一种口服固体制剂及其应用的制作方法
本发明涉及医药制剂领域,具体涉及一种口服固体制剂及其应用。
背景技术:
一般情况下,口服固体制剂的吸收速度取决于活性物质(活性成分)从固体制剂溶出(释放)的速度,而活性成分的溶出(释放)速度往往取决于固体制剂的崩解速度,因此基本上可以认为,口服固体制剂的崩解速度是活性成分被机体吸收速度的决定步骤。崩解剂大都具有良好的吸水性和膨胀性,在口服固体制剂中加入崩解剂的作用是消除或破坏因粘合剂或者高度压缩而产生的结合力,使其在溶出介质中迅速裂碎成细小颗粒状物质,从而使活性成分被迅速溶解吸收,发挥治疗作用。崩解剂的崩解机理主要为毛细管作用和膨胀作用,崩解剂在口服固体制剂中形成易于润湿的毛细管通道,当固体制剂置于溶出介质中时,溶出介质迅速的随毛细管进入固体制剂内部,使整个固体制剂润湿,从而口服固体制剂的崩解经历润湿、虹吸、膨胀和破碎过程。
常用的崩解剂有干淀粉、羧甲基淀粉钠、低取代羟丙基纤维素、交联聚维酮、交联羧甲基纤维素钠(cmc-na)等,主要通过毛细管作用和膨胀作用起到促使固体制剂崩解的功效。但有些活性物质吸湿性强,且在吸湿后会变粘,使得常规崩解剂无法有效使之崩解,导致其溶出释放性能较差。
高血压(hypertension)是最常见的心血管疾病,也是导致充血性心力衰竭、脑卒中、冠心病、肾功能衰竭、主动脉瘤的发病率和病死率升高的主要危险因素。抗高血压药物在高血压的治疗与防治中起着关键作用。随着对高血压发病机制认识的不断深入,许多具有较佳疗效的抗高血压药物,比如利尿剂、β-受体阻滞剂、钙通道拮抗剂、血管紧张素转化酶抑制剂(acei,普利类)、血管紧张素iiat1受体拮抗剂(arb,沙坦类),不断被发现并成功应用于临床。经过多年的临床实践,确证at1受体拮抗剂沙坦类药物由于其降血压平稳、疗效佳、作用时间长、患者耐受性好,尤其在预防卒中、延缓糖尿病和非糖尿病肾病的肾功能不全、改善左心室肥厚、对靶器官的保护作用等方面具有较多优势,且不影响缓激肽降解和前列腺素合成,从而不引起干咳和血管神经性水肿,现已成为全球抗高血压药物市场的主流品种。但是沙坦类抗高血压药物的降血压有效率仅约为50-60%,且具有一定程度的不良反应。因此,开发降压效果更强、不良反应更少,并对靶器官有较佳保护作用的小剂量长效降压药已成为一个热门的研究方向。
公开号为cn103709154a的中国专利申请首次公开了结构如下式(b)所示的化合物:
上述化合物是偶联了川芎嗪的沙坦类药物,是血管紧张素ii受体拮抗剂阿齐沙坦(tak-536)的前药,该化合物在体内释放出羟基川芎嗪,从而与阿齐沙坦发生有效的协同作用,增强抗高血压疗效,具有一定的降心率作用,减少不良反应,对患者心肾也具有较理想的保护作用。
申请人在进一步的研究中发现式(b)化合物的钾盐,即如下式(a)所示的化合物,溶解性能更好,生物利用度更高,具有更强效和更长效的降压效果,同时具有更明显且持久的降心率效果,且安全性高,对患者心肾功能具有较理想的保护作用,能够用于预防和/或治疗高血压、慢性心衰、糖尿病肾病等,
为了改善化合物的稳定性和成药性,申请人在进一步研究中得到了该化合物的晶型i、ii、iii、iv。在将上述晶型制成口服固体制剂的过程中,使用各种常用崩解剂都无法有效使之崩解,导致活性成分的溶出释放性能较差。进一步研究,申请人发现这些晶型具有吸湿性强,且在吸湿后会变粘的特性,使得常规崩解剂无法有效使之崩解,导致活性成分的溶出释放性能较差。
技术实现要素:
本发明提供了一种口服固体制剂,其活性成分含有式(a)所示化合物的晶型。本发明的口服固体制剂迅速崩解、提高其溶出速率,从而提高生物利用度,并特别能够解决吸湿性强且在吸湿后会变粘的活性物质(活性成分)所存在的采用常规崩解剂无法有效使之崩解的问题。
常规崩解剂主要靠毛细管作用将水分导入固体制剂内部,同时崩解剂吸水后膨胀,消除固体制剂中因为粘合剂或高度压缩而产生的结合力,达到促使固体制剂崩解的目的。当药物活性物质吸湿性强,且在吸湿后会变粘时,在采用常规崩解剂将其制成口服固体制剂后,口服固体制剂将难以崩解,例如体现在模拟胃酸ph条件下固体制剂无法崩解释放药物。导致这种结果的原因可能是活性物质吸湿性强,会与崩解剂竞争吸收水分,同时吸水后变粘,阻碍崩解剂的膨胀,阻断了水进入的通道,从而导致常规崩解剂难以发挥其崩解性能。
本发明首先提供了一种口服固体制剂,其活性成分含有式(a)所示化合物的晶型:
式(a)化合物的晶型可以选自晶型i、ii、iii、iv中的任一种、两种或者更多种的混合物或者混晶。
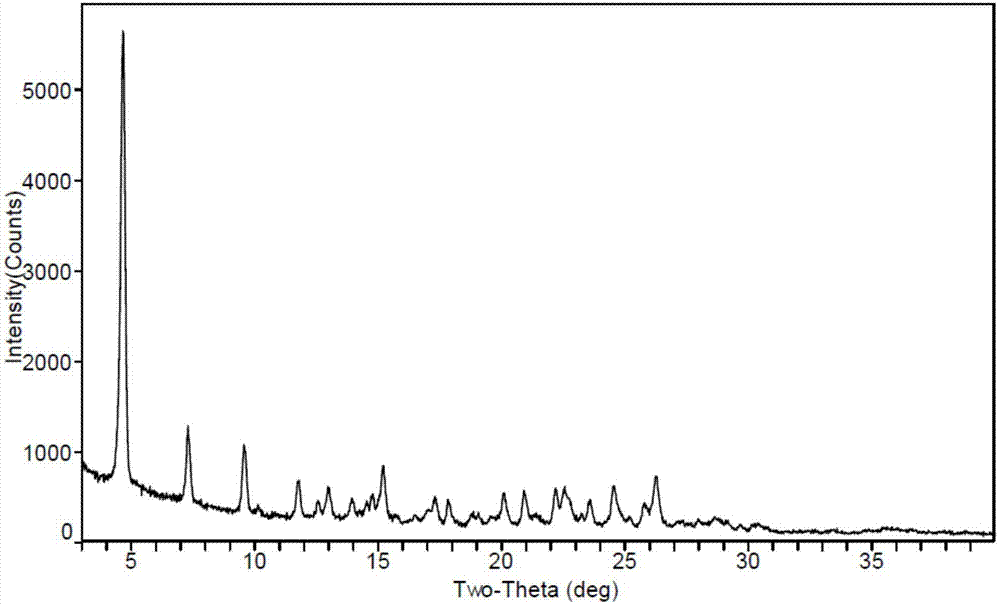
根据本发明的晶型,晶型i的x-射线粉末衍射图在衍射角2θ为5.3±0.2、8.6±0.2度位置有特征峰;优选,晶型i的x-射线粉末衍射图进一步在衍射角2θ为13.3±0.2、20.1±0.2度位置有特征峰;进一步优选,晶型i的x-射线粉末衍射图进一步在衍射角2θ为6.3±0.2、10.6±0.2、26.3±0.2度位置有特征峰;更优选,晶型i的x-射线粉末衍射图进一步在衍射角2θ为12.7±0.2度位置有特征峰;最优选,晶型i的x-射线粉末衍射图基本上如图1所示。
根据本发明的晶型,晶型ii的x-射线粉末衍射图在衍射角2θ为4.7±0.2度位置有特征峰;优选,晶型ii的x-射线粉末衍射图进一步在衍射角2θ为7.3±0.2、9.6±0.2、15.2±0.2、26.3±0.2度位置有特征峰;进一步优选,晶型ii的x-射线粉末衍射图进一步在衍射角2θ为11.8±0.2、24.6±0.2度位置有特征峰;更优选,晶型ii的x-射线粉末衍射图进一步在衍射角2θ为22.6±0.2度位置有特征峰;最优选,晶型ii的x-射线粉末衍射图基本上如图2所示。
根据本发明的晶型,晶型iii的x-射线粉末衍射图在衍射角2θ为5.2±0.2、8.0±0.2度位置有特征峰;优选,晶型iii的x-射线粉末衍射图进一步在衍射角2θ为12.4±0.2、13.6±0.2度位置有特征峰;进一步优选,晶型iii的x-射线粉末衍射图进一步在衍射角2θ为19.2±0.2度位置有特征峰;更优选,晶型iii的x-射线粉末衍射图进一步在衍射角2θ为10.3±0.2、12.2±0.2、21.4±0.2度位置有特征峰;最优选,晶型iii的x-射线粉末衍射图基本上如图3所示。
根据本发明的晶型,晶型iv的x-射线粉末衍射图在衍射角2θ为7.4±0.2、14.7±0.2、16.0±0.2度位置有特征峰;优选,晶型iv的x-射线粉末衍射图进一步在衍射角2θ为8.4±0.2、22.6±0.2、23.2±0.2、29.7±0.2度位置有特征峰;更优选,晶型iv的x-射线粉末衍射图进一步在衍射角2θ为24.0±0.2度位置有特征峰;最优选,晶型iv的x-射线粉末衍射图基本上如图4所示。
根据本发明,优选地,晶型i的dsc图谱显示熔点为184±5℃;晶型ii的dsc图谱显示熔点为145±5℃;晶型iii的dsc图谱显示熔点为187±5℃;晶型iv的dsc图谱显示熔点为145±5℃。
根据本发明,优选地,tga图谱显示晶型i的分解温度为180±5℃;晶型ii的分解温度为148±5℃;晶型iii的分解温度为183±5℃;晶型iv的分解温度为149±5℃。
根据本发明,优选地,晶型i具有基本如图10所示的dsc-tga图谱;晶型ii具有基本如图11所示的dsc-tga图谱;晶型iii具有基本如图12所示的dsc-tga图谱;晶型iv具有基本如图13所示的dsc-tga图谱。
根据本发明的实施方案,式(a)所示化合物的晶型可以选自晶型i、ii、iii、iv中的任一种、两种或更多种的任意比例的混合物或者混晶。
优选,所述组合物或混合物含有任意比例的晶型i和晶型ii,更优选,为晶型i和晶型ii的任意比例的混合物。
由于晶型i和晶型ii在一些条件下可以互相转化,本领域技术人员可以理解,对于晶型i和晶型ii的混合物中晶型i和晶型ii的比例没有特别限制。例如,晶型i和晶型ii的重量比可以为1:99~99:1,如5:95~95:5。作为实例,晶型i和晶型ii的重量比可以为1:9~9:1,2:8~8:2、3:7~7:3、4:6~6:4或5:5。
式(a)化合物无定型的化学稳定性较差,放置后会产生杂质。晶型i、ii、iii和iv的化学性质稳定,放置后所产生杂质均明显低于无定型。在四种晶型中,晶型iv相对不稳定,室温下干燥会转晶为晶型i。晶型ii在高温、高湿和压片条件下晶型相对稳定。在室温条件下,晶型i、ii、iii的晶型稳定性依次为:晶型ii>晶型i>晶型iii。
本发明还提供如上所述式(a)所示化合物的晶型i、ii、iii和iv的制备方法。
式(b)化合物可以通过本领域已知的方法,例如cn103709154a中公开的方法制备得到,在此将cn103709154a全文引入。式(a)化合物可以通过将式(b)化合物与钾盐试剂反应制备得到。
本发明制备晶型i的方法可以通过将式(a)化合物的悬浊液搅拌、在式(a)化合物的溶液中加入抗溶剂、将式(a)化合物的溶液冷却、将式(a)化合物在溶剂气氛中扩散结晶,或将式(a)化合物晶型iii和/或晶型iv的悬浊液搅拌得到。具体来说,可以包括选自下列的一种或多种方法:
(1)取式(a)化合物,加入溶剂,得悬浊液,搅拌,得到晶型i。优选,溶剂为乙醇/异丙醚混合溶液、乙醇/正庚烷混合溶液、异丙醇/正庚烷混合溶液、或四氢呋喃/正庚烷混合溶液;更优选,混合溶液中两种溶剂的体积比为1:8-8:1,最优选1:5-5:1。优选,室温搅拌0.5-3天,最优选,室温搅拌1-2天。
(2)将式(a)化合物溶于良溶剂中,得澄清溶液,搅拌下加入抗溶剂,得到晶型i。优选,良溶剂为甲醇、乙醇、或正丁醇,抗溶剂为异丙醚、甲基叔丁基醚、或甲基环己烷。
(3)将式(a)化合物在加热条件下溶于溶剂中,得澄清溶液,冷却结晶,得到晶型i。优选,溶剂为乙醇/异丙醚混合溶液、乙醇/乙酸乙酯混合溶液、乙醇/甲基叔丁基醚混合溶液、乙醇/正庚烷混合溶液、乙醇/甲基环己烷混合溶液、或正丁醇/正庚烷混合溶液;更优选,混合溶液中两种溶剂的体积比为1:8-8:1,最优选1:5-5:1。优选,加热至温度为40-90,最优选,加热至温度为50-70℃。
(4)将式(a)化合物置于乙醇的溶剂气氛中扩散1-3天,得到晶型i。
(5)将式(a)化合物晶型iii和/或晶型iv加入溶剂,形成悬浊液,搅拌,干燥,得到晶型i;优选地,加入的溶剂选自酯类溶剂,例如乙酸乙酯,乙酸异丙酯或其混合物。
本发明制备晶型ii的方法可以通过将式(a)化合物的溶液挥发干、将式(a)化合物的溶液、饱和溶液、过饱和溶液或悬浊液搅拌、在式(a)化合物的溶液中加入抗溶剂、将式(a)化合物的溶液冷却、将式(a)化合物的饱和溶液在溶剂气氛中扩散、或将式(a)化合物在溶剂气氛中扩散结晶得到。具体来说,可以包括选自下列的一种或多种方法:
(1)将式(a)化合物溶于溶剂中,得澄清溶液,室温挥发干,得到晶型ii。优选,溶剂为乙醇/乙酸乙酯混合溶液、丙酮/乙酸乙酯混合溶液、丙酮/异丙醚混合溶液、或丙酮/正庚烷混合溶液;更优选,混合溶液中两种溶剂的体积比为1:8-8:1,最优选1:5-5:1。
(2)取式(a)化合物,加入溶剂,得澄清溶液、饱和溶液、过饱和溶液或悬浊液,搅拌,得到晶型ii。优选,溶剂为异丙醇、仲丁醇、乙酸乙酯、甲苯、乙酸异丙酯或者乙醇/乙酸乙酯混合溶液、乙醇/乙酸异丙酯混合溶液、乙醇/甲苯混合溶液、丙酮/正庚烷混合溶液、或1,4-二氧六环/正庚烷混合溶液;更优选,混合溶液中两种溶剂的体积比为1:8-8:1,最优选1:5-5:1。优选,室温搅拌10分钟-5天,最优选,室温搅拌3小时-3天。
(3)将式(a)化合物溶于良溶剂中,得澄清溶液,搅拌下加入抗溶剂,得到晶型ii。优选,良溶剂为丁酮、二甲基亚砜、或1,4-二氧六环,抗溶剂为正庚烷、异丙醚、或乙酸异丙酯。
(4)将式(a)化合物在加热条件下溶于溶剂中,得澄清溶液,冷却结晶,得到晶型ii。优选,溶剂为仲丁醇、硝基甲烷、丙酮、或四氢呋喃。优选,加热至温度为40-90,最优选,加热至温度为50-70℃。
(5)将式(a)化合物的乙醇饱和溶液,置于异丙醚或乙酸异丙酯的溶剂气氛中扩散,直至析出固体,得到晶型ii。
(6)将式(a)化合物置于甲苯、异丙醇、四氢呋喃、或乙酸乙酯的溶剂气氛中扩散1-3天,得到晶型ii。
本发明制备晶型iii的方法可以通过将式(a)化合物的悬浊液搅拌、或将式(a)化合物的饱和溶液在溶剂气氛中扩散结晶得到。具体来说,可以包括选自下列的一种或两种方法:
(1)取式(a)化合物,加入四氢呋喃,得悬浊液,搅拌,得到晶型iii。优选,室温搅拌12小时至5天,最优选,室温搅拌1-3天。
(2)将式(a)化合物的四氢呋喃饱和溶液,置于异丙醚的溶剂气氛中扩散,直至析出固体,得到晶型iii。
本发明制备晶型iv的方法可以通过在式(a)化合物的溶液中加入抗溶剂结晶得到。具体来说:
将式(a)化合物溶于正丁醇中,得澄清溶液,搅拌下加入正庚烷,得到晶型iv。
本发明制备晶型i和晶型ii的混合物(或者混晶)的方法可以通过将晶型ii的悬浊液在室温或高温条件下搅拌结晶得到。具体来说,可以包括选自下列的一种或多种方法:
(1)取晶型ii,加入乙酸异丙酯,得悬浊液,50-90℃下搅拌,得到晶型i和晶型ii的混合物。优选,50-90℃下搅拌3小时至3天,最优选,60-90℃下搅拌5小时至1天;
(2)取晶型ii湿品,粉碎过筛后,于40~60℃(如50℃)真空烘料,优选真空烘料3小时至3天,例如24h;
(3)取晶型ii湿品,于40~60℃(如50℃)真空烘料3小时至3天后微粉化,优选真空烘料24小时后微粉化;
(4)取晶型ii,加入溶剂,得悬浊液,室温搅拌,得到晶型i和晶型ii的混合物。优选,溶剂为甲基叔丁基醚、或乙醇/甲基环己烷混合溶液;更优选,混合溶液中两种溶剂的体积比为1:8-8:1,最优选1:6-5:1。优选,搅拌3小时至3天,最优选,搅拌1-3天。
作为选择,还可以将本发明的晶型i作为原料用于制备晶型ii;优选地,将晶型i在酯类溶剂(例如乙酸乙酯、乙酸异丙酯或其混合物)中形成悬浊液,室温搅拌过夜获得晶型ii;
或者,还可以将本发明的晶型iii作为原料用于制备晶型ii;优选地,将晶型iii在酯类溶剂(例如乙酸乙酯、乙酸异丙酯或其混合物)中形成悬浊液,室温搅拌过夜获得晶型ii;
或者,还可以将本发明的晶型iii作为原料用于制备晶型i;优选地,晶型iii在酯类溶剂(例如乙酸乙酯、乙酸异丙酯或其混合物)中形成悬浊液,室温搅拌过夜获得晶型i;
或者,还可以将本发明的晶型iv作为原料用于制备晶型i;优选地,晶型iv在室温下干燥过夜获得晶型i。
根据本发明的口服固体制剂的实施方案,除含有活性成分外,还可以进一步含有崩解剂、助崩剂、赋形剂和润滑剂。在优选的技术方案中,以重量百分比计,活性成分含量为约5%-50%,崩解剂含量为约1%-20%,助崩剂含量为约0.1%-35%,赋形剂含量为约20%-80%,润滑剂含量为约0.25%-10%,崩解剂和助崩剂的重量比为约10:1-1:10。
进一步优选,活性成分含量为约8-30%,崩解剂含量为约2-18%,助崩剂含量为约0.5-30%,赋形剂含量为约30-80%,润滑剂含量为约0.5-8%,崩解剂和助崩剂的重量比为约8:1-1:8。更优选,活性成分含量为约10-20%,崩解剂含量为约4-15%,助崩剂含量为约1-25%,赋形剂含量为约50-80%,润滑剂含量为约1-5%,崩解剂和助崩剂的重量比为约5:1-1:5。
更进一步优选,活性成分含量为约12-16%,崩解剂含量为约6-10%,助崩剂含量为约2-5%,赋形剂含量为约65-78%,润滑剂含量为约2-4%。
在本发明中,崩解剂可以为吸湿膨胀型崩解剂。优选,吸湿膨胀型崩解剂选自干淀粉、交联羧甲基纤维素钠、羧甲基纤维素钠、羧甲基纤维素钙、羧甲基淀粉钠、甲基纤维素、低取代羟丙基纤维素、交联聚维酮、壳聚糖、微晶纤维素中的一种或多种的混合物。
在本发明中,助崩剂为小分子易溶性物质或产气型盐。优选,产气型盐选自碳酸盐、碳酸氢盐中的一种或多种的混合物。更优选,产气型盐选自碳酸钠、碳酸钙、碳酸钾、碳酸钙镁、碳酸锌、碳酸镁、碳酸铵、甘氨酸碳酸钠、倍半碳酸钠、碳酸氢钠、碳酸氢钙、碳酸氢钾、碳酸氢铵中的一种或多种的混合物。优选,所述小分子易溶性物质选自氯化钠、葡萄糖、果糖、木糖醇中的一种或多种的混合物;更优选,所述小分子易溶性物质选自氯化钠、葡萄糖中的一种或两种的混合物。
本发明的活性成分具有吸湿性强,且在吸湿后会变粘的特性,常规崩解剂无法有效使之崩解,导致活性成分的溶出释放性能较差。本发明的口服固体制剂中添加了助崩剂,在溶出介质被引入后,或者作为助崩剂的小分子易溶性物质迅速溶解,一方面形成毛细管通道,另一方面使固体制剂内外产生渗透压差,两者结合将溶出介质引入固体制剂内部,促进崩解剂吸水膨胀产生崩解作用,从而促使固体制剂崩解;或者作为助崩剂的产气型盐与溶出介质接触反应释放气体,形成引入水的空间,一方面促进崩解剂吸水膨胀产生崩解作用,另一方面在制剂内部产生气体压力促进崩解,从而促使固体制剂崩解。本发明中所提及的溶出介质一般指的是胃液、肠液、模拟胃液、或模拟肠液。
在本发明中,赋形剂没有特别限制。优选,赋形剂选自淀粉、乳糖、甘露醇、纤维素乳糖、微晶纤维素、磷酸氢钙、甘露醇-淀粉复合物中的一种或多种的混合物。
在本发明中,润滑剂没有特别限制。优选,润滑剂选自滑石粉、硬脂酸镁、硬脂酸钙、胶体二氧化硅、水合二氧化硅、十八烷基富马酸钠、聚乙二醇、硬脂酰富马酸钠、单硬脂酸甘油酯、氢化植物油中的一种或多种的混合物。
本发明的口服固体制剂中还可以进一步含有粘合剂和/或稀释剂。
在本发明中,粘合剂没有特别限制。优选,粘合剂选自淀粉及其衍生物类(包括但不限于淀粉、预胶化淀粉、糊精、麦芽糊精等)、纤维素衍生物类(包括但不限于甲基纤维素、羧甲基纤维素钠、羟丙基纤维素、羟丙甲纤维素、乙基纤维素、微晶纤维素等)、天然胶及合成胶(包括但不限于明胶、阿拉伯胶、角豆胶、桃胶等)、聚乙二醇、聚维酮、二山嵛酸甘油酯、卡波姆、聚乙烯醇、聚(甲基)丙烯酸树脂、糖醇类(包括但不限于蔗糖、液体葡萄糖、麦芽糖醇等)、玉米朊、海藻酸钠、单月桂酸酯中的一种或多种的混合物。本领域技术人员能够根据活性成分和各辅料性能选择粘合剂的合适用量。一般来说,粘合剂含量为约0-15%。
在本发明中,稀释剂没有特别限制。优选,稀释剂选自乳糖(例如,单水合物、喷雾干燥的单水合物、无水物及其类似物)、甘露糖醇、木糖醇、葡萄糖、蔗糖、山梨糖醇、微晶纤维素、淀粉及二水合磷酸氢二钙中的一种或多种的混合物。本领域技术人员能够根据活性成分和各辅料性能选择稀释剂的合适用量。
本发明的口服固体制剂还可以进一步任选含有表面活性剂、抗氧化剂、着色剂、调味剂、防腐剂和/或掩味剂等。本领域技术人员能够根据活性成分的含量和各辅料性能选择表面活性剂、抗氧化剂、着色剂、调味剂、防腐剂和/或掩味剂等的具体物质和合适用量。
本发明的口服固体制剂可以是片剂、胶囊剂、散剂、颗粒剂、滴丸剂、膜剂等。优选,本发明的口服固体制剂为片剂。本发明的口服固体制剂可用于制备血管紧张素ii受体拮抗剂,或者用于制备预防和/或治疗高血压、慢性心衰、糖尿病肾病的药物。
本发明的片剂单位制剂总重为约90mg-600mg;硬度为约3kg-20kg。活性成分的含量为约10mg-100mg/单位制剂。
在优选的实施方案中,本发明的片剂中活性成分可以为例如晶型i、晶型ii或二者的任意比例的混合物。根据本发明的实施方案,赋形剂为甘露醇,崩解剂为交联羧甲基纤维素钠,助崩剂为碳酸氢钠,润滑剂为硬脂酸镁;优选,以重量百分比计,活性成分含量为约5%-50%,交联羧甲基纤维素钠含量为约1%-20%,碳酸氢钠含量为约0.1%-35%,甘露醇含量为约20%-80%,硬脂酸镁含量为约0.25%-10%,崩解剂和助崩剂的重量比为10:1-1:10;进一步优选,活性成分含量为约8-30%,交联羧甲基纤维素钠含量为约2-18%,碳酸氢钠含量为约0.5-30%,甘露醇含量为约30-80%,硬脂酸镁含量为约0.5-8%,崩解剂和助崩剂的重量比为8:1-1:8;更优选,活性成分含量为约10-20%,交联羧甲基纤维素钠含量为约4-15%,碳酸氢钠含量为约1-25%,甘露醇含量为约50-80%,硬脂酸镁含量为约1-5%,崩解剂和助崩剂的重量比为5:1-1:5;最优选,活性成分含量为约12-16%,交联羧甲基纤维素钠含量为约6-10%,碳酸氢钠含量为约2-5%,甘露醇含量为约65-78%,硬脂酸镁含量为约2-4%。
作为选择,本发明的口服固体制剂中还可以含有其他活性成分,例如其他用于预防和/或治疗高血压的活性成分,如钙离子拮抗剂(二氢吡啶类、芳烷基胺类、苯硫氮革类和三苯哌嗪类)。
本发明还提供如上所述口服固体制剂的制备方法,包括将活性成分与上述其他组分混合。
根据本发明所述口服固体制剂的制备方法,可以采用粉末压片法、湿法制粒法或干法制粒法进行。
本发明还提供如上所述口服固体制剂或片剂在制备血管紧张素ii受体拮抗剂中的应用,或者在制备用于预防和/或治疗高血压、慢性心衰、糖尿病肾病的药物中的应用。
本发明还提供如上所述口服固体制剂作为血管紧张素ii受体拮抗剂的应用,或者用于预防和/或治疗高血压、慢性心衰、糖尿病肾病的应用。
本发明还提供一种用于口服固体制剂的复合崩解剂体系,包括崩解剂和助崩剂。优选,该用于口服固体制剂的复合崩解剂体系由崩解剂和助崩剂组成。
其中,所述崩解剂和助崩剂具有如上所述的定义。
在本发明中,优选,崩解剂和助崩剂的重量比为10:1-1:10,更优选8:1-1:8,最优选5:1-1:5。
在本发明中,助崩剂是指能够帮助吸湿性强且在吸湿后会变粘的活性物质(活性成分)崩解的助剂。这一类的活性物质无法采用常规崩解剂进行有效崩解。本发明发现加入小分子易溶性物质或者产气型盐,能够将这一类无法有效崩解的活性物质予以崩解。因而本发明中将其定义为助崩剂。
在本发明中,复合崩解剂体系也可以被称为复合崩解剂系统、复合崩解剂组合物或复合崩解剂。本发明的复合崩解剂体系在制备口服固体制剂时,在溶出介质被引入后,或者作为助崩剂的小分子易溶性物质迅速溶解,一方面形成毛细管通道,另一方面使口服固体制剂内外产生渗透压差,两者结合将溶出介质引入口服固体制剂内部,促进崩解剂吸水膨胀产生崩解作用,从而促使固体制剂崩解;或者作为助崩剂的产气型盐与溶出介质接触反应释放气体,形成引入水的空间,一方面促进崩解剂吸水膨胀产生崩解作用,另一方面在制剂内部产生气体压力促进崩解,从而促使固体制剂崩解。本发明中所提及的溶出介质指的是胃液、肠液、模拟胃液、或模拟肠液。
因此,本发明所提供的复合崩解剂体系,特别适用采用常规崩解剂无法有效使之崩解的活性成分,例如吸湿性强,且在吸湿后会变粘的活性成分,包括但不限于如上所述式(a)所示化合物的晶型。
本发明的有益效果是:
1、本发明的固体制剂能有效克服活性成分遇水产生的粘性,从而导致无法崩解的问题,促进固体制剂崩散,释放药物。
2、本发明的固体制剂不需要添加额外的酸,仅仅利用酸性环境即可实现快速崩解,使活性成分溶出,提高了生物利用度。
3、本发明通过加入小分子易溶性物质或者产气型盐,实现了吸湿性强且在吸湿后会变粘的活性物质(活性成分)的有效崩解,从而使得活性成分溶出,提高了生物利用度。
4、本发明的固体制剂可用于预防和/或治疗高血压、慢性心衰、糖尿病肾病。
5、本发明的固体制剂通过口服途径给药,使用方便。
6、本发明的固体制剂能够改善活性成分的成药性和患者的顺应性。
术语解释
术语或“晶体”或“晶型”指具有相同化学组成但有不同的形成结晶的分子和/或离子的空间排布的晶型。
术语“无定型”指不是结晶的分子和/或离子的固体形式。无定型固体不显示确定的具有清晰最大值的x-射线粉末衍射图形。
术语“x-射线粉末衍射图基本上如图所示”是指x-射线粉末衍射图所示的主要峰中至少50%,或至少60%,或至少70%,或至少80%,或至少90%,或至少95%,或至少99%的峰出现在x-射线粉末衍射图中;其主要峰指以最高峰作为参照(最高峰的相对强度指定为100%),相对强度大于10%、优选大于30%的峰。
附图说明
图1为晶型i的x-射线粉末衍射图;
图2为晶型ii的x-射线粉末衍射图;
图3为晶型iii的x-射线粉末衍射图;
图4为晶型iv的x-射线粉末衍射图;
图5为式(a)化合物的1h-nmr图谱;
图6为式(a)化合物无定型形式的x-射线粉末衍射图谱;
图7为晶型iv室温干燥前后x-射线粉末衍射图谱(从上到下依次为晶型i、晶型iv室温干燥后、晶型iv);
图8为晶型ii在压片条件下的x-射线粉末衍射图(从上到下依次为初始样品、25公斤压力压片后的样品、20公斤压力压片后的样品);
图9为晶型ii在高温高湿条件下10天后的x-射线粉末衍射图(从上到下依次为初始样品、60℃条件下的样品、85%相对湿度下的样品);
图10为晶型i的dsc-tga图谱;
图11为晶型ii的dsc-tga图谱;
图12为晶型iii的dsc-tga图谱;
图13为晶型iv的dsc-tga图谱。
具体实施方式
下面将结合具体实施例对本发明做更进一步的详细说明。本领域技术人员可以借鉴本文内容,适当更改工艺参数实现。特别需要指出的是,所有类似的替换和改动均应被视为包括在本发明的保护范围内。本发明的产品已经通过较佳实施例进行了描述,相关人员能在不脱离本发明内容、精神和范围内对本文所述的产品进行改动或适当变更与组合,来实现和应用本技术。
实施例1:式(a)化合物的制备
将式(b)化合物(1.0g)溶于二氯甲烷(5ml)中,室温搅拌形成溶液,往溶液中加入邻苯二甲酰亚胺钾(0.27g),保温反应4h,冷却至-50℃,过滤,溶剂旋干所得固体为式(a)化合物(无定型)。
熔点:135-145℃。
ms/hrmsm/z:717[m+h]+;677[m-k]-。
1h-nmr(400mhz,dmso-d6)δ:1.44(t,3h),1.46(t,3h),2.38(s,3h),2.41(s,3h),2.44(s,3h),4.64(q,2h),5.29(d,1h),5.32(d,1h),5.52(d,1h),5.56(d,1h),6.86(q,1h),6.90(d,2h),7.18(m,2h),7.22(d,2h),7.33(m,1h),7.36(m,1h),7.46(d,1h),7.52(dd,1h),7.75(d,1h)。
1h-nmr图谱和x射线粉末衍射图谱分别见图5和图6。
实施例2:式(a)化合物在自发性高血压大鼠上的抗高血压药效试验
取12周龄的自发性高血压大鼠(以下简称shr,购自北京维通利华实验动物技术有限公司),以2.5%的戊巴比妥钠进行腹腔注射麻醉,将血压植入子的血压感应导管插入腹主动脉,植入子固定于腹壁,缝合后进行术后日常看护。选取收缩压超过160mmhg的动物进入分组,每组8只动物,共3组。对照组给予0.5%的羧甲基纤维素钠(下称cmc-na);式(b)化合物组和式(a)化合物组采用0.5%cmc-na溶解,给药剂量均以1mg/kg阿齐沙坦有效剂量计,给药体积为4ml/kg,均灌胃给药,以给药前动物的收缩压和心率为基准值,比较给药前、后各时间点shr的收缩压和心率变化,每个时间点测三次取平均值。结果见下表1和表2。
表1.式(b)化合物、式(a)化合物口服给药前、后各时间点收缩压变化(平均值(mmhg)±标准误差)
*p<0.01(相对于对照组)。
从表1结果可以看出,给药3小时后各给药组与对照组比较收缩压均显著下降,在给药5-7小时药效达峰,式(a)化合物组具有比式(b)化合物组更强效和长效的降压效果。
表2.式(b)化合物、式(a)化合物口服给药前、后各时间点心率变化(平均值(次/分钟)±标准误差)
*p<0.05(相对于对照组单因素方差比较)。
从表2结果可以看出,式(a)化合物组具有比式(b)化合物组更强效和长效的降心率效果。
实施例3:晶型i的制备
(1)取15mg式(a)化合物,加入0.2ml乙醇/异丙醚(体积比为1:5)混合溶液,得悬浊液,室温搅拌1天,过滤,干燥,得到晶型i。xrd检测图谱见附图1;dsc:184℃。按照相同的方法,用乙醇/正庚烷(体积比为1:5)混合溶液、异丙醇/正庚烷(体积比为1:5)混合溶液、或四氢呋喃/正庚烷(体积比为1:5)混合溶液同样制备得到了晶型i。
(2)将15mg式(a)化合物溶于0.1ml甲醇中,得澄清溶液,搅拌下加入1.0ml异丙醚,析出固体,继续搅拌,过滤,干燥,得到晶型i。按照相同的方法,用良溶剂乙醇/抗溶剂异丙醚、良溶剂乙醇/抗溶剂甲基叔丁基醚、良溶剂乙醇/抗溶剂甲基环己烷、良溶剂正丁醇/抗溶剂异丙醚同样制备得到了晶型i。
(3)将10mg式(a)化合物在60℃溶于乙醇/异丙醚(0.2ml:0.5ml)混合溶液中,得澄清溶液,冷却结晶,得到晶型i。按照相同的方法,用乙醇/乙酸乙酯(0.1ml:0.5ml)混合溶液、乙醇/甲基叔丁基醚(0.2ml:0.5ml)混合溶液、乙醇/正庚烷(0.2ml:0.5ml)混合溶液、乙醇/甲基环己烷(0.2ml:0.5ml)混合溶液、或正丁醇/正庚烷(0.2ml:0.5ml)混合溶液同样制备得到了晶型i。
(4)将8mg式(a)化合物置于乙醇的溶剂气氛中(即置于装有乙醇的大容器中)扩散1天,干燥,得到晶型i。
(5)将15mg式(a)化合物晶型iii样品,加入0.2ml乙酸乙酯形成悬浊液,搅拌过夜,干燥,得到晶型i。
(6)将15mg式(a)化合物晶型iv样品,加入0.2ml乙酸乙酯形成悬浊液,搅拌过夜,室温干燥,得到晶型i。
实施例4:晶型ii的制备
(1)取1.1g式(a)化合物,加入10ml乙酸乙酯,得澄清溶液,室温搅拌3小时,过滤,干燥,得到产品0.88g,所得晶型ii的xrd检测图谱见附图2;dsc:145.4℃。逐渐减少乙酸乙酯的用量,发现饱和溶液、过饱和溶液、悬浊液室温搅拌均能够得到晶型ii。
按照相同的方法,用异丙醇、仲丁醇、乙酸异丙酯、或甲苯,或者乙醇/乙酸乙酯(体积比为1:5)混合溶液、乙醇/乙酸异丙酯(体积比为1:5)混合溶液、乙醇/甲苯(体积比为1:5)混合溶液、丙酮/正庚烷(体积比为1:5)混合溶液、或1,4-二氧六环/正庚烷(体积比为1:5)混合溶液,同样制备得到了晶型ii。
(2)将5mg式(a)化合物溶于乙醇/乙酸乙酯(0.2ml:0.5ml)混合溶液中,得澄清溶液,室温挥发干,得到晶型ii。按照相同的方法,用丙酮/乙酸乙酯混合溶液(1.0ml:0.5ml)、丙酮/异丙醚混合溶液(2.0ml:0.5ml)、或丙酮/正庚烷混合溶液(2.0ml:0.5ml)同样制备得到了晶型ii。
(3)将15mg式(a)化合物溶于0.8ml丁酮中,得澄清溶液,搅拌下加入4.0ml正庚烷,析出固体,过滤,干燥,得到晶型ii。按照相同的方法,用良溶剂为丁酮/抗溶剂异丙醚、良溶剂二甲基亚砜/抗溶剂乙酸异丙酯、或良溶剂1,4-二氧六环/抗溶剂异丙醚同样制备得到了晶型ii。
(4)将10mg式(a)化合物在60℃溶于仲丁醇中,得澄清溶液,冷却结晶,得到晶型ii。按照相同的方法,用硝基甲烷、丙酮、或四氢呋喃同样制备得到了晶型ii。
(5)将5mg式(a)化合物溶于适量乙醇中,得饱和溶液,置于异丙醚的溶剂气氛中(即置于装有异丙醚的大容器中)扩散,直至析出固体,过滤,干燥,得到晶型ii。按照相同的方法,置于乙酸异丙酯的溶剂气氛中扩散同样制备得到了晶型ii。
(6)将8mg式(a)化合物置于甲苯的溶剂气氛中(即置于装有甲苯的大容器中)扩散3天,干燥,得到晶型ii。按照相同的方法,置于异丙醇、四氢呋喃、乙酸乙酯的溶剂气氛中扩散同样制备得到了晶型ii。
实施例5:晶型iii的制备
(1)取100mg式(a)化合物,加入1.0ml四氢呋喃,得悬浊液,室温搅拌1天,过滤,干燥,所得晶型iii的xrd检测图谱见附图3;dsc:187.3℃。
(2)将5mg式(a)化合物溶于适量四氢呋喃中,得饱和溶液,置于异丙醚的溶剂气氛中(即置于装有异丙醚的大容器中)静置,直至析出固体,过滤,干燥,得到晶型iii。
实施例6:晶型iv的制备
将50mg式(a)化合物溶于1.0ml正丁醇中,得澄清溶液,搅拌下加入5.0ml正庚烷,析出固体,过滤,所得晶型iv的xrd检测图谱见附图4;dsc熔点:144.7℃。
晶型iv的晶型在室温下干燥会转变为晶型i,详见附图7。
实施例7:晶型i和晶型ii的混合物的制备
(1)取100mg晶型ii,加入2.5ml乙酸异丙酯,得悬浊液,于80℃水浴中搅拌8小时,过滤,干燥,经xrd检测结果可知,晶型i含量约为95%,晶型ii含量约为5%。
(2)取100mg晶型ii湿品,粉碎过筛后,于50℃真空烘料24h,经xrd检测结果可知,晶型i含量约为10%,晶型ii含量约为90%。
(3)取100g晶型ii湿品,于50℃真空烘料24h,微粉机微粉化后,经xrd检测结果可知,晶型i含量约为30%,晶型ii含量约为70%。
(4)取15mg晶型ii,加入0.5ml甲基叔丁基醚得到悬浊液,在室温下搅拌3天,过滤,干燥,得到晶型i和晶型ii的混合物。按照相同的方法,用乙醇/甲基环己烷(体积比为1:5)混合溶液同样制备得到了晶型i和晶型ii的混合物。
实施例8:室温竞争试验
(1)晶型i与晶型ii的竞争实验
取等量晶型i和晶型ii样品混合均匀,加入0.8ml乙酸乙酯形成悬浊液,搅拌过夜,xrd检测,按照等量递增的方式配制晶型i和ii的混合物,与过夜样品对比得到,晶型i含量约为5%。基于晶型i的特征峰有明显的减弱趋势,可以预见,只要给予足够的时间,晶型i会完全转晶为晶型ii。从而可知室温条件下,晶型ii稳定性优于晶型i。
(2)晶型ii与晶型iii的竞争实验
取等量晶型ii和晶型iii样品混合均匀,加入0.2ml乙酸乙酯形成悬浊液,搅拌过夜,xrd检测为晶型ii。由此可见,混合物中晶型iii完全转晶为晶型ii,从而可知室温条件下,晶型ii稳定性优于晶型iii。
(3)晶型i与晶型iii的竞争实验
取等量晶型i和晶型iii样品混合均匀,加入0.2ml乙酸乙酯形成悬浊液,搅拌过夜,xrd检测为晶型i。由此可见,混合物中晶型iii完全转晶为晶型i,从而可知室温条件下,晶型i稳定性优于晶型iii。
(4)晶型iii与晶型iv稳定性对比实验
分别取晶型iii和晶型iv样品,室温干燥过夜,xrd检测显示晶型iii仅有不超过30%的量转化为晶型i。然而,晶型iv全部转晶为晶型i,稳定性较差。由上述实验可知,室温条件下,晶型i、ii和iii的稳定性均优于晶型iv。
实施例9:考察压片对晶型ii的影响
压片方法:取50mg的晶型ii,于单冲压片机下进行20公斤和25公斤压力压片。
对压片后样品粉末进行xrd检测,与初始样品进行比较,见图8,从上到下依次为初始样品、25公斤压力压片后的样品、20公斤压力压片后的样品,由图中看出压片后晶型保持不变。
实施例10:考察高温和高湿对晶型ii的影响
取两份20mg的晶型ii,分别在60℃(避光密封)和85%相对湿度下(室温避光敞口)放置,10天后进行xrd检测,与初始样品进行比较,样品晶型未发生改变,详见图9(从上到下依次为初始样品、60℃条件下的样品、85%相对湿度下的样品)。
实施例11:稳定性对比试验
申请人对式(a)化合物的无定型、晶型i和ii的混合物和晶型ii的稳定性进行了考察,取无定型、晶型i和晶型ii的混合物(重量比为1:2)、晶型ii和晶型ⅲ各200mg,采用如下方式进行考察:
包装:内层pvc材质自封袋抽真空,中层铝箔抽真空,外层铝箔干燥剂抽真空充氮气;考察条件:25℃,60%相对湿度。考察指标为总杂质含量,于不同时间点取样检测,每批次取样检测三次,取平均值,取样时间为:0、1、2、3和6个月。试验结果见下表3。
表3:稳定性试验
结论:从上表可见,晶型i,晶型ii,晶型iii及晶型i和晶型ii的混合物的化学稳定性明显优于无定型,晶型化合物对比无定型更有利于药物的质量控制及成药性。
实施例12:口服固体制剂的制备和崩解实验
按照以下三种制备方法分别制备了制备例1-11和对比例1-11的片剂,各制备例和对比例的处方组成、压片硬度和片重差异见下表3。
制备例1、7、8和9,对比例1、7、8和9采用粉末压片法:根据物料性状选择60目筛,将物料过筛备用(制备例7和对比例7中,将活性成分与赋形剂共同过筛,崩解剂与助崩剂混合过筛备用;其余的制备例和对比例分别过筛);将活性成分、赋形剂、崩解剂、助崩剂(对比例中无助崩剂)倒入三维混合机中混合,再加入润滑剂进行总混;总混物料于旋转压片机中压片。
制备例2、4、5和10,对比例2、4、5和10采用湿法制粒法:根据物料性状选择60目筛,将物料过筛备用;将活性成分、赋形剂、崩解剂、助崩剂(对比例中无助崩剂)倒入制粒机中混合,加入粘合剂水溶液(其中,制备例2和对比例2中采用5%聚维酮水溶液,制备例4和对比例4中采用10%淀粉浆,制备例5和对比例5中采用8%淀粉浆),制粒,流化床干燥;将干燥后的颗粒放入三维混合机,再加入润滑剂进行总混;总混物料于旋转压片机中压片。
制备例3、6和11,对比例3、6和11采用干法制粒法:根据物料性状选择60目筛,将物料过筛备用;将活性成分、赋形剂、崩解剂、助崩剂(对比例中无助崩剂)倒入三维混合机中混合,在干法制粒机中制粒;将制得颗粒放入三维混合机,再加入润滑剂进行总混;总混物料于旋转压片机中压片。
按照如下实验条件进行崩解实验,各制备例和对比例的崩解时间见下表4。
仪器:zbs-6e智能崩解试验仪(天津天大天发科技有限公司)
方法:吊篮法
介质:含0.5%吐温80的0.1mhcl介质
往返频率:每分钟30-32次
温度:37℃
表4片剂处方及崩解时间
结果表明,本发明的口服固体制剂具有优异的崩解速度和简便的制备方法。
实施例13:药代动力学对比试验
药品与试剂:本研究中受试样品为本发明式(a)化合物的晶型i、晶型ii、混晶(晶型i和晶型ii的混合物,其中晶型i和晶型ii的重量比为1:3)、晶型iii和无定型,样品起始物质纯度为99%以上,羧甲基纤维素钠为药用级辅料。
试验动物:sd大鼠被随机分为6组,每组6只动物,雌雄各半。
药物配制:根据各样品的重量加入适量体积的0.5%(w/v)的羧甲基纤维素钠水溶液,使药物终浓度为0.15mg/ml,置于磁力搅拌器下搅拌备用。
给药和样品采集:将各受试物按10ml/kg的剂量灌胃给予禁食的sd大鼠。在给药后的15min、30min、1h、2h、4h、6h、8h、10h和24h分别收集0.3ml血液至edta-k2的抗凝管中,于4℃下3000g离心10min后取上清液保存至-80℃待检。
样品通过lc-ms/ms(absciex,api3500qtrap)分析血浆中化合物b和阿齐沙坦。在各时间点取样的动物血浆中检测不到化合物b,给药后各受试物在动物体内会快速转化为阿齐沙坦,给予受试物后动物血浆中阿齐沙坦的药代动力学参数如下表5:
表5:药代动力学对比试验结果
结论:上述实验中,本发明的口服固体制剂中的活性成分晶型或晶型混合物的生物利用度良好,优于无定型。
- 还没有人留言评论。精彩留言会获得点赞!