囊中使用以随着时间的过去缓慢溶解和释放活性成分的机制。延长释放的片剂或胶囊的优势在于,它们通常能够比同样药物的立即释放制剂更少频次的施用,而且,它们在血流中保持更稳定的药物水平,从而延长药物作用的持续时间并且降低药物在血流中的峰值量。
在此使用的述语“延迟释放”指的是在施用药物组合物后的特定时段(迟滞期)内药物组合物的活性成分的释放被延迟或推迟的药物释放曲线。
在此使用的术语“立即释放”参照不含有溶解速率控制材料的药物制剂。施用立即释放制剂之后,活性剂的释放基本没有延迟。立即释放包衣可以包含施用后立即溶解的合适材料,从而释放其中的药物内含物。在一些实施方式中,在此使用的术语“立即释放”参照在施用2小时内释放活性成分的药物制剂。
本申请的一个方面涉及一种通过向有此需要的病人施用一种药物组合物来治疗遗尿的方法。所述药物组合物包含一种或多种镇痛剂和任选地,一种或多种抗毒蕈碱剂、一种或多种抗利尿剂、一种或多种解痉剂和/或一种或多种5型磷酸二酯酶抑制剂(pde5抑制剂)。所述药物组合物被配制成立即释放、延长释放、延迟释放或其组合。
在一个实施方式中,所述药物组合物通过在不溶物质(如丙烯酸酯类或甲壳质)的基质中包埋活性成分而被配制成延长释放的。延长释放形式被设计为通过在特定时段内维持恒定的药物水平而以预定的速率释放镇痛剂化合物。这可以通过不同的制剂实现,包括,但不限于,脂质体和药物-聚合物共轭体,如水凝胶。
延长释放的制剂可被设计为以预定的速率释放活性剂以维持特定的延长的时段内的恒定的药物水平,例如,在施用后或在与药物延迟释放相关的迟滞期后的最高至约12小时、约11小时、约10小时、约9小时、约8小时、约7小时、约6小时、约5小时、约4小时、约3小时、约2小时或约1小时。
在某些实施方式中,活性剂在约2小时至约12小时之间的时间间隔期间被释放。或者,活性剂可以在约3小时、约4小时、约5小时、约6小时、约7小时、约8小时、约9小时、约10小时、约11小时或约12小时内被释放。在其他一些实施方式中,活性剂在施用后约5小时至约8小时之间的时段内被释放。
在一些实施方式中,延长释放制剂包括活性核,所述活性核由一种或多种惰性粒子组成,各自以其表面上包被有药物(例如,以含药物的包衣或成膜组合物的形式(使用例如流化床技术或本领域的技术人员公知的其他方法))的珠、丸剂、药丸、颗粒粒子、微胶囊、微球、微颗粒、纳米胶囊或纳米球的形式。所述惰性粒子可以是不同大小的,只要其足够大以保持不易溶解。或者,所述活性核可以通过含有药物成分的聚合物组合物的造粒和碾磨和/或通过挤出和滚圆来制备。
所述活性剂可以通过本领域技术人员公知的技术被引入到惰性载体,例如药物分层、粉末包衣、挤出/滚圆、滚压或造粒。在所述核中的药物的量将取决于所需要的剂量,且通常为从约5至90wt%变化。通常,基于包衣粒子的重量,根据所需的延滞时间和/或所选择的聚合物和包衣溶剂,在活性核上的聚合物包衣为1至50%。本领域的技术人员将能够选择在核上包被或引入核合适量的药物以实现所需的剂量。在一个实施方式中,无活性的核可以是糖球或缓冲晶体或封装缓冲晶体,如碳酸钙、碳酸氢钠、富马酸、酒石酸等,它们改变药物的微环境以促进其释放。
所述延长释放的制剂可使用各种延长释放包衣或有助于活性剂随时间逐渐释放的机制。在一些实施方式中,延长释放的制剂包含通过控制溶解释放而控制释放的聚合物。在特别的实施方式中,活性剂被并入含有不溶的聚合物和由不同厚度的聚合物材料包被的药物粒子或颗粒的基质中。聚合物材料可包括含有蜡状材料的类脂屏障,如巴西棕榈蜡、蜂蜡、鲸蜡、小烛树蜡、紫胶蜡(shellacwax)、可可豆脂、十六十八醇(cetostearylalcohol)、部分氢化的植物油、地蜡、石蜡、地蜡、肉豆蔻醇、硬脂醇、鲸蜡醇和硬脂酸,同时存在的还有表面活性剂,如聚氧乙烯失水山梨醇单油酸酯(polyoxyethylenesorbitanmonooleate)。当与水性介质(如生物体液)接触时,根据聚合物包衣的厚度,在预定的滞后时间以后,聚合物包衣被乳化或侵蚀。所述滞后时间与胃肠蠕动、ph值或在胃内的滞留时间(gastricresidence)无关。
在其他实施方式中,延长释放的制剂包含实现控制扩散释放的聚合物基质。所述基质可包含一种或多种亲水的和/或水溶胀性的形成基质的聚合物,依赖于ph值的聚合物和/或不依赖于ph值的聚合物。
在一个实施方式中,所述延长释放的制剂包含水溶性的或水溶胀性的形成基质的聚合物,非必须地包含一种或多种溶解度增强剂和/或释放促进剂。随着水溶性聚合物的增溶作用,活性剂溶解(如果可溶),且通过基质的含水部分逐渐扩散。由于更多的水渗入到基质核中,凝胶层随时间生长,增加了凝胶层的厚度并提供了药物释放的扩散屏障。随着外层变得完全水化,聚合物链完全舒展,且不能再保持凝胶层的完整性,导致在基质的表面上的外层水化的聚合物解开缠结和侵蚀。水继续通过凝胶层向核渗入,直到其完全被侵蚀。可溶的药物通过这种扩散和侵蚀的联合作用释放,而对于不溶性药物,不论剂量如何,侵蚀都是主要机制。
与此相似的,水溶胀性聚合物通常在生物体液中水化并溶胀形成匀质的基质结构,该结构在药物释放期间保持其形状,并作为用于药物的载体、溶解度增强剂和/或释放促进剂。初始的基质聚合物水化阶段导致了药物的缓慢释放(迟滞阶段)。一旦水溶胀性聚合物完全水化并溶胀,基质中的水可以同样地溶解药物物质,并使其通过基质包衣扩散出来。
此外,由于依赖ph值的释放促进剂的浸出,基质的孔隙率能够增加,从而以更快的速率释放药物。随后,药物释放速率变为恒定,且其成为通过水合的聚合物凝胶的药物扩散的函数。基质的释放速率依赖于不同的参数,包括聚合物类型和等级;药物溶解性和剂量;聚合物与药物的比例;填料类型和等级;聚合物与填料的比例;药物和聚合物的粒径;以及基质的孔隙率和形状。
示例性的亲水性的和/或水溶胀性的形成基质的聚合物包括,但不限于,纤维素聚合物,包括羟烷基纤维素和羧烷基纤维素,如羟丙基甲基纤维素(hpmc)、羟丙基纤维素(hpc)、羟乙基纤维素(hec)、甲基纤维素(mc)、羧甲基纤维素(cmc);粉状纤维素,如微晶纤维素、乙酸纤维素、乙基纤维素、其盐、及其组合物;藻酸盐;树胶,包括杂多糖胶以及同多糖胶,如黄原胶、黄芪胶、果胶、阿拉伯胶、梧桐胶、藻酸盐、琼脂、瓜尔胶、羟丙基瓜尔胶、硅酸镁铝、角叉藻聚糖、豆角胶、胶凝糖胶及其衍生物;丙烯酸树脂,包括丙烯酸、甲基丙烯酸、丙烯酸甲酯和甲基丙烯酸甲酯的聚合物和共聚物,以及交联的聚丙烯酸衍生物,如卡波姆(例如,
所述延长释放制剂可进一步包含至少一种粘合剂,该粘合剂能够使亲水性化合物交联以在水性介质(包含生物体液)中形成亲水性聚合物基质(即,凝胶基质)。
示例性的粘合剂包括同多糖,如半乳甘露聚糖胶、瓜尔胶、羟丙基瓜尔胶、羟丙基纤维素(hpc;如klucelexf)以及豆角胶。在其他实施方式中,所述粘合剂为海藻酸衍生物、hpc或微晶纤维素(mcc)。其他的粘合剂包括,但不限于,淀粉、微晶纤维素、羟丙基纤维素、羟乙基纤维素、羟丙基甲基纤维素和聚乙烯吡咯烷酮。
在一个实施方式中,引入方法为通过向惰性载体上喷射活性剂和粘合剂的悬浮液的药物分层。
所述粘合剂可以在珠型制剂中以约0.1wt%至约15wt%,且优选约0.2wt%至约10wt%的量存在。
在一些实施方式中,亲水性聚合物基质可进一步包含离子聚合物、非离子聚合物或不溶于水的疏水性聚合物,以提供更强大的凝胶层和/或减少基质中孔隙的数量和尺寸,从而减慢扩散和侵蚀速度以及伴随的活性剂的释放。这可以额外抑制最初爆发效果,并产生更稳定的活性剂的“零级释放”。
示例性的用于减慢溶出速率的离子聚合物包含阴离子聚合物和阳离子聚合物。示例性的阴离子聚合物包括,例如,羧甲基纤维素钠(nacmc);海藻酸钠;丙烯酸或卡波姆的聚合物(如
示例性的用于减慢溶出速率的非离子聚合物包括,例如,羟丙基纤维素(hpc)和聚氧化乙烯(peo)(例如,polyoxtm)。
示例性的疏水性聚合物包括乙基纤维素(例如,ethoceltm,
所述溶胀性聚合物可以以1wt%至50wt%,优选5wt%至40wt%,最优选5wt%至20wt%的比例并入制剂中。所述可溶胀性聚合物和粘合剂可以在造粒前或者造粒后并入制剂中。所述聚合物也可以分散在有机溶剂或水性醇中并在造粒期间被喷射。
示例性的释放促进剂包括依赖于ph值的肠溶聚合物,所述依赖于ph值的肠溶聚合物在ph值低于约4.0时保持完整,且在ph值高于4.0,优选高于5.0,最优选高于6.0时溶解,且认为其在本发明中作为释放促进剂是有用的。示例性的依赖ph值的聚合物包括,但不限于,甲基丙烯酸共聚物、甲基丙烯酸-甲基丙烯酸甲酯共聚物(如德国rohm股份有限公司的
这些聚合物可以单独使用或者组合使用,或者与上述以外的聚合物一起使用。优选的依赖ph值的肠溶聚合物为药学上可接受的甲基丙烯酸共聚物。这些共聚物为基于甲基丙烯酸和甲基丙烯酸甲酯的阴离子聚合物,且其优选具有约135,000的平均分子量。在这些共聚物中,自由羧基与甲酯化的羧基的比例的范围为,例如,1:1至1:3,如,大约1:1或1:2。此聚合物在商业上以
在一些实施方式中,所述基质可包括释放促进剂和溶解度增强剂的组合。所述溶解度增强剂可以是离子型或非离子型表面活性剂、络合剂、亲水性聚合物和ph值调节剂(如酸化剂和碱化剂)以及通过分子包埋增加难溶性药物的溶解度的分子。几种溶解度增强剂可以同时使用。
所述溶解度增强剂可包括表面活性剂,如多库酯钠;硫酸月桂酯钠;硬脂酰醇富马酸钠;吐温类
所述溶解度增强剂通常构成剂型的1wt%至80wt%,优选1wt%至60wt%,更优选1wt%至50wt%,且其可以以不同的方式并入。它们可以在造粒前以干燥或湿润的方式并入制剂中。它们也可以在其他材料造粒或者其他工艺以后加入制剂中。在造粒期间,溶解度增强剂可以以添加或者不添加粘合剂的溶液形式喷射。
在一个实施方式中,所述延长释放制剂包含不溶于水的透水聚合物包衣或者基质,其包括形成在活性核上的一种或多种不溶于水的透水膜。所述包衣可额外包括一种或多种水溶性的聚合物和/或一种或多种增塑剂。不溶于水的聚合物包衣包含用于释放核中的活性剂的隔离包衣,其中,与较高粘度等级相比,较低分子量(粘度)等级显示出更快的释放速率。
在一些实施方式中,不溶于水的成膜聚合物包括一种或多种烷基纤维素醚,如乙基纤维素及其混合物,(例如,等级为pr100、pr45、pr20、pr10和pr7的乙基纤维素;
在一些实施方式中,不溶于水的聚合物在不需要增塑剂的情况下提供了合适的性能(例如,延长释放特性、机械性能和包被性能)。例如,可以使用含有如下物质的包衣而无需增塑剂:聚乙酸乙烯酯(pva)、丙烯酸酯/甲基丙烯酸酯的中性共聚物(如,由evonikindustries的商业可用的eudragitne30d)、乙基纤维素与羟丙基纤维素共同应用、蜡等。
在另一个实施方式中,不溶于水的聚合物基质可进一步包括增塑剂。所需的增塑剂的含量取决于增塑剂、不溶于水聚合物的性能以及最终所需的包衣的性能。相对于包衣的总重量,增塑剂的合适的水平为约1wt%至约20wt%、约3wt%至约20wt%、约3wt%至约5wt%、约7wt%至约10wt%、约12wt%至约15wt%、约17wt%至约20wt%、或约1wt%、约2wt%、约3wt%、约4wt%、约5wt%、约6wt%、约7wt%、约8wt%、约9wt%、约10wt%、约15wt%或约20wt%,包括其中的所有范围和子范围。
示例性的增塑剂包括,但不限于,三乙酸甘油酯、乙酰单酸甘油乙酯、油(蓖麻油、氢化蓖麻油、葡萄籽油、芝麻油、橄榄油等)、柠檬酸酯、柠檬酸三乙酯、柠檬酸乙酰基三乙酯、柠檬酸乙酰基三丁酯、柠檬酸三丁酯、柠檬酸乙酰基三正丁酯、邻苯二甲酸二乙酯、邻苯二甲酸二丁酯、邻苯二甲酸二辛酯、对羟基苯甲酸甲酯、对羟基苯甲酸丙酯、对羟基苯甲酸丙酯、对羟基苯甲酸丁酯、癸二酸二乙酯、癸二酸二丁酯、甘油三丁酸酯、取代甘油三酯和甘油脂、单乙酰化和双乙酰化甘油脂(如
增塑剂可以是用于赋予其他的硬或脆的聚合物材料弹性的高沸点的有机溶剂,且其能影响活性剂的释放曲线。增塑剂通常会引起沿聚合物链的内聚的分子间作用力的减少,从而导致多种聚合物性能的变化,包括聚合物的抗拉强度的降低、伸长率的增加以及玻璃化转变温度或软化温度的下降。增塑剂的含量和选择可以影响片剂的硬度,例如,甚至可以影响其溶解或崩解性能,以及其物理和化学稳定性。一些增塑剂可以增加包衣的弹性和/或可挠性,从而降低包衣的脆性。
在另一个实施方式中,所述延长释放制剂包含至少两种形成凝胶的聚合物的组合,其包括至少一种非离子型形成凝胶的聚合物和/或至少一种阴离子型形成凝胶的聚合物。由形成凝胶的聚合物的组合形成的凝胶提供了控制释放,使得当制剂被摄入并接触到胃肠液时,最接近表面的聚合物水合以形成粘性凝胶层。由于高粘度,粘性层仅能逐渐溶解掉,以相同的过程露出下面的材料。因此,物质缓慢溶解掉,从而慢慢将活性成分释放到胃肠液。至少两种形成凝胶的聚合物的组合使得产生的凝胶的性能(如粘性)能够被操纵以提供所需的释放曲线。
在一个特定的实施方式中,所述制剂包含至少一种非离子型形成凝胶的聚合物和至少一种阴离子型形成凝胶的聚合物。在另一个实施方式中,所述制剂包含两种不同的非离子型形成凝胶的聚合物。在另一个实施方式中,所述制剂包含除了溶解度、粘度和/或分子量之外具有相同化学性质的非离子型的形成凝胶的聚合物的组合(例如不同粘度等级的羟丙基甲基纤维素的组合,如hpmck100和hpmck15m或hpmck100m)。
示例性的阴离子型形成凝胶的聚合物包括但不限于,羧甲基纤维素钠(nacmc);羧甲基纤维素(cmc);阴离子型多糖,如海藻酸钠、海藻酸、果胶、聚葡糖醛酸(聚α-和-β-1,4-葡萄糖醛酸)、聚半乳糖醛酸(果胶酸)、硫酸软骨素、角叉藻聚糖、帚叉藻聚糖(furcellaran);阴离子胶,如黄原胶;丙烯酸或卡波姆的聚合物(如
示例性的非离子型的形成凝胶的聚合物包括,但不限于,聚维酮(pvp,聚乙烯吡咯烷酮)、聚乙烯醇、pvp和聚乙酸乙烯酯的共聚物、hpc(羟丙基纤维素)、hpmc(羟丙基甲基纤维素)、羟乙基纤维素、羟甲基纤维素、明胶、聚环氧乙烷、阿拉伯树胶、糊精、淀粉、聚甲基丙烯酸羟乙酯(phema)、水溶性非离子型聚甲基丙烯酸酯及其共聚物、改性纤维素、改性多糖、非离子型胶、非离子型多糖和/或其混合物。
所述制剂可非必须地包含上述的肠溶聚合物,和/或至少一种赋形剂,如填料、粘合剂(如上所述)、崩解剂、和/或流动性助剂或助流剂。
示例性的填料包括但不限于,乳糖;葡萄糖;果糖;蔗糖;磷酸二钙;糖醇,也称“糖多醇”,如山梨醇、甘露醇(manitol)、拉克替醇、木糖醇、异麦芽酮糖醇(isomalt)、赤藓糖醇和氢化淀粉水解物(多种糖醇的混合物)、玉米淀粉、马铃薯淀粉、羧甲基纤维素钠、乙基纤维素、乙酸纤维素、肠溶聚合物,或其混合物。
示例性的粘合剂包括但不限于,水溶性亲水性聚合物,如聚维酮(pvp:聚乙烯吡咯烷酮),共聚维酮(聚乙烯吡咯烷酮和聚乙酸乙烯酯的共聚物),低分子量的hpc(羟丙基纤维素),低分子量的hpmc(羟丙基甲基纤维素),低分子量的羧甲基纤维素,乙基纤维素,明胶,聚环氧乙烷,阿拉伯树胶,糊精,硅酸镁铝,以及淀粉和聚甲基丙烯酸酯类,如eudragitne30d、eudragitrl、eudragitrs、eudragite,聚乙酸乙烯酯,肠溶聚合物,或其混合物。
示例性的崩解剂包括但不限于,低取代的羧甲基纤维素钠,交聚维酮(交联聚乙烯吡咯烷酮),羧甲基淀粉钠(淀粉乙醇酸钠),交联羧甲基纤维素钠(交联羧甲纤维素),预糊化淀粉(淀粉1500),微晶纤维素,不溶于水的淀粉,羧甲基纤维素钙,低取代羟丙基纤维素,以及硅酸镁或硅酸铝。
示例性的助流剂包括但不限于,镁、二氧化硅、滑石、淀粉、二氧化钛等。
在另一个实施方式中,所述延长释放制剂通过用包被材料以及非必须的成孔物和其他赋形剂包被含有水溶性/水分散性药物的颗粒而形成,所述颗粒如珠或珠群体(如上所述)。所述包被材料优选选自纤维素聚合物,如乙基纤维素(如,
在此,适合用在控制释放包衣中的成孔物可为有机或无机试剂,且包括能够在使用环境中从包衣溶解、提取或浸出的材料。示例性的成孔剂包括但不限于,有机化合物,如单糖、寡糖和多糖类,包括蔗糖、葡萄糖、果糖、甘露醇、甘露糖、半乳糖、山梨醇、支链淀粉和葡聚糖;在使用环境中可溶的聚合物,如水溶性亲水聚合物,羟基烷基纤维素,羧基烷基纤维素,羟丙基甲基纤维素,纤维素醚,丙烯酸类树脂,聚乙烯吡咯烷酮,交联聚乙烯吡咯烷酮,聚环氧乙烷,碳蜡(carbowaxes),聚羧乙烯等,二醇,多元醇,多元醇,聚亚烷基二醇,聚乙二醇,聚丙二醇,或其嵌段聚合物,聚二醇和聚(α-ω)亚烷基二醇;以及无机化合物,如碱金属盐类,碳酸锂、氯化钠、溴化钠、氯化钾、硫酸钾、磷酸钾、醋酸钠、柠檬酸钠、适当的钙盐、其组合物等。
所述控制释放包衣可进一步包含本领域公知的其它添加剂,如增塑剂、抗粘剂、助流剂(或流动性助剂)以及消泡剂。
在一些实施方式中,包被的颗粒或珠可能另外包括“外包衣”,以提供例如,防潮、减少静电、味道掩蔽、调味、着色和/或磨光或其它对珠的装饰作用。对于这样的外包衣,合适的包衣材料是本领域公知的,且包括,但不限于,纤维素聚合物,如羟丙基甲基纤维素、羟丙基纤维素和微晶纤维素或其组合(例如,多种
包被的颗粒或珠可额外包含增强剂,例如,其可示例性但不限制地为增强溶解剂、增强溶出剂、吸收促进剂、渗透促进剂、稳定剂、络合剂、酶抑制剂、p-糖蛋白抑制剂以及多药耐药蛋白抑制剂。或者,所述制剂也可包含与包衣的颗粒分开的增强剂,例如在单独的珠的群体中或者作为粉末。在另一个实施方式中,增强剂可以包含在包被颗粒上的单独的层中,可以在控制释放包衣的下方或上方。
在其他实施方式中,所述延长释放制剂被配制成通过渗透机制释放活性剂。例如,胶囊可制成单渗透单元,或者其可包含2、3、4、5或6个封装在硬明胶胶囊内的推拉单元,藉此,每个双层推拉单元包含渗透推层和药物层,且两者都被半透膜包围。在与药物层相邻的膜上钻通一个或多个孔。这层膜可另外被依赖ph值的肠溶包衣覆盖,以防止释放,直到胃排空后。明胶胶囊在摄入后立即溶解。随着推拉单元进入小肠,肠溶包衣分解,然后让液体流动以通过半透膜,使渗透推部分溶胀,从而迫使药物以一定速率通过小孔,所述速率由水通过半透膜的速率而精确控制。药物的释放可以以恒定速率进行长达24小时以上。
所述渗透推层包含一种或多种产生用于使水通过半透膜进入递送载体的核的驱动力的渗透剂。一类渗透剂包括水溶胀性亲水性聚合物,也被称为“渗透聚合物(osmopolymers)”和“水凝胶”,其包括但不限于,亲水性乙烯基和丙烯酸聚合物、如海藻酸钙的多糖,聚氧化乙烯(peo)、聚乙二醇(peg)、聚丙二醇(ppg)、聚(甲基丙烯酸2-羟乙酯)、聚(丙烯酸)、聚(甲基丙烯酸)、聚乙烯吡咯烷酮(pvp)、交联pvp、聚乙烯醇(pva)、pva/pvp共聚物、具有如甲基丙烯酸甲酯和醋酸乙烯酯的疏水单体的pva/pvp共聚物、含有大peo嵌段的亲水性聚氨酯、交联甲羧纤维素钠、角叉藻聚糖、羟乙基纤维素(hec)、羟丙基纤维素(hpc)、羟丙基甲基纤维素(hpmc)、羧甲基纤维素(cmc)和羧乙基纤维素(cec)、海藻酸钠、聚卡波非、明胶、黄原胶和淀粉羟乙酸钠。
另一类渗透剂包括酶原,其能够吸取水分以实现穿过半透膜的渗透压梯度。酶原的例子包括但不限于,无机盐,如硫酸镁、氯化镁、氯化钙、氯化钠、氯化锂、硫酸钾、磷酸钾、碳酸钠、亚硫酸钠、硫酸锂、氯化钾、和硫酸钠;糖,如右旋糖(dextrose)、果糖、葡萄糖(glucose)、肌醇、乳糖、麦芽糖、甘露醇、棉子糖、山梨醇、蔗糖、海藻糖和木糖醇;有机酸,如抗坏血酸、苯甲酸、富马酸、柠檬酸、马来酸、癸二酸、山梨酸、己二酸、依地酸、谷氨酸、对甲苯磺酸、琥珀酸和酒石酸;尿素;以及它们的混合物。
有助于形成半透膜的材料包括不同级别的丙烯酸树脂、乙烯树脂、醚类、聚酰胺类、聚酯类和纤维素衍生物,这些材料在生理相关的ph值下透水且不溶于水,或者,这些材料易于通过化学改变,例如交联而呈现不溶于水的性能。
在一些实施方式中,所述延长释放制剂包括抵抗胃和肠内的侵蚀的多糖包衣。这种聚合物只能在包含了大量的微生物的结肠中降解,所述微生物含有可生物降解的酶,所述可生物降解的酶分解例如多糖包衣,从而以可控的依赖时间的方式释放药物内容物。示例性的多糖包衣可包括,例如,直链淀粉、阿拉伯半乳聚糖、壳聚糖、硫酸软骨素、环糊精、葡聚糖、瓜尔豆胶、果胶、木聚糖及其组合或衍生物。
在一些实施方式中,所述药物组合物被配制成延迟的延长释放。在此使用的术语“延迟的延长释放”参照具有这样的释放曲线的药物制剂使用:在所述释放曲线中,在施用后药物的释放中具有预设定的延迟,并且一旦开始,药物就在延长的时段内被连续释放。在一些实施方式中,延迟的延长释放制剂包括由肠溶包衣包被的延长释放制剂,这是应用于口服药物的隔离,以防止在药物到达小肠之前释放。如肠溶包衣的延迟释放制剂防止对胃有刺激作用的药物(如阿司匹林)在胃中溶解。这种包衣还用于保护对酸不稳定的药物,防止其暴露在胃的酸性环境中,而是将其递送到碱性ph环境中(肠道的ph值为5.5以上),在所述碱性环境下其不会降解,并给予他们所需的作用。术语“脉冲式释放”是延迟释放的一种,其在此参照以下使用:在预定的迟滞期以后,在短时期内立即提供迅速且瞬时的药物释放的药物制剂,从而产生在施用药物后药物的“脉冲”血浆分布。制剂可以被设计为在施用后预定的时间间隔下提供单脉冲式释放或多脉冲式释放,或者在一个时段的延长释放(例如,活性成分剩余部分的连续释放)后提供脉冲式释放(例如,活性成分的20-60%)。延迟释放或脉冲式释放制剂通常包括一种或多种由隔离包衣覆盖的成分,其在一个特定的迟滞期以后溶解、侵蚀或破裂。
根据目的,用于延迟释放的隔离包衣可以由各种不同的材料组成。此外,制剂可包括多个隔离包衣以助于以时间方式释放。该包衣可以是糖包衣、薄膜包衣(例如,基于羟丙基甲基纤维素、甲基纤维素、甲基羟乙基纤维素、羟丙基纤维素、羧甲基纤维素、丙烯酸酯共聚物、聚乙二醇和/或聚乙烯吡咯烷酮)或基于甲基丙烯酸共聚物、邻苯二甲酸乙酸纤维素、羟丙基甲基纤维素邻苯二甲酸酯、羟丙基甲基纤维素醋酸酯琥珀酸酯、聚醋酸乙烯邻苯二甲酸酯、虫胶和/或乙基纤维素的包衣。此外,所述制剂可另外包括时间延迟材料,例如,单硬脂酸甘油酯或二硬脂酸甘油酯。
在一些实施方式中,所述延迟的延长释放制剂包括肠溶包衣,所述肠溶包衣包含有助于活性剂在胃肠道的近端或远端区域中释放的一种或多种聚合物。在此使用的术语“肠溶聚合物包衣”是指包含具有依赖ph或者不依赖ph值的释放曲线的一种或多种聚合物的包衣。肠溶包衣的药丸将不溶于酸性胃液中(ph值~3),但它们会溶于小肠或结肠中存在的碱性(ph值7-9)环境中。肠溶聚合物包衣通常抑制活性剂释放,直到在施药后约3~4小时的胃排空的迟滞期后的某时。
依赖ph值的肠溶包衣包含一种或多种依赖ph值或对ph值敏感的聚合物,其能在较低ph值的条件下(如在胃部)保持它们的结构完整,而在胃肠道更远端的区域(如小肠)的较高ph值的环境下溶解,从而释放出药物内容物。对于本发明的目的,“依赖ph值”被定义为具有根据环境ph值而变化的特性(例如,溶解)。早期已描述示例性的依赖ph值的聚合物。依赖ph值的聚合物通常显示出特有的对于溶解的最适ph值。在一些实施方式中,依赖ph的聚合物显示出在约5.0和5.5之间、约5.5和6.0之间、约6.0和6.5之间或约6.5和7.0之间的最适ph值。在另一些的实施方式中,依赖ph的聚合物显示出≥5.0、≥5.5、≥6.0、≥6.5或≥7.0的最适ph值。
在一些实施方式中,包衣方法采用一种或多种依赖ph值和一种或多种不依赖ph值的聚合物的混合。一旦可溶性聚合物到达了其溶解的最适ph值,依赖ph值和不依赖ph值的聚合物的混合可以减小活性成分的释放速率。
在一些实施方式中,“时间控制的”或“依赖时间的”释放曲线可以使用包含一种或多种活性剂的不溶于水的胶囊体而获得,其中,所述胶囊体在其一端以不溶的、但可渗透的且可溶胀的水凝胶塞封闭。当与胃肠液或溶解介质接触时,所述塞溶胀,将其自身推出胶囊,并在预定的延滞时间(该时间可通过,例如,塞的位置和尺寸控制)以后,释放药物。所述胶囊体可以进一步由保持胶囊完整的外部的依赖ph值的肠溶包衣包被,直到其到达小肠。合适的塞的材料包含,例如,聚甲基丙烯酸酯类、可侵蚀的压缩聚合物(例如,hpmc,聚乙烯醇)、凝结的熔融聚合物(如甘油单油酸酯)和酶控制的可侵蚀的聚合物(例如,多糖,如直链淀粉、阿拉伯半乳聚糖、壳聚糖、硫酸软骨素、环糊精、葡聚糖、瓜尔豆胶、果胶和木聚糖)。
在另一些的实施方式中,胶囊或双层片可被配制成包含含有药物的核,其由溶胀层以及外部不溶但可半渗透的聚合物包衣或膜覆盖。在破裂前的延滞时间可通过聚合物包衣的渗透和机械性能,以及溶胀层的溶胀行为所控制。通常,溶胀层包含一种或多种溶胀剂,如溶胀且在其结构中保留水分的可溶胀的亲水聚合物。
在延迟释放的包衣中使用的示例性的可水溶胀的材料包含,但不限于,聚环氧乙烷(例如,平均分子量在1,000,000和7,000,000之间,例如,如
或者,药物的释放时间可通过崩解延滞时间来控制,所述崩解延滞时间取决于不溶于水聚合物膜(如乙基纤维素,ec)的耐受性和厚度之间的平衡,所述不溶于水聚合物膜包含在主体底部的预定的微孔,以及一定量的可溶胀的辅料,如低级取代的羟丙基纤维素(l-hpc)和乙醇酸钠。口服给药后,胃肠液渗透通过微孔,造成可溶胀的辅料的溶胀,这样产生使胶囊部分崩解的内部压力,所述胶囊部分包含含有可溶胀的材料的第一胶囊体、含有药物的第二胶囊体和附着在第一胶囊体上的外盖。
肠溶层可进一步包含抗粘剂,如滑石或甘油单硬脂酸酯和/或增塑剂。肠溶层可进一步包含一种或多种增塑剂,所述增塑剂包括但不仅限于,柠檬酸三乙酯、柠檬酸乙酰基三乙酯、柠檬酸乙酰基三丁酯、聚乙二醇乙酰化甘油一酯、甘油、三醋酸甘油酯、丙二醇、邻苯二甲酸酯(如邻苯二甲酸二乙酯、邻苯二甲酸二丁酯)、二氧化钛、氧化铁、蓖麻油、山梨醇和癸二酸二丁酯。
在另一个实施方式中,延迟释放制剂采用了透水但不溶的薄膜包衣以封装活性成分,以及渗透剂。随着水从肠道通过薄膜慢慢扩散进入核,所述核溶胀直到膜破裂,从而释放活性成分。可以调整膜包衣,以获得不同速率的水渗透或释放时间。
在另一个实施方式中,延迟释放制剂采用了不透水的片状包衣,藉此,水通过包衣中的控制孔进入,直到核突然破裂。当片剂突然破裂时,药物内容物立即释放,或者经过较长的时间释放。可修改这些和其他技术,以在药物开始释放前允许形成预定的迟滞期。
在另一个实施方式中,活性剂以制剂形式被递送,从而提供延迟释放和延长释放(延迟-持续)。术语“延迟-延长-释放”在此参照以下使用:在施用后的一个预定的时间或者迟滞期,提供活性剂的脉冲式释放的药物制剂,然后是活性剂的延长释放。
在一些实施方式中,立即释放、延长释放、延迟释放或延迟-延长释放制剂包括活性核,所述活性核由一种或多种惰性粒子组成,各自以其表面上包被有药物(例如,以含药物的成膜组合物的形式(使用例如流化床技术或本领域的技术人员公知的其他方法))的珠、丸剂、药丸、颗粒粒子、微胶囊、微球、微颗粒、纳米胶囊或纳米球的形式。所述惰性粒子可以是不同大小的,只要其足够大而保持不易溶解。或者,所述活性核可以通过含有药物成分的聚合物组合物的造粒和碾磨和/或通过挤出和滚圆来制备。
在核中的药物量将取决于所需要的剂量,且通常从约5至90wt%变化。通常,基于包衣粒子的重量,根据所需的延滞时间和释放曲线的类型和/或所选择的聚合物和包衣溶剂,在活性核上的聚合物包衣将为约1至50%。本领域的技术人员将能够选择合适量的在核上包被或引入核以实现所需的剂量的药物。在一个实施方式中,无活性的核可以是糖球或缓冲晶体或封装缓冲晶体,如碳酸钙、碳酸氢钠、富马酸、酒石酸等,它们改变药物的微环境以促进其释放。
在一些实施方式中,例如,延迟释放或延迟-延长释放组合物是通过以不溶于水的聚合物和肠溶聚合物的混合物包被水溶性/可分散的含药颗粒(如珠)而形成,其中,不溶于水的聚合物和肠溶聚合物可以4:1至1:1的重量比存在,且基于包被的珠的总重量,包衣的总重量为10至60wt%。药物分层的珠可非必须地包括控制内溶出率的乙基纤维素膜。优化外层的组合物,以及聚合物膜的内层和外层的单独的重量,以对于给定的活性实现理想的昼夜节律释放曲线,这基于体外/体内的相关性而预计得到。
在另一些实施方式中,所述制剂可包含含有立即释放的药物的颗粒(没有控制溶出速率的聚合物膜)以及延迟-延长释放的珠(其显示出,例如在口服施药以后2至4小时的延滞时间)的混合物,从而提供双脉冲释放曲线。
在一些实施方式中,活性核由一种或多种控制溶出速率的聚合物的层包被,从而获得理想的释放曲线(有或没有延滞时间)。内层膜可以在吸取水或体液进入核后在很大程度上控制药物释放速率,而外层膜可以提供所需的延滞时间(在吸取水或体液进入核后没有或很少的药物释放的时期)。内层膜可包括不溶于水的聚合物,或不溶于水的聚合物和水溶性聚合物的混合物。
如上所述,在很大程度上控制延滞时间最长达6小时的适合于外膜的聚合物包括肠溶聚合物,以及10至50wt%的不溶于水的聚合物。不溶于水的聚合物与肠溶聚合物的比可从4:1至1:2变化;优选所述聚合物以约1:1的比例存在。通常使用的不溶于水的聚合物为乙基纤维素。
不溶于水的聚合物的例子包括乙基纤维素、聚醋酸乙烯酯(来自basf的kollicoatsr#0d)、基于丙烯酸乙酯和甲基丙烯酸甲酯的中性共聚物、具有季铵基团的丙烯酸酯和甲基丙烯酸酯的共聚物(如
在一些实施方式中,使用amberlitetmirp69树脂作为延长释放的载体。amberlitetmirp69是一种不溶的强酸性钠型阳离子交换树脂,其适合作为阳离子(碱性)物质的载体。在其他实施方式中,使用duolitetmap143/1093树脂作为延长释放的载体。duolitetmap143/1093是一种不溶的强碱性阴离子交换树脂,其适合作为阴离子(酸性)物质的载体。
当作为药物载体使用时,amberliteirp69或/和duolitetmap143/1093树脂提供了向不溶性聚合物基质上粘合药剂的方法。通过形成树脂-药物复合物(药物树脂酸盐(酯))实现延长释放。随着药物与高电解质浓度达到平衡,药物在体内从树脂中释放,这是典型的胃肠道药物释放。由于与阳离子交换系统的芳香结构的疏水相互作用,通常,更多的疏水性药物会以较低的速率从树脂中洗脱。
在一些实施方式中,所述药物组合物被配制成口服施用。口服剂型包括,例如,片剂、胶囊和锭剂,并且也可以包括多个可被胶囊封装或者不被胶囊封装颗粒、珠、粉末或丸剂。片剂和胶囊代表最方便的口服剂型,在此情况下使用固体的药物载体。
在延迟释放制剂中,可以向丸剂、片剂或胶囊应用一个或多个隔离包衣,以助于减缓药物在肠道中的溶解和相伴释放。通常情况下,隔离包衣包含一种或多种聚合物,在药物组合物或活性核周围,包围、围绕或形成层或膜。
在一些实施方式中,在制剂中活性剂被递送,从而在施用后的预定的时间提供延迟释放。延迟可能高达约10分钟、约20分钟、约30分钟、约1小时、约2小时、约3小时、约4小时、约5小时、约6小时或更长的时间。
不同的包衣技术可应用于含有活性剂的颗粒、珠、粉末或丸剂、片剂、胶囊或其结合,以产生不同的和区别的释放曲线。在一些实施方式中,药物组合物是以包含单包衣层的片剂或胶囊的形式。在另一些实施方式中,药物组合物是以包含多包衣层的片剂或胶囊的形式。在一些实施方式中,本申请的药物组合物被配制成100%的活性成分的延长释放或延迟延长释放。
在其他实施方式中,本申请的药物组合物被配制成以在施用两小时内释放的“立即释放”组分和在2至12小时的时段内释放的“延长释放”组分为特征的两段延长释放或延迟两段延长释放。在一些实施方式中,“立即释放”组分提供活性剂的总剂量的约20-60%,且“延长释放”组分提供要由药物制剂递送的活性剂的总剂量的40-80%。例如,立即释放组分可提供要由药物制剂递送的活性剂的总剂量的约20-60%,或约20%、25%、30%、35%、40%、45%、50%、55%、60%。延长释放组分提供要由制剂递送的活性剂的总剂量的约40%、45%、50%、55%、60%、65%、70%、75%或80%。在一些实施方式中,立即释放组分和延长释放组分包含相同的活性成分。在其他实施方式中,立即释放组分和延长释放组分包含不同活性成分(例如,镇痛剂在一个组分中而抗毒蕈碱剂在另一个组分中)。在一些实施方式中,立即释放组分和延长释放组分各自包含选自阿司匹林、布洛芬、萘普生钠、吲哚美辛、萘丁美酮和对乙酰氨基酚的镇痛剂。在其他实施方式中,立即释放组分和/或延长释放组分进一步包含一种或多种选自抗毒蕈碱剂、抗利尿剂和解痉剂的附加的活性剂。
在一些实施方式中,所述药物组合物包含多种选自镇痛剂、抗毒蕈碱剂、抗利尿剂、解痉剂和5型磷酸二酯酶(pde5)抑制剂的活性成分。抗毒蕈碱剂的例子包括,但不限于:奥昔布宁、索非那新、达非那新、非索罗定、托特罗定、托斯必姆、阿托品和三环抗忧郁药。抗利尿剂的例子包括,但不限于,抗利尿激素(adh)、血管紧张素ⅱ、醛固酮、血管加压素、血管加压素类似物(例如,去氨加压素精氨加压素、赖氨加压素、苯赖加压素、鸟氨加压素、特利加压素);血管加压素受体激动剂、心房利钠肽(anp)和c型利钠肽(cnp)受体(即,npr1、npr2和npr3)拮抗剂(例如,hs-142-1、靛红、[asu7,23’]b-anp-(7-28)]、安南汀(anantin)、来自天蓝色链霉菌(streptomycescoerulescens)的环肽,以及3g12单克隆抗体);促生长素抑制素2型受体拮抗剂(如,促生长素抑制素),其药学上可接受的衍生物、类似物、盐、水合物和溶剂合物。解痉剂的例子包括,但不限于,卡立普多、苯(并)二氮卓类、巴氯芬、环苯扎珠、美他沙酮、美索巴莫、可乐定、可乐定类似物和丹曲洛林。5型磷酸二酯酶(pde5)抑制剂的例子包括,但不限于,他达拉非、西地那非和伐地那非。
在一些实施方式中,所述药物组合物包含一种或多种镇痛剂。在其它一些实施方式中,所述药物组合物包含:(1)一种或多种镇痛剂,和(2)一种或多种选自抗毒蕈碱剂、抗利尿剂、解痉剂和pde5抑制剂的其他活性成分。在另一个实施方式中,所述药物组合物包含(1)一种或多种镇痛剂和(2)一种或多种抗毒蕈碱剂。在另一个实施方式中,所述药物组合物包含(1)一种或多种镇痛剂和(2)一种或多种抗利尿剂。在另一个实施方式中,所述药物组合物包含(1)一种或多种镇痛剂和(2)一种或多种解痉剂。在另一个实施方式中,所述药物组合物包含(1)一种或多种镇痛剂和(2)一种或多种pde5抑制剂。在另一个实施方式中,所述药物组合物包含(1)一种或两种镇痛剂,(2)一种或两种抗毒蕈碱剂,和(3)一种或两种抗利尿剂。在另一个实施方式中,所述药物组合物包含(1)一种或两种镇痛剂,(2)一种或两种抗毒蕈碱剂,和(3)一种或两种解痉剂。在另一个实施方式中,所述药物组合物包含(1)一种或两种镇痛剂,(2)一种或两种抗毒蕈碱剂,和(3)一种或两种pde5抑制剂。在另一个实施方式中,所述药物组合物包含(1)一种或多种镇痛剂,(2)一种或多种抗利尿剂,和(3)一种或多种解痉剂。在另一个实施方式中,所述药物组合物包含(1)一种或多种镇痛剂,(2)一种或多种抗利尿剂,和(3)一种或多种pde5抑制剂。在另一个实施方式中,所述药物组合物包含(1)一种或多种镇痛剂,(2)一种或多种解痉剂,和(3)一种或多种pde5抑制剂。
在一个实施方式中,多数活性成分被配制成立即释放。在另外一些实施方式中,多数活性成分被配制成延长释放。在另外一些实施方式中,多数活性成分被配制成立即释放和延长释放(例如,每个活性成分的第一部分被配制成立即释放,而每个活性成分的第二部分被配制成延长释放)。在又一个实施方式中,多数活性成分中的一些被配制成立即释放,而多数活性成分中的一些被配制成延长释放(例如,活性成分a、b、c被配制成立即释放,而活性成分c和d被配制成延长释放)。在另外的一些实施方式中,立即释放组分和/或延长释放组分被进一步包被上延迟释放包衣(如肠溶包衣)。
在某些实施方式中,所述药物组合物包含立即释放组分和延长释放组分。该立即释放组分可包含一种或多种选自镇痛剂、抗毒蕈碱剂、抗利尿剂、解痉剂和pde5抑制剂的活性成分。该延长释放组分可包含一种或多种选自镇痛剂、抗毒蕈碱剂、抗利尿剂、解痉剂和pde5抑制剂的活性成分。在一些实施方式中,该立即释放组分和延长释放组分恰好具有相同的活性成分。在另外一些实施方式中,该立即释放组分和延长释放组分具有不同的活性成分。在另外一些实施方式中,该立即释放组分和延长释放组分具有一种或多种共同的活性成分。在一些实施方式中,立即释放组分和/或延长释放组分被进一步包被上延迟释放包衣(如肠溶包衣)。
在一个实施方式中,所述药物组合物包含两种或更多种被配制成在大约相同的时间立即释放的活性成分(例如,两种或更多种的镇痛剂,或者一种或多种的镇痛剂和一种或多种抗毒蕈碱剂或抗利尿剂或解痉剂或pde5抑制剂的混合物)。在另一个实施方式中,所述药物组合物包含两种或更多种被配制成在大约相同的时间延长释放的活性成分。在另一个实施方式中,所述药物组合物包含两种或更多种活性成分,该活性成分被配制成两种延长释放组分,各延长释放组分提供不同的延长释放曲线。例如,第一延长释放组分在第一释放速率下释放第一活性成分,而第二延长释放组分在第二释放速率下释放第二活性成分。在另一个实施方式中,所述药物组合物包含两种或更多种均被配制成延迟释放的活性成分。
在另一个实施方式中,所述药物组合物包含两种或更多种被配制成延迟释放的活性成分。在另一个实施方式中,所述药物组合物包含两种或更多种被配制成两种延迟释放组分的活性成分,各延迟释放组分提供不同的延迟释放曲线。例如,第一延迟释放组分在第一时间点释放第一活性成分,而第二延迟释放组分在第二时间点释放第二活性成分。
在其它一些实施方式中,所述药物组合物包含被配制成立即释放的两种活性成分(例如,两种镇痛剂,或一种镇痛剂和一种抗毒蕈碱剂或抗利尿剂或解痉剂或pde5抑制剂的混合物),和(2)被配制成延长释放的两种活性成分(例如,两种镇痛剂,或一种镇痛剂和一种抗毒蕈碱剂或抗利尿剂或解痉剂或pde5抑制剂的混合物)。在其它一些实施方式中,所述药物组合物包含被配制成立即释放的三种活性成分,和(2)被配制成延长释放的三种活性成分。在其它一些实施方式中,所述药物组合物包含被配制成立即释放的四种活性成分,和(2)被配制成延长释放的四种活性成分。在这些实施方式中,在立即释放组分中的活性成分可以与在延长释放组分中的活性成分相同或不同。在其它一些实施方式中,所述立即释放组分和/或所述延长释放组分可以进一步被延迟释放包衣(例如肠溶包衣)包被。
在一些实施方式中,所述药物组合物包含一种或多种镇痛剂;和一种抗利尿剂,其中,所述一种或多种镇痛剂被配制成延迟释放,以及其中,所述抗利尿剂被配制成立即释放。在其它一些实施方式中,所述药物组合物进一步包含选自抗毒蕈碱剂、抗利尿剂、解痉剂和pde5抑制剂的附加剂,其中,所述附加剂被配制成延迟释放。在一些实施方式中,所述延迟释放制剂延迟所述活性成分(例如,镇痛剂、抗毒蕈碱剂、抗利尿剂、解痉剂和/或pde5抑制剂)的释放达1、2、3、4或5小时。
立即释放组合物可包含在单一单位剂量中施用的给定活性剂的总剂量的100%。或者,可以包含一种立即释放组分作为组合释放曲线制剂中的组分,其中所述组合释放制剂可以提供将通过药物制剂递送的所述活性剂的总剂量的约1%至约90%,例如所述立即释放组分可以提供将通过制剂递送的所述活性剂的总剂量的=约5%-90%、约5%-80%、约5%-70%、约5%-60%、约5%-50%、约5%-40%、约5%-30%、约5%-20%、约10%-90%、约10%-80%、约10%-70%、约10%-60%、约10%-50%、约10%至约40%、约10%至约30%、约10%至约20%、约20%至约90%、约20%-80%、约20%-70%、20%至约60%、约20%至约50%、约20%至约30%、30%至约90%、约30%-80%、约30%-70%、约30%至约60%、约30%至约50%、约30%至约40%、约40%至约90%、约40%至约80%、约40%至约70%、约40%至约60%、约40%至约50%、约50%至约90%、约50%至约80%、约50%至约70%、约50%至约60%、约60%至约90%、约60%至约80%、约60%至约70%、约70%至约90%、约70%至约80%或约80%至约90%。在另外一些实施方式中,立即释放组分提供将要通过制剂递送的活性剂的总剂量的约2、4、5、10、15、20、25、30、35、40、45、50、55、60、65、70、75、80、85、90%。
在一些实施方式中,立即释放、延迟释放、延长释放或延迟延长释放制剂包括活性核,所述活性核由一种或多种惰性粒子组成,所述惰性粒子各自以其表面上包被有药物(例如,以含药物的成膜组合物的形式(使用例如流化床技术或本领域的技术人员公知的其他方法))的珠、丸剂、药丸、颗粒粒子、微胶囊、微球、微颗粒、纳米胶囊或纳米球的形式。所述惰性粒子可以是不同大小的,只要其足够大而保持不易溶解即可。或者,所述活性核可以通过含有药物成分的聚合物组合物的造粒和碾磨和/或通过挤出和滚圆来制备。在一些实施方式中,本申请的药物组合物的立即释放、延迟释放、延长释放或延迟延长释放制剂被配制成易于儿童(特别是,吞服药丸困难的幼儿)施用的液体剂型。在一些实施方式中,本申请的药物组合物被配制成延迟释放、延长释放或延迟延长释放的口服液。在一些实施方式中,所述口服液包含在液体介质中悬浮的含药物的颗粒粒子、微胶囊、微球、微颗粒、纳米胶囊或纳米球。在一些实施方式中,含药物的颗粒粒子、微胶囊、微球、微颗粒、纳米胶囊或纳米球可被配制成或包被成延迟释放、延长释放或延迟延长释放。在一个实施方式中,在药物组合物中的活性成分被质子化并与阳离子交换聚合物形成离子复合物。然后所述离子复合物被包被成延迟释放、延长释放或延迟延长释放。
在其他实施方式中,本申请的药物组合物的立即释放、延迟释放、延长释放或延迟延长释放制剂被配制成在口中迅速溶化的药丸。在一些实施方式中,所述药物组合物包含被配制或包被成延迟释放、延长释放或延迟延长释放的药物的含药物的颗粒粒子、微胶囊、微球、微颗粒、纳米胶囊或纳米球。当口服给药时,药丸在口中迅速溶化并释放所述含药物的颗粒粒子、微胶囊、微球、微颗粒、纳米胶囊或纳米球,基于预期的药物释放曲线(例如,延迟释放或延长释放)轮流释放他们携带的药物。
在所述核中的药物量将取决于所需要的剂量,且通常从约5至90wt%变化。通常,基于包衣粒子的重量,根据所需的延滞时间和释放曲线的类型和/或所选择的聚合物和包衣溶剂,在活性核上的聚合物包衣将为约1至50%。本领域的技术人员将能够选择合适量的在核上包被或引入核的药物以实现所需的剂量。在一个实施方式中,无活性的核可以是糖球或缓冲晶体或封装缓冲晶体,如碳酸钙、碳酸氢钠、富马酸、酒石酸等,它们改变药物的微环境以促进其释放。
在一些实施方式中,延迟释放制剂是通过以不溶于水的聚合物和肠溶聚合物的混合物包被水溶性/可分散的含药物的颗粒(如珠)而形成,其中,不溶于水的聚合物和肠溶聚合物可以4∶1至1∶1的重量比存在,且基于包被的珠的总重量,包衣的总重量为10至60wt%。药物分层的珠可非必须地包括控制内溶出率的乙基纤维素膜。优化外层的组合物,以及聚合物膜的内层和外层的单独的重量,以对于给定的活性实现理想的昼夜节律释放曲线,这基于体外/体内的相关性而预计得到。
在另一些实施方式中,所述制剂包含含有立即释放药物的颗粒(没有控制溶出速率的聚合物膜)和延迟释放的珠(其显示出,例如在口服施药以后2至4小时的延滞时间)的混合物,从而提供双脉冲释放曲线。在另一些实施方式中,所述制剂包括两种类型的延迟释放珠的混合物:显示出1至3小时延滞时间的第一种类型和显示出4至6小时延滞时间的第二种类型。在另一些实施方式中,所述制剂包括两种类型的释放珠的混合物:显示出立即释放的第一种类型和延长释放后显示出1-4小时延滞时间的第二种类型。
在另外一些实施方式中,制剂被设计为具有这样的释放曲线:使得药物的一部分(例如,20-60%)在施用后立即或两小时之内释放,剩余部分在延长的时段内释放。所述药物组合物可以日常施用或根据需要施用。在某些实施方式中,所述药物组合物被睡前施用于受试者。在一些实施方式中,所述药物组合物在睡前立即施用。在一些实施方式中,所述药物组合物在睡前大约两小时之内,优选睡前大约一小时之内施用。在另一个实施方式中,所述药物组合物在睡前大约两小时施用。在一个进一步的实施方式中,所述药物组合物在睡前至少两小时施用。在另一个实施方式中,所述药物组合物在睡前大约一小时施用。在一个进一步的实施方式中,所述药物组合物在睡前至少一小时施用。在又一个实施方式中,所述药物组合物在睡前立即施用。优选地,所述药物组合物口服施用。
在立即释放组分、延长释放组分、延迟释放组分或延迟延长释放组分中的活性剂的合适的剂量(“治疗有效量”),将取决于例如病情的严重程度和进程、施药方式、特别制剂的生物利用度、病人的年龄和体重、病人的临床病史和对活性剂的反应、医嘱,等等。
通常建议,无论一次或多次施用,立即释放组分、延长释放组分或延迟-延长-释放组分中的镇痛剂的治疗有效量在约10μg/kg体重/日至约100mg/kg体重/日的范围内施用。在一些实施方式中,以单剂或多剂每日施用的各活性剂的范围在约10μg/kg体重/日至约100mg/kg体重/日、10μg/kg体重/日至约30mg/kg体重/日、10μg/kg体重/日至约10mg/kg体重/日、10μg/kg体重/日至约3mg/kg体重/日、10μg/kg体重/日至约1mg/kg体重/日、10μg/kg体重/日至约300μg/kg体重/日、10μg/kg体重/日至约100μg/kg体重/日、10μg/kg体重/日至约30μg/kg体重/日、30μg/kg体重/日至约100mg/kg体重/日、30μg/kg体重/日至约30mg/kg体重/日、30μg/kg体重/日至约10mg/kg体重/日、30μg/kg体重/日至约3mg/kg体重/日、30μg/kg体重/日至约1mg/kg体重/日、30μg/kg体重/日至约300μg/kg体重/日、30μg/kg体重/日至约100μg/kg体重/日、100μg/kg体重/日至约100mg/kg体重/日、100μg/kg体重/日至约30mg/kg体重/日、100μg/kg体重/日至约10mg/kg体重/日、100μg/kg体重/日至约3mg/kg体重/日、100μg/kg体重/日至约1mg/kg体重/日、100μg/kg体重/日至约300μg/kg体重/日、300μg/kg体重/日至约100mg/kg体重/日、300μg/kg体重/日至约30mg/kg体重/日、300μg/kg体重/日至约10mg/kg体重/日、300μg/kg体重/日至约3mg/kg体重/日、300μg/kg体重/日至约1mg/kg体重/日、1mg/kg体重/日至约100mg/kg体重/日、1mg/kg体重/日至约30mg/kg体重/日、1mg/kg体重/日至约10mg/kg体重/日、1mg/kg体重/日至约3mg/kg体重/日、3mg/kg体重/日至约100mg/kg体重/日、3mg/kg体重/日至约30mg/kg体重/日、3mg/kg体重/日至约10mg/kg体重/日、10mg/kg体重/日至约100mg/kg体重/日、10mg/kg体重/日至约30mg/kg体重/日或30mg/kg体重/日至约100mg/kg体重/日。
此处所述的活性剂可被包含在日常单剂或多剂口服施用的立即释放组分或延长释放组分、延迟释放组分、延迟-延长释放组分或其联合中,该单剂或多剂的范围在1mg至1000mg,1mg至300mg,1mg至100mg,1mg至30mg,1mg至10mg,1mg至3mg,3mg至1000mg,3mg至300mg,3mg至100mg,3mg至30mg,3mg至10mg,10mg至1000mg,10mg至300mg,10mg至100mg,10mg至30mg,30mg至1000mg,30mg至300mg,30mg至100mg,100mg至1000mg,100mg至300mg或300mg至1000mg。正如所料,所述剂量将取决于患者的病情、尺寸、年龄和病情。
在一些实施方式中,所述药物组合物包含单一镇痛剂。在一个实施方式中,所述单一镇痛剂是阿司匹林。在另一个实施方式中,所述单一镇痛剂是布洛芬。在另一个实施方式中,所述单一镇痛剂是萘普生或萘普生钠。在另一个实施方式中,所述单一镇痛剂是吲哚美辛。在另一个实施方式中,所述单一镇痛剂是萘丁美酮。在另一个实施方式中,所述单一镇痛剂是对乙酰氨基酚。
在另一些实施方式中,所述药物组合物包含一对镇痛剂。这种配对的镇痛剂的例子包括,但不限于,乙酰水杨酸和布洛芬、乙酰水杨酸和萘普生钠、乙酰水杨酸和萘丁美酮、乙酰水杨酸和对乙酰氨基酚、乙酰水杨酸和吲哚美辛、布洛芬和萘普生钠、布洛芬和萘丁美酮、布洛芬和对乙酰氨基酚、布洛芬和吲哚美辛、萘普生、萘普生钠和萘丁美酮、萘普生钠和对乙酰氨基酚、萘普生钠和吲哚美辛、萘丁美酮和对乙酰氨基酚、萘丁美酮和吲哚美辛、以及对乙酰氨基酚和吲哚美辛。所述配对的镇痛剂以0.1∶1至10∶1、0.2∶1至5∶1或0.3∶1至3∶1的重量比混合。在一个实施方式中,所述配对的镇痛剂以1∶1的重量比混合。
在另一些实施方式中,本申请的所述药物组合物进一步包含一种或多种抗毒蕈碱剂。所述抗毒蕈碱剂的例子包括,但不限于奥昔布宁、索非那新、达非那新、非索罗定、托特罗定、托斯必姆、阿托品和三环抗忧郁药。抗毒蕈碱剂的日常剂量范围在1μg至100mg、1μg至30mg;1μg至10mg、1μg至3mg、1μg至1mg、1μg至300μg、1μg至100μg、1μg至30μg、1μg至10μg、1μg至3μg、3μg至100mg、3μg至30mg;3μg至10mg、3μg至3mg、3μg至1mg、3μg至300μg、3μg至100μg、3μg至30μg、3μg至10μg、10μg至100mg、10μg至30mg;10μg至10mg、10μg至3mg、10μg至1mg、10μg至300μg、10μg至100μg、10μg至30μg、30μg至100mg、30μg至30mg;30μg至10mg、30μg至3mg、30μg至1mg、30μg至300μg、30μg至100μg、100μg至100mg、100μg至30mg;100μg至10mg、100μg至3mg、100μg至1mg、100μg至300μg、300μg至100mg、300μg至30mg;300μg至10mg、300μg至3mg、300μg至1mg、1mg至100mg、1mg至30mg、1mg至3mg、3mg至100mg、3mg至30mg、3mg至10mg、10mg至100mg、10mg至30mg或30mg至100mg。
在另一些实施方式中,本申请的所述药物组合物进一步包含一种或多种抗利尿剂。所述抗利尿剂的例子包括,但不限于,抗利尿激素(adh)、血管紧张素ⅱ、醛固酮、血管加压素、血管加压素类似物(例如,去氨加压素精氨加压素、赖氨加压素、苯赖加压素、鸟氨加压素、特利加压素);血管加压素受体激动剂、心房利钠肽(anp)和c型利钠肽(cnp)受体(即,npr1、npr2、npr3)拮抗剂(例如,hs-142-1、靛红、[asu7,23’]b-anp-(7-28)]、安南汀(anantin)、来自天蓝色链霉菌(streptomycescoerulescens)的环肽,以及3g12单克隆抗体);促生长素抑制素2型受体拮抗剂(如,促生长素抑制素),其药学上可接受的衍生物、和类似物、盐、水合物及其溶剂合物。在一些实施方式中,一种或多种抗利尿剂包括去氨加压素。在其他实施方式中,一种或多种抗利尿剂是去氨加压素。抗利尿剂的日常剂量范围在1μg至100mg、1μg至30mg;1μg至10mg、1μg至3mg、1μg至1mg、1μg至300μg、1μg至100μg、1μg至30μg、1μg至10μg、1μg至3μg、3μg至100mg、3μg至30mg;3μg至10mg、3μg至3mg、3μg至1mg、3μg至300μg、3μg至100μg、3μg至30μg、3μg至10μg、10μg至100mg、10μg至30mg;10μg至10mg、10μg至3mg、10μg至1mg、10μg至300μg、10μg至100μg、10μg至30μg、30μg至100mg、30μg至30mg;30μg至10mg、30μg至3mg、30μg至1mg、30μg至300μg、30μg至100μg、100μg至100mg、100μg至30mg;100μg至10mg、100μg至3mg、100μg至1mg、100μg至300μg、300μg至100mg、300μg至30mg;300μg至10mg、300μg至3mg、300μg至1mg、1mg至100mg、1mg至30mg、1mg至3mg、3mg至100mg、3mg至30mg、3mg至10mg、10mg至100mg、10mg至30mg或30mg至100mg。
在另外一些实施方式中,本申请所述药物组合物进一步包含一种或多种解痉剂。解痉剂的例子包括,但不限于,卡立普多、苯(并)二氮卓类、巴氯芬、环苯扎珠、美他沙酮、美索巴莫、可乐定、可乐定类似物和丹曲洛林。在一些实施方式中,解痉剂以每日0.1mg至1000mg、0.1mg至300mg、0.1mg至100mg、0.1mg至30mg、0.1mg至10mg、0.1mg至3mg、0.1mg至1mg、0.1mg至0.3mg、0.3mg至1000mg、0.3mg至300mg、0.3mg至100mg、0.3mg至30mg、0.3mg至10mg、0.3mg至3mg、0.3mg至1mg、1mg至1000mg、1mg至300mg、1mg至100mg、1mg至30mg、1mg至10mg、1mg至3mg、3mg至1000mg、3mg至300mg、3mg至100mg、3mg至30mg、3mg至10mg、10mg至1000mg、10mg至300mg、10mg至100mg、10mg至30mg、30mg至1000mg、30mg至300mg、30mg至100mg、100mg至1000mg、100mg至300mg或300mg至1000mg的剂量使用。
在另一些实施方式中,本申请的所述药物组合物进一步包含一种或多种pde5抑制剂。pde5抑制剂的例子包括,但不限于,他达拉非、西地那非和伐地那非。在一些实施方式中,一种或多种pde5抑制剂包括他达拉非。在其他实施方式中,一种或多种pde5抑制剂是他达拉非。在一些实施方式中,pde5抑制剂以每日0.1mg至1000mg、0.1mg至300mg、0.1mg至100mg、0.1mg至30mg、0.1mg至10mg、0.1mg至3mg、0.1mg至1mg、0.1mg至0.3mg、0.3mg至1000mg、0.3mg至300mg、0.3mg至100mg、0.3mg至30mg、0.3mg至10mg、0.3mg至3mg、0.3mg至1mg、1mg至1000mg、1mg至300mg、1mg至100mg、1mg至30mg、1mg至10mg、1mg至3mg、3mg至1000mg、3mg至300mg、3mg至100mg、3mg至30mg、3mg至10mg、10mg至1000mg、10mg至300mg、10mg至100mg、10mg至30mg、30mg至1000mg、30mg至300mg、30mg至100mg、100mg至1000mg、100mg至300mg或300mg至1000mg的剂量使用。
所述抗毒蕈碱剂、抗利尿剂、解痉剂和/或pde5抑制剂可以单独配制成或与药物组合物中的其他活性成分一起被配制成立即释放、延长释放、延迟释放、延迟-延长-释放或其组合。
在某些实施方式中,所述药物组合物被配制成延长释放并包含(1)选自乙酰水杨酸、布洛芬、萘普生、萘普生钠、萘丁美酮、对乙酰氨基酚和吲哚美辛的镇痛剂,以及(2)抗利尿剂(如,去氨加压素)。
在一些实施方式中,所述药物组合物被配制成延长释放并包含两种或更多种镇痛剂。
在其它一些实施方式中,所述药物组合物被配制成延长释放并包含一种或多种的镇痛剂和一种或多种的抗毒蕈碱剂。
在一些实施方式中,所述药物组合物包含单一镇痛剂和单一抗利尿剂。在一个实施方式中,所述单一镇痛剂是阿司匹林。在另一个实施方式中,所述单一镇痛剂是布洛芬。在另一个实施方式中,所述单一镇痛剂是萘普生或萘普生钠。在另一个实施方式中,所述单一镇痛剂是吲哚美辛。在另一个实施方式中,所述单一镇痛剂是萘丁美酮。在另一个实施方式中,所述单一镇痛剂是对乙酰氨基酚。在另一个实施方式中,所述单一抗利尿剂是去氨加压素。所述镇痛剂和抗利尿剂可以以上述范围内的剂量给药。
在一些实施方式中,所述药物组合物包含一种或多种单独或联合的用量为10-1000mg,10-800mg,10-600mg,10-500mg,10-400mg,10-300mg,10-250mg,10-200mg,10-150mg,10-100mg30-1000mg,30-800mg,30-600mg,30-500mg,30-400mg,30-300mg,30-250mg,30-200mg,30-150mg,30-100mg,100-1000mg,100-800mg,100-600mg,100-400mg,100-250mg,300-1000mg,300-800mg,300-600mg,300-400mg,400-1000mg,400-800mg,400-600mg,600-1000mg,600-800mg或800-1000mg的镇痛剂,其中,所述组合物被配制成具有一种释放曲线的延长释放,在该释放曲线中,所述一种或多种镇痛剂在2-12小时或5-8小时的时段内被连续释放。
在一些实施方式中,所述组合物被配制成具有一种释放曲线的延长释放,在该释放曲线中,所述一种或多种镇痛剂的至少90%在2-12小时或5-8小时的时段内被连续释放。
在一些实施方式中,所述组合物被配制成具有一种释放曲线的延长释放,在该释放曲线中,所述一种或多种镇痛剂在5、6、7、8、10或12小时的时段内被连续释放。在一些实施方式中,所述药物组合物进一步包含抗毒蕈碱剂、抗利尿剂、解痉剂或pde5抑制剂。
在另外一些实施方式中,所述组合物被配制成具有一种释放曲线的延长释放,在该释放曲线中,所述镇痛剂在2-12小时或5-8小时的时段内以稳定的速率释放。在另外一些实施方式中,所述组合物被配制成具有释放曲线的延长释放,在该释放曲线中,所述镇痛剂在5、6、7、8、10或12小时的时段内以稳定的速率释放。此处使用的“在一个时段以稳定的速率”定义为一种释放曲线,在该释放曲线中,在给定时段的任意点的释放速率在该给定时段的平均释放速率的30%-300%内。例如,如果80mg阿司匹林在8小时时段内以稳定的速率释放,该时段内的平均释放速率为10mg/hr,那么在这个阶段任意时间的实际释放速率为在3mg/hr到30mg/hr的范围内(即,在8小时时段之内在10mg/hr平均释放速率的30%-300%内)。在一些实施方式中,所述药物组合物进一步包含抗毒蕈碱剂、抗利尿剂、解痉剂或pde5抑制剂。
在一些实施方式中,所述镇痛剂选自阿司匹林、布洛芬、萘普生钠、萘普生、吲哚美辛、萘丁美酮和对乙酰氨基酚。在一个实施方式中,所述镇痛剂是对乙酰氨基酚。所述药物组合物被配制成提供镇痛剂的小量稳定释放,从而维持血液中有效药物浓度,使得相比于立即释放制剂,单剂用药中的药物总量就减少了。
在另外的一些实施方式中,所述药物组合物包含一种或多种单独或联合的用量为10-1000mg,10-800mg,10-600mg,10-500mg,10-400mg,10-300mg,10-250mg,10-200mg,10-150mg,10-100mg,30-1000mg,30-800mg,30-600mg,30-500mg,30-400mg,30-300mg,30-250mg,30-200mg,30-150mg,30-100mg,100-1000mg,100-800mg,100-600mg,100-400mg,100-250mg,300-1000mg,300-800mg,300-600mg,300-400mg,400-1000mg,400-800mg,400-600mg,600-1000mg,600-800mg或800-1000mg的镇痛剂,其中,所述镇痛剂被配制成以一种两段释放曲线为特征的延长释放,在该释放曲线中,镇痛剂的20-60%在施用2小时内释放,而剩余部分在2-12小时或5-8小时的时段内连续地或以稳定的速率释放。在另外的一个实施方式中,所述镇痛剂被配制成具有一种两段释放曲线的延长释放,在该释放曲线中,镇痛剂的20%、30%、40%、50%或60%在施用2小时内释放,而剩余部分在2-12小时或5-8小时的时段内连续地或以稳定的速率释放。在一个实施方式中,所述镇痛剂选自阿司匹林、布洛芬、萘普生钠、萘普生、吲哚美辛、萘丁美酮和对乙酰氨基酚。在一个实施方式中,所述镇痛剂是对乙酰氨基酚。在另外的一个实施方式中,所述镇痛剂为对乙酰氨基酚。在一些实施方式中,所述药物组合物进一步包含抗毒蕈碱剂、抗利尿剂、解痉剂和/或pde5抑制剂。
所述药物组合物可被配制成片剂、胶囊剂、锭剂、粉剂、颗粒、液体、凝胶或乳液的形式。所述液体、凝胶或乳液可由受试者以直接形式或者包含在胶囊中的形式摄入。在一些实施方式中,本申请的药物组合物的立即释放、延迟释放、延长释放或延迟-延长释放制剂被配制成易于儿童(特别是,吞服药丸困难的幼儿)施用的液体剂型。
本申请的另一个方面涉及一种通过向有此需要的受试者可选择地施用两种或更多种镇痛剂来防止发生耐药性以治疗遗尿的方法。在一个实施方式中,该方法包括施用针对第一时段的第一镇痛剂,然后施用针对第二时段的第二镇痛剂。在另一个实施方式中,该方法进一步包括施用针对第三时段的第三镇痛剂。所述第一、第二和第三镇痛剂彼此不同,且其中至少一种被配制成延长释放或延迟的延长释放。在一个实施方式中,所述第一镇痛剂是对乙酰氨基酚,所述第二镇痛剂是布洛芬且所述第三镇痛剂是萘普生钠。各个时段的长短可以根据受试者对各个镇痛剂的反应有所不同。在一些实施方式中,每个时段持续时间从三天至三周。在另一个实施方式中,第一、第二和第三镇痛剂都被配制成延长释放或延迟的延长释放。
本申请的另一个方面涉及治疗受试者遗尿的方法,其包含向有此需要的受试者施用一种药物组合物,该药物组合物包含:活性成分,所述活性成分以每剂1-1000mg的量包含一种或多种镇痛剂,其中,一种或多种镇痛剂选自阿司匹林、布洛芬、萘普生、萘普生钠、吲哚美辛、萘丁美酮和对乙酰氨基酚。
在一个实施方式中,所述药物组合物被配制成延长释放,使得活性成分在2-12小时的时段内持续释放。在一个相关的实施方式中,所述药物组合物进一步用肠溶包衣包被。
在另一个实施方式中,所述药物组合物被配制成以一种两阶段释放曲线为特征的延长释放,在该释放曲线中,活性成分的20-60%在施用两小时内被释放,而活性成分的剩余部分在2-12小时的时段内被连续地释放。在一个相关实施方式中,所述药物组合物更进一步用肠溶包衣包被。
在另一个实施方式中,所述药物组合物被配制成延迟释放或立即释放。
在另一个实施方式中,一种或多种镇痛剂由对乙酰氨基酚组成。
在另一个实施方式中,一种或多种镇痛剂由3-100mg量的对乙酰氨基酚组成。
在另一个实施方式中,一种或多种镇痛剂由对乙酰氨基酚和选自阿司匹林、布洛芬、萘普生、萘普生钠、吲哚美辛和萘丁美酮的另一种镇痛剂组成。
在另一个实施方式中,所述活性成分进一步包含抗毒蕈碱剂。
在另一个实施方式中,所述活性成分进一步包含抗利尿剂。在一个相关实施方式中,抗利尿剂是去氨加压素。
在另一个实施方式中,所述活性成分进一步包含解痉剂。
在另一个实施方式中,所述活性成分进一步包含pde5抑制剂。在一个相关实施方式中,pde5抑制剂是他达拉非。
在另一个实施方式中,所述活性成分进一步包含选自抗毒蕈碱剂、抗利尿剂、解痉剂和pde5抑制剂的两种附加剂。
本申请的另一方面涉及一种治疗受试者遗尿的方法,该方法包含向有此需要的受试者施用一种药物组合物,该药物组合物包含:第一活性成分,其包含选自镇痛剂、抗毒蕈碱剂、抗利尿剂、解痉剂和pde5抑制剂的一种或多种;以及第二活性成分,其包含选自镇痛剂、抗毒蕈碱剂、抗利尿剂、解痉剂和pde5抑制剂的一种或多种,其中所述第一活性成分被配制成立即释放,并且其中,所述第二活性成分被配制成延长释放。
在一个实施方式中,所述药物组合物进一步用肠溶包衣包被。
在另一个实施方式中,所述第一活性成分包含镇痛剂。在一个相关实施方式中,所述第一活性成分进一步包含抗毒蕈碱剂。在另一个相关实施方式中,所述活性成分进一步包含抗利尿剂,如去氨加压素。在另一个相关实施方式中,所述第一活性成分进一步包含解痉剂。在另一个相关实施方式中,所述第一活性成分进一步包含pde5抑制剂,如他达拉非。
本申请的另一个方面涉及一种药物组合物,该药物组合物包含一种或多种镇痛剂、去氨加压素和药学可接受的载体。
在一个实施方式中,一种或多种镇痛剂被配制成延长释放,并且其中,去氨加压素被配制成立即释放。
在另一个实施方式中,一种或多种镇痛剂和去氨加压素被配制成在2-12小时的时段内的延长释放。
在另一个实施方式中,去氨加压素与一种或多种镇痛剂各自的20-60%被配制成立即释放,并且一种或多种镇痛剂各自的剩余部分被配制成延长释放。在一个相关的实施方式中,所述药物组合物用肠溶包衣包被。
本发明将通过以下的非限制性实施例进一步说明。在本申请中引用的所有参考文件、专利和公开的专利申请的内容都通过引用方式并入本申请。
实施例1:排尿冲动的抑制
招收男女都参加的20名志愿受试者,他们各自都经历了过早的排尿冲动或排尿需求,这干扰了他们足以感到充分休息的一段时间的睡眠的能力。每个受试者在就寝前以单剂量摄入400至800mg的布洛芬。至少有14个受试者报告说,因为没有被频繁的排尿冲动唤醒,他们能够更好地休息。
有几名受试者报告说,在夜间使用布洛芬几个星期以后,不再能实现排尿冲动不频繁的裨益。然而,所有这些受试者都进一步报告说,在放弃服用药剂几天以后,又获得了这样的裨益。更新近的测试确定了类似的结果可以在更低剂量下实现而无任何随之减少的裨益。
实施例2:镇痛剂、肉毒杆菌神经毒素和抗毒蕈碱剂对巨噬细胞对炎症和非炎症性刺激的反应的影响
实验设计
本研究旨在确定镇痛剂和抗毒蕈碱剂在控制巨噬细胞对由cox2和前列腺素(pge、pgh等)介导的炎症和非炎症性刺激的反应中的剂量和体外效力。它建立了对膀胱细胞中炎症和非炎症性效应物的基线(剂量和动力学)的反应。简言之,在不存在或存在各种效应物的情况下,使培养细胞暴露于镇痛剂和/或抗毒蕈碱剂。
所述效应物包括:脂多糖(lps)、发炎剂和cox2诱导物,作为炎症性刺激物;卡巴胆碱或乙酰胆碱、平滑肌收缩刺激剂,作为非炎症性刺激物;肉毒杆菌神经毒素a,一种已知的乙酰胆碱释放的抑制剂,作为阳性对照;花生四烯酸(aa),伽玛亚麻酸(dgla)或二十碳五烯酸(epa),作为前列腺素的前体,他们是在通过环氧合酶(cox1和cox2)和终端前列腺素合酶在细胞内依次氧化aa、dgla或epa之后而产生的。
所述镇痛剂包括:水杨酸盐,如阿司匹林;异丁基丙酸酚酸衍生物(布洛芬),如雅维(advil)、布洛芬制剂(motrin)、磺胺二甲噁唑(nuprin)和medipren;萘普生钠,如萘普生钠(aleve)、萘普生制剂(anaprox)、antalgin、feminaxultra、萘普生(flanax)、inza、midolextendedrelief、nalgesin、naposin、萘普生缓释片剂(naprelan)、naprogesic、萘普生(naprosyn)、萘普生(naprosyn)混悬液、ec-萘普生(ec-naprosyn)、narocin、萘普生(proxen)、synflex和xenobid;醋酸衍生物,如吲哚美辛(indocin);1-萘乙酸衍生物,如萘丁美酮或瑞力芬;n-乙酰基对氨基苯酚(apap)衍生物,如对乙酰氨基酚或扑热息痛(泰诺林);和塞来考昔。
所述抗毒蕈碱剂包括:奥昔布宁、索非那新、达非那新和阿托品。
使巨噬细胞受到以下物质的短期(1-2小时)或长期(24-48小时)刺激:
(1)不同剂量的每种单独的镇痛剂。
(2)在lps存在的情况下不同剂量的每种镇痛剂。
(3)在卡巴胆碱或乙酰胆碱存在的情况下不同剂量的每种镇痛剂。
(4)在aa、dgla或epa存在的情况下不同剂量的每种镇痛剂。
(5)不同剂量的单独的肉毒杆菌神经毒素a。
(6)在lps存在的情况下不同剂量的肉毒杆菌神经毒素a。
(7)在卡巴胆碱或乙酰胆碱存在的情况下不同剂量的肉毒杆菌神经毒素a。
(8)在aa、dgla或epa存在的情况下不同剂量的肉毒杆菌神经毒素a。
(9)不同剂量的单独的每种抗毒蕈碱剂。
(10)在lps存在的情况下不同剂量的每种抗毒蕈碱剂。
(11)在卡巴胆碱或乙酰胆碱存在的情况下不同剂量的每种抗毒蕈碱剂。
(12)在aa、dgla或epa存在的情况下不同剂量的每种抗毒蕈碱剂。
然后分析细胞的pgh2、pge、pge2、前列腺环素、血栓烷、il-1β、il-6、tnf-α的释放;cox2活性;camp和cgmp的产生;il-1β、il-6、tnf-α和cox2mrna的产生;以及cd80、cd86和mhcii类分子的表面表达。
巨噬细胞
在此研究中使用鼠科raw264.7或j774巨噬细胞(由atcc获得)。将细胞保持在含有rpmi1640的培养基中,并补充有10%胎牛血清(fbs)、15mmhepes、2mm左旋谷酰胺、100u/ml青霉素和100μg/ml的链霉素。将细胞在37℃,5%的co2气氛下培养,且每星期分离(传代)一次。
巨噬细胞以镇痛剂的体外治疗
将raw264.7巨噬细胞以1.5x105个细胞/孔(在100μl培养基中)的细胞密度接种在96孔板中。将细胞用以下物质处理:(1)不同浓度的镇痛剂(对乙酰氨基酚、阿司匹林、布洛芬或萘普生),(2)不同浓度的脂多糖(lps),它是对巨噬细胞的炎症性刺激的效应物,(3)不同浓度的卡巴胆碱或乙酰胆碱,它们是非炎症性刺激的效应物,(4)镇痛剂和lps或(5)镇痛剂和卡巴胆碱或乙酰胆碱。简言之,将镇痛剂溶解在无fbs的培养基中(即,补充有15mmhepes、2mm左旋谷酰胺、100u/ml青霉素和100μg/ml的链霉素的rpmi1640)并通过用相同介质的连续稀释稀释到所需浓度。对于在没有lps存在的情况下以镇痛剂处理的细胞,向每个孔中加入50μl的镇痛剂溶液和50μl的无fbs的培养基。对于在有lps存在的情况下以镇痛剂处理的细胞,向每个孔中加入50μl的镇痛剂溶液和50μl的在无fbs的培养基中的lps(来自鼠伤寒沙门氏菌(salmonellatyphimurium))。所有的条件重复测试两次。
在培养24或48小时后,收集150μl的培养上清液,在4℃,8,000rpm下旋转2分钟以除去细胞和碎片,并在-70℃储存以用于通过elisa分析细胞因子的反应。通过在500μl的磷酸盐缓冲液(pbs)中离心(在4℃,1,500rpm下5分钟)收集和洗涤细胞。然后将一半的细胞在液氮中快速冻结,并在-70℃下储存。将剩余的细胞用荧光单克隆抗体染色并通过流式细胞计分析。
辅刺激分子表达的流式细胞计分析
对于流式细胞计分析,将巨噬细胞在100μl的facs缓冲液(具有2%的牛血清白蛋白(bsa)和0.01%nan3的磷酸盐缓冲液(pbs))中稀释,并通过添加fitc-结合的抗cd40、pe-结合的抗cd80、pe-结合的抗-cd86抗体、抗mhcii类(i-ad)pe(bd生物科学)而在4℃下染色30分钟。然后将细胞通过在300μl的facs缓冲液中离心(在4℃,1,500rpm下5分钟)清洗。在第二次洗涤后,细胞重新悬浮在200μl的facs缓冲液中,且借助accuric6流式细胞计(bd生物科学)分析表达给定标记(单阳性)或者标记的组合(双阳性)的细胞的百分比。
通过elisa分析细胞因子的反应
对培养上清液进行细胞因子特异性elisa,以确定在用镇痛剂、lps单独处理或者lps和镇痛剂结合处理的巨噬细胞的培养物中的il-1β,il-6和tnf-α反应。这些测定是在用在0.1m的碳酸氢钠缓冲液(ph9.5)中的100μl的抗小鼠il-6、tnf-αmabs(bd生物科学)或il-1βmab(r&d系统)包被过夜的nuncmaxisorpimmunoplates(nunc)上进行的。在用pbs(每孔200μl)清洗两次以后,向每个孔(区)中添加200μl的pbs3%bsa,且在室温下孵育板2小时。通过每孔添加200μl,再次清洗板两次,重复添加100μl的细胞因子标准品和连续稀释的培养上清液,并将该板在4℃下孵育过夜。最后,将该板清洗两次,并用100μl生物素化的抗鼠il-6、tnfαmabs(bd生物科学)或il-1β(r&d系统)的二抗,随后用过氧化物酶标记的羊抗生物素mab(vector实验室)孵育。通过添加2,2′-连氮-双(3-乙基苄基噻唑啉-6-磺酸)(abts)底物和h2o2(sigma)而使比色反应显影,且吸光度使用
cox2的活性测定和camp和cgmp的生成
在培养的巨噬细胞中的cox2的活性通过顺序竞争elisa(r&d系统)确定。camp和cgmp的生成通过camp测定和cgmp测定来确定。这些测定在本领域中是通常进行的。
结果
表1总结了由raw264巨噬细胞株进行的实验以及在镇痛剂对辅刺激分子cd40和cd80的细胞表面表达的影响方面的主要发现。这些分子的表达是通过cox2和炎症信号来刺激的,且因此评估这些分子的表达以确定cox2的抑制的功能后果。
如表2所示,除了最高剂量(即,5x106nm)(其表现出增强,而不是抑制辅刺激分子的表达)以外,对乙酰氨基酚、阿司匹林、布洛芬和萘普生在所有的测试剂量(即,5x105nm、5x104nm、5x103nm、5x102nm、50nm和5nm)下抑制巨噬细胞的辅刺激分子cd40和cd80的基础表达。如图1a和1b所示,镇痛剂用量为低至0.05nm(即,0.00005μm)时观察到对cd40和cd50表达的这样的抑制效果。这一发现支持了这样的观点:小剂量的镇痛剂的控制释放比大剂量的急性递送更优选。实验还表明,对乙酰氨基酚、阿司匹林、布洛芬和萘普生对lps诱导的cd40和cd80的表达具有类似的抑制效果。
表1.实验总结
表2.主要发现的总结
*nd:未进行(毒性)
表3总结了几项研究的结果,这些研究测量了成人在口服治疗剂量后的镇痛剂的血清水平。如表3中所示,在口服治疗剂量后镇痛剂的最大血清水平在104至105nm范围内。因此,在表2中的体外测试的镇痛剂剂量覆盖了人体内可实现的浓度范围。
表3.口服治疗剂量后人血液中的镇痛剂的血清水平
实施例3:镇痛剂、肉毒杆菌神经毒素和抗毒蕈碱剂对小鼠膀胱平滑肌细胞对炎症和非炎症性刺激的反应的影响
实验设计
本研究旨在说明在实施例2中确定的镇痛剂的最优剂量如何影响在细胞培养或组织培养中的膀胱平滑肌细胞,并论述不同类的镇痛剂是否能够协同以更有效地抑制cox2和pge2反应。
在实施例2中描述了效应物、镇痛剂和抗毒蕈碱剂。
使小鼠膀胱平滑肌细胞的原代培养物受到以下物质的短期(1-2小时)或长期(24-48小时)刺激:
(1)不同剂量的每种单独的镇痛剂。
(2)在lps存在的情况下不同剂量的每种镇痛剂。
(3)在卡巴胆碱或乙酰胆碱存在的情况下不同剂量的每种镇痛剂。
(4)在aa、dgla或epa存在的情况下不同剂量的每种镇痛剂。
(5)不同剂量的单独的肉毒杆菌神经毒素a。
(6)在lps存在的情况下不同剂量的肉毒杆菌神经毒素a。
(7)在卡巴胆碱或乙酰胆碱存在的情况下不同剂量的肉毒杆菌神经毒素a。
(8)在aa、dgla或epa存在的情况下不同剂量的肉毒杆菌神经毒素a。
(9)不同剂量的单独的每种抗毒蕈碱剂。
(10)在lps存在的情况下不同剂量的每种抗毒蕈碱剂。
(11)在卡巴胆碱或乙酰胆碱存在的情况下不同剂量的每种抗毒蕈碱剂。
(12)在aa、dgla或epa存在的情况下不同剂量的每种抗毒蕈碱剂。
然后分析细胞的pgh2、pge、pge2、前列腺环素、血栓烷、il-1β、il-6、tnf-α的释放;cox2活性;camp和cgmp的产生;l-1β、il-6、tnf-α和cox2mrna的产生;以及cd80、cd86和mhcii类分子的表面表达。
材料和方法
小鼠膀胱细胞的分离和纯化
从被安乐死的动物c57bl/6小鼠(8-12周龄)取出膀胱细胞,且将细胞通过酶消化分离,随后用percoll梯度纯化。简言之,将从10只小鼠得到的膀胱用剪刀切碎为在10ml的消化缓冲液(rpmi1640、2%胎牛血清、0.5mg/ml胶原酶、30μg/ml的dna酶)中的精制浆液。将膀胱浆液在37℃下酶消化30分钟。将未消化的碎片通过细胞训练器(cell-trainer)进一步分散。使细胞混悬液沉淀,并加入到不连续的20%、40%和75%percoll梯度以纯化单核细胞。每个实验使用50-60个膀胱。
在用rpmi1640清洗后,将膀胱细胞再悬浮到补充有10%胎牛血清、15mmhepes、2mm左旋谷酰胺、100u/ml青霉素和100μg/ml的链霉素的rpmi1640中,并以3x104个细胞/孔(100μl)的细胞密度接种到澄清底部的黑色96孔细胞培养微培养平板中。将细胞在37℃,5%的co2气氛下培养。
细胞以镇痛剂的体外治疗
将膀胱细胞用镇痛剂溶液(50μl/孔)单独或者与卡巴胆碱(10摩尔,50μl/孔)(作为非炎症性刺激物的例子)共同处理,或者与鼠伤寒沙门氏菌的脂多糖(lps)(1μg/ml,50μl/孔)(作为非炎症性刺激物的例子)共同处理。当没有其他的效应物加入细胞中时,向孔中加入50μl的无胎牛血清的rpmi1640以调整最终体积为200μl。
在培养24小时后,收集150μl的培养上清液,在4℃,8,000rpm下旋转2分钟以除去细胞和碎片,并在-70℃储存以用于通过elisa分析前列腺素e2(pge2)的反应。将细胞固定、透化并封闭以使用荧光底物检测环氧合酶-2(cox-2)。在选定的实验中,细胞在体外刺激12小时以用于cox2反应的分析。
cox2反应分析
cox2反应通过使用人/小鼠总cox2免疫测定法(r&d系统)的基于细胞的elisa分析,所述分析根据制造商的说明书进行。简言之,在细胞固定和透化以后,向澄清底部的黑色96孔细胞培养微培养平板的孔中加入小鼠抗总cox2和兔抗总gapdh。经过培育和清洗后,向孔中加入hrp结合的抗小鼠igg和ap结合的抗兔igg。在另一个培育和清洗后,加入hrp-荧光底物和ap-荧光底物。最后,使用
pge2反应分析
前列腺素e2的反应通过顺序竞争elisa(r&d系统)分析。具体而言,向由山羊抗小鼠多克隆抗体包被的96孔聚苯乙烯微孔板的孔中加入培养上清液或pge2标准样。在微孔板振荡器上孵育一小时后,加入hrp结合的pge2,并将板在室温下额外孵育两小时。然后清洗板,并向每个孔中加入hrp底物溶液。容许显色30分钟,并通过在450nm(在570nm处校正波长)处读取板之前加入硫酸以停止反应。结果表示为pge2平均pg/ml。
其他实验
pgh2、pge、前列腺环素(prostacydin)、血栓烷、il-1β、il-6和tnf-α的释放;camp和cgmp的产生;il-1β、il-6、tnf-α和cox2mrna的产生;以及cd80、cd86和mhcii类分子的表面表达采用如实施例2中所述的方法确定。
镇痛剂抑制小鼠膀胱细胞对炎症性刺激的cox2反应
对几种镇痛剂(对乙酰氨基酚、阿司匹林、布洛芬和萘普生)在5μm或50μm的浓度下对小鼠膀胱细胞进行测试,以确定镇痛剂是否能诱发cox2反应。24小时培养的分析表明,所测试的镇痛剂均未诱导在体外小鼠膀胱细胞中的cox2反应。
表4.在体外刺激和镇痛剂处理后小鼠膀胱细胞的cox2表达
还测试了在体外这些镇痛剂对小鼠膀胱细胞对卡巴胆碱或lps刺激的cox2反应的影响。如表1所示,测试的卡巴胆碱的剂量对于小鼠膀胱细胞中的cox-2水平没有显著影响。另一方面,lps显著增加总cox2水平。值得注意的是,对乙酰氨基酚、阿司匹林、布洛芬和萘普生均能抑制lps对cox2水平的影响。当这些药物在5μm或50μm测试时,可以看出镇痛剂的抑制效果(表4)。
镇痛剂抑制小鼠膀胱细胞对炎症性刺激的pge2反应
测量在小鼠膀胱细胞培养上清液中的pge2的分泌,以确定因镇痛剂的小鼠膀胱细胞cox2水平改变的生物学意义。如表5所示,在未刺激的膀胱细胞或在卡巴胆碱的存在下培养的膀胱细胞的培养上清液中未检测到pge2。与上述的cox2反应相一致的,用lps刺激小鼠膀胱细胞诱导pge2的高水平分泌。镇痛剂对乙酰氨基酚、阿司匹林、布洛芬和萘普生的添加抑制了lps对pge2分泌的影响,且在用5或50μm剂量的镇痛剂处理的细胞反应之间并未观察到区别。
表5.在体外刺激和镇痛剂处理后小鼠膀胱细胞的pge2分泌
总之,这些数据表明仅用镇痛剂在5μm或50μm下不会诱导小鼠膀胱细胞中的cox2和pge2反应。然而,在5μm或50μm下,镇痛剂显著抑制体外由lps(1μg/ml)刺激的小鼠膀胱细胞的cox2和pge2反应。未观察到镇痛剂对由卡巴胆碱(1mm)刺激的小鼠膀胱细胞的cox2和pge2反应的显著影响。
实施例4:镇痛剂、肉毒杆菌神经毒素和抗毒蕈碱剂对小鼠膀胱平滑肌细胞收缩的影响
实验设计
使培养的小鼠或大鼠膀胱平滑肌细胞和小鼠或大鼠的膀胱平滑肌组织在不同浓度的镇痛剂和/或抗毒蕈碱剂的存在下暴露于炎症性刺激物和非炎症性刺激物。测量刺激诱导的肌肉收缩以评估镇痛剂和/或抗毒蕈碱剂的抑制效果。
在实施例2中描述了效应物、镇痛剂和抗毒蕈碱剂。
使小鼠膀胱平滑肌细胞的原代培养物受到以下物质的短期(1-2小时)或长期(24-48小时)刺激:
(1)不同剂量的每种单独的镇痛剂。
(2)在lps存在的情况下不同剂量的每种镇痛剂。
(3)在卡巴胆碱或乙酰胆碱存在的情况下不同剂量的每种镇痛剂。
(4)在aa、dgla或epa存在的情况下不同剂量的每种镇痛剂。
(5)不同剂量的单独的肉毒杆菌神经毒素a。
(6)在lps存在的情况下不同剂量的肉毒杆菌神经毒素a。
(7)在卡巴胆碱或乙酰胆碱存在的情况下不同剂量的肉毒杆菌神经毒素a。
(8)在aa、dgla或epa存在的情况下不同剂量的肉毒杆菌神经毒素a。
(9)不同剂量的单独的每种抗毒蕈碱剂。
(10)在lps存在的情况下不同剂量的每种抗毒蕈碱剂。
(11)在卡巴胆碱或乙酰胆碱存在的情况下不同剂量的每种抗毒蕈碱剂。
(12)在aa、dgla或epa存在的情况下不同剂量的每种抗毒蕈碱剂。
材料和方法
如实施例3所述分离原代小鼠膀胱细胞。在选定的实验中,使用膀胱组织的培养物。使用grass多道记录仪(美国quincymass)记录膀胱平滑肌细胞收缩。
实施例5:口服镇痛剂和抗毒蕈碱剂对小鼠膀胱平滑肌细胞的cox2和pge2反应的影响。
实验设计
对正常小鼠和患有膀胱过度活跃综合征的小鼠给予口服剂量的阿司匹林、萘普生钠、布洛芬、吲哚美辛、萘丁美酮、泰诺林、塞来考昔、奥昔布宁、索非那新、达非那新、阿托品及其组合。对照组包括未处理的正常小鼠和未处理的患有膀胱过度活跃综合征的oab小鼠。最后剂量三十(30)分钟后,收集膀胱并用卡巴胆碱或乙酰胆碱离体刺激。在选定的实验中,膀胱在用卡巴胆碱刺激之前用肉毒杆菌神经毒素a处理。将动物保留在代谢笼中,并评估排尿频率(和体积)。通过监测水摄取和笼窝重(cagelitterweight)确定膀胱排出量。通过elisa测定血清pgh2、pge、pge2、前列腺环素、血栓烷、il-1β、il-6、tnf-α、camp和cgmp水平。在全血细胞中的cd80、cd86和mhcii类的表达通过流式细胞计检测。
在实验结束后,将动物安乐处死并用grass多道记录仪记录离体膀胱收缩。将膀胱部分固定在福尔马林中,且通过免疫组织化学分析cox2反应。
实施例6:镇痛剂、肉毒杆菌神经毒素和抗毒蕈碱剂对人膀胱平滑肌细胞对炎症和非炎症性刺激的反应的影响
实验设计
设计本研究以表征在实施例1至5中确定的镇痛剂的最优剂量如何影响在细胞培养或组织培养中的人膀胱平滑肌细胞,并论述不同类的镇痛剂是否能够协同以更有效地抑制cox2和pge2反应。
在实施例2中描述了效应物、镇痛剂和抗毒蕈碱剂。
使人膀胱平滑肌细胞受到以下物质的短期(1-2小时)或长期(24-48小时)刺激:
(1)不同剂量的每种单独的镇痛剂。
(2)在lps存在的情况下不同剂量的每种镇痛剂。
(3)在卡巴胆碱或乙酰胆碱存在的情况下不同剂量的每种镇痛剂。
(4)在aa、dgla或epa存在的情况下不同剂量的每种镇痛剂。
(5)不同剂量的单独的肉毒杆菌神经毒素a。
(6)在lps存在的情况下不同剂量的肉毒杆菌神经毒素a。
(7)在卡巴胆碱或乙酰胆碱存在的情况下不同剂量的肉毒杆菌神经毒素a。
(8)在aa、dgla或epa存在的情况下不同剂量的肉毒杆菌神经毒素a。
(9)不同剂量的单独的每种抗毒蕈碱剂。
(10)在lps存在的情况下不同剂量的每种抗毒蕈碱剂。
(11)在卡巴胆碱或乙酰胆碱存在的情况下不同剂量的每种抗毒蕈碱剂。
(12)在aa、dgla或epa存在的情况下不同剂量的每种抗毒蕈碱剂。
然后分析细胞的pgh2、pge、pge2、前列腺环素、血栓烷、il-1β、il-6、tnf-α的释放;cox2活性;camp和cgmp的产生;il-1β、il-6、tnf-α和cox2mrna的产生;以及cd80、cd86和mhcii类分子的表面表达。
实施例7:镇痛剂、肉毒杆菌神经毒素和抗毒蕈碱剂对人膀胱平滑肌细胞收缩的影响
实验设计
使培养的人膀胱平滑肌细胞在不同浓度的镇痛剂和/或抗毒蕈碱剂的存在下暴露于炎症性刺激物和非炎症性刺激物。测量刺激诱导的肌肉收缩以评估镇痛剂和/或抗毒蕈碱剂的抑制效果。
在实施例2中描述了效应物、镇痛剂和抗毒蕈碱剂。
使人膀胱平滑肌细胞受到以下物质的短期(1-2小时)或长期(24-48小时)刺激:
(1)不同剂量的每种单独的镇痛剂。
(2)在lps存在的情况下不同剂量的每种镇痛剂。
(3)在卡巴胆碱或乙酰胆碱存在的情况下不同剂量的每种镇痛剂。
(4)在aa、dgla或epa存在的情况下不同剂量的每种镇痛剂。
(5)不同剂量的单独的肉毒杆菌神经毒素a。
(6)在lps存在的情况下不同剂量的肉毒杆菌神经毒素a。
(7)在卡巴胆碱或乙酰胆碱存在的情况下不同剂量的肉毒杆菌神经毒素a。
(8)在aa、dgla或epa存在的情况下不同剂量的肉毒杆菌神经毒素a。
(9)不同剂量的单独的每种抗毒蕈碱剂。
(10)在lps存在的情况下不同剂量的每种抗毒蕈碱剂。
(11)在卡巴胆碱或乙酰胆碱存在的情况下不同剂量的每种抗毒蕈碱剂。
(12)在aa、dgla或epa存在的情况下不同剂量的每种抗毒蕈碱剂。
使用grass多道记录仪(美国quincymass)记录膀胱平滑肌细胞收缩。
实施例8:镇痛剂对正常人膀胱平滑肌细胞对炎症和非炎症性信号的反应的影响
实验设计:
正常的人膀胱平滑肌细胞的培养
将正常的人膀胱平滑肌细胞通过酶消化从人膀胱的宏观正常部分分离。将细胞在体外通过在37℃下在5%co2的气氛中在补充有10%胎牛血清、15mmhepes、2mm左旋谷酰胺、100u/ml青霉素和100mg/ml的链霉素的rpmi1640中培养而扩大,并通过用胰蛋白酶处理以分离细胞随后在新的培养瓶中再接种而每星期传代一次。培养的第一星期,培养基补充有0.5ng/ml表皮生长因子、2ng/ml成纤维细胞生长因子和5μg/ml胰岛素。
体外以镇痛剂处理正常的人膀胱平滑肌细胞
将受胰蛋白酶消化的,并以3x104个细胞/孔(100μl)的细胞密度接种在微培养平板中的膀胱平滑肌细胞用镇痛剂溶液(50μl/孔)单独或者与卡巴胆碱(10摩尔,50μl/孔)(作为非炎症性刺激物的例子)共同处理,或者与鼠伤寒沙门氏菌的脂多糖(lps)(1μg/ml,50μl/孔)(作为非炎症性刺激物的例子)共同处理。当没有其他的效应物加入细胞中时,向孔中加入50μl的无胎牛血清的rpmi1640以调整最终体积为200μl。
在培养24小时后,收集150μl的培养上清液,在4℃,8,000rpm下旋转2分钟以除去细胞和碎片,并在-70℃储存以用于通过elisa分析前列腺素e2(pge2)的反应。将细胞固定、透化并封闭以使用荧光底物检测cox2。在选定的实验中,细胞在体外刺激12小时以用于cox2、pge2和细胞因子反应的分析。
cox2、pge2和细胞因子反应分析
如在实施例3中所述,分析cox2和pge2反应。如在实施例2中所述,分析细胞因子反应。
镇痛剂抑制正常的人膀胱平滑肌细胞对炎症性和非炎症性刺激物的cox2反应-在培养24小时以后的细胞和培养上清液的分析表明,无单独测试的镇痛剂诱导正常的人膀胱平滑肌细胞中的cox2反应。然而,如表6所总结的,在正常的人膀胱平滑肌细胞中,卡巴胆碱诱导低的但显著的cox2反应。另一方面,lps处理导致在正常的人膀胱平滑肌细胞中较高水平的cox2反应。对乙酰氨基酚、阿司匹林、布洛芬和萘普生均能抑制卡巴胆碱和lps对cox2水平的影响。当这些药物在5μm或50μm测试时,可以看出镇痛剂对lps诱导的反应的抑制效果。
表6.在体外用炎症性和非炎症性刺激物刺激和用镇痛剂处理后正常的人膀胱平滑肌细胞的cox2表达
#数据以重复两次的平均值表达
镇痛剂抑制正常的人膀胱平滑肌细胞对炎症性和非炎症性刺激物的pge2反应-与上述的对cox2反应的诱导相一致,卡巴胆碱和lps诱导正常的人膀胱平滑肌细胞的pge2的产生。还发现对乙酰氨基酚、阿司匹林、布洛芬和萘普生也在5μm或50μm下抑制lps诱导的pge2反应(表7)。
表7.在体外用炎症性和非炎症性刺激物刺激和用镇痛剂处理后的正常的人膀胱平滑肌细胞的pge2分泌
#数据以重复两次的平均值表达
镇痛剂抑制正常的人膀胱细胞对炎症性刺激物的细胞因子反应-在培养24小时以后的细胞和培养上清液的分析表明,无单独测试的镇痛剂诱导正常的人膀胱平滑肌细胞中的il-6或tnfα的分泌。如表8和9中所示,测试的卡巴胆碱的剂量对正常的人膀胱平滑肌细胞中诱导低但显著的tnfα和il-6反应。另一方面,lps处理导致这些促炎症反应细胞因子的大量诱导。对乙酰氨基酚、阿司匹林、布洛芬和萘普生均能抑制卡巴胆碱和lps对tnfα和il-6反应的影响。当这些药物在5μm或50μm测试时,可以看出镇痛剂对lps诱导的反应的抑制效果。
表8.在体外用炎症性和非炎症性刺激物刺激和用镇痛剂处理后正常的人膀胱平滑肌细胞的tnfα分泌
#数据以重复两次的平均值表达
表9.在体外用炎症性和非炎症性刺激物刺激和用镇痛剂处理后正常的人膀胱平滑肌细胞的il-6分泌
#数据以重复两次的平均值表达
将原代的正常的人膀胱平滑肌细胞分离、培养并评价其在非炎症性(卡巴胆碱)和炎症性(lps)刺激物的存在下对镇痛剂的反应。此研究的目的是确定正常的人膀胱平滑肌细胞是否能重现前述的由鼠科膀胱细胞得到的现象。
以延迟释放或延长释放制剂或者延迟和延长释放制剂的镇痛剂和/或抗毒蕈碱剂重复上述实验。
上述说明书是用于教导本领域的普通技术人员如何实践本发明目的,其并不意欲详细描述对于本领域的普通技术人员来说阅读了说明书以后能显而易见的那些明显的修改和变化。但是,意欲将所有的明显的修改和变化包括在本发明的范围内,这将通过以下权利要求定义。除非文中有明确的相反指示,权利要求意图覆盖以任何顺序的能够有效实现其所需目的的所要求的组分和步骤。
- 还没有人留言评论。精彩留言会获得点赞!