延迟释放口服盐酸坦索罗辛的制作方法
本申请要求2016年5月4日提交的美国临时申请no.62/331,599的优先权,其全部内容通过引用并入本文。
背景技术:
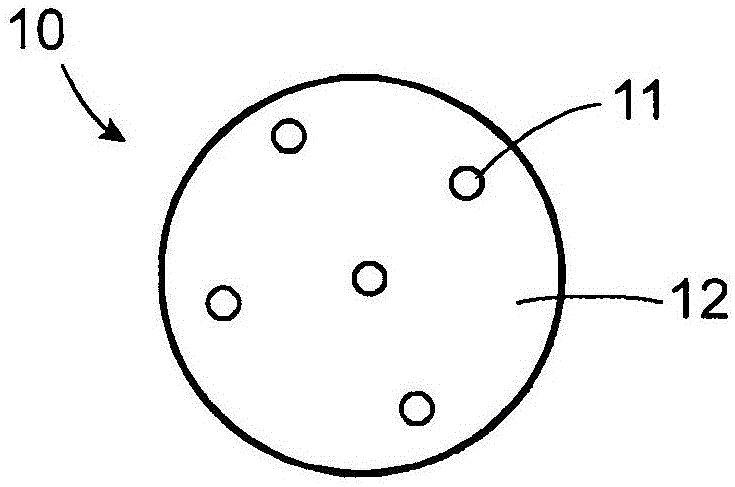
:1.发明领域在一个实施方案中,本发明涉及盐酸坦索罗辛的制剂,和更具体地涉及盐酸坦索罗辛的口服受控释放制剂,特别是涉及盐酸坦索罗辛的口服受控释放悬浮液制剂,其用于患有吞咽困难的患者。2.相关技术描述坦索罗辛是一种α1肾上腺素受体拮抗剂,经fda批准用于治疗良性前列腺增生(bph)的体征和症状。批准的产品在美国以((r)-5-(2-{[2-(2-乙氧基苯氧基)乙基]氨基}丙基)-2-甲氧基苯-1-磺酰胺盐酸盐)销售,是含有0.4mg盐酸坦索罗辛的口服胶囊(延长释放)。批准的处方信息的剂量和施用部分表明胶囊不应被压碎,咀嚼或打开。良性前列腺增生(bph)是前列腺中的基质细胞和上皮细胞扩大的病症。随着增生恶化,尿道受到影响,导致尿液流过尿道的阻力增加。这导致尿液排泄不完全,排尿困难,并且可能导致膀胱壁肌肉萎缩,以及膀胱壁的不稳定和虚弱。bph是男性的常见病,其发病率和患病率随年龄增长而增加。有一部分人群无法吞服丸剂(胶囊和片剂)。无法吞咽或吞咽困难称为吞咽困难。吞咽困难可能是由于窒息或害怕窒息或可能是由于共病现象、例如干燥的咽喉,口腔或喉咙阻塞,以及许多其他疾病和疾病。导致吞咽困难的共病现象导致老年人吞咽困难的发生率增加,但该病情不仅限于老年人,并且可能影响任何年龄的人。目前,坦索罗辛仅以胶囊形式(延长释放)提供,并且在批准的产品的处方信息中明确指示不要压碎,咀嚼或打开胶囊。因此,患有吞咽困难而需要坦索罗辛来治疗良性前列腺增生的体征和症状的人将找不到能使他们服用坦索罗辛的治疗或制剂选择。坦索罗辛首先在美国专利no.4,703,063中描述。在现有技术中还描述了受控释放的坦索罗辛制剂。例如,美国专利no.4,772,475描述了坦索罗辛,微晶纤维素和释放控制剂,例如(甲基)丙烯酸共聚物,例如(甲基丙烯酸酯共聚物)的受控释放制剂,其被颗粒化并形成片剂和胶囊。美国专利no.7,018,658描述了一种类似的产品,区别在于'658专利的颗粒包含坦索罗辛的核心,其涂覆有释放控制剂的壳。根据'658专利,'475专利的剂型的释放特征(释放曲线)不足以用于延长释放剂型(参见'658专利,第2栏,第2-12行),和由释放控制剂的外壳包围的粒料内核提供有效的延长释放(参见'658专利,第3栏,第5-25行)转向提到“小药囊”(参见'658专利,第6栏,第14-16行),但没有提供如何构建小药囊,或者如何以这种形式施用颗粒的详细信息。因此,胶囊本身有时包装在单剂量小药囊中,这可以是鉴于'658专利第6栏第30-39行中后面的讨论,合适的包装包含单位剂量的坦索罗辛,包括泡罩包装和塑料或玻璃瓶。在任何时候都没有表明坦索罗辛以液体形式施用,或者这种液体是将坦索罗辛粉末悬浮在前体液体中的结果。实际上,所有实例都涉及涂覆的粒料。不是片剂或胶囊并且可以更容易吞咽的坦索罗辛悬浮液制剂将为患有吞咽困难的患者提供制剂选择,使他们能够服用坦索罗辛并接受他们需要的药物治疗。还具有独特的ph依赖性药物释放。食物摄取前对人类的施用导致生物利用度增加30%,与食物摄取后相比,cmax增加40-70%(physiciansdeskreference2002)。因此,上的标签指示片剂每天在同一餐后大约半小时服用。不受这些进餐时间(“食物效应”)限制的坦索罗辛制剂应该增加患者的依从性并提供施用灵活性。盐酸坦索罗辛在一些国家也可作为ocas((r)-5-(2-{[2-(2-乙氧基苯氧基)乙基]氨基}丙基)-2-甲氧基苯-1-磺酰胺盐酸盐口服控制吸收系统)而获得。ocas是一种薄膜包衣的延长释放片剂,含有聚乙二醇7,000,000,聚乙二醇8,000,硬脂酸镁,丁基化羟基甲苯,无水胶体二氧化硅,羟丙甲纤维素和氧化铁黄。据称,ocas片剂仅受食物轻微影响,且受影响的方式不太可能具有临床意义。对于患有吞咽困难的男性,ocas片剂提出了与片剂相同的挑战。技术实现要素:本发明的某些实施方案涉及药物剂型,其包含坦索罗辛游离碱或其药学上可接受的盐,尤其是盐酸坦索罗辛,以受控释放基质提供,例如以小药囊(sachet)的形式,所述药物剂型当与饮用水或其他合适的液体混合时对于患有吞咽困难的患者而言是可以饮用的。在本发明的一个实施方案中,盐酸坦索罗辛作为延长释放悬浮液提供,以便于施用。如本文所用,术语“受控释放”是指从组合物或剂型中释放药剂如药物,其中药剂在延长的时间段内根据所需的性质释放。受控释放特征(释放曲线)包括例如延长释放,持续释放,延长释放,脉冲释放和延迟释放特征(释放曲线)。在某些实施方案中,本发明还涉及受控释放制剂,其包含小药囊,所述小药囊在受控释放基质中包含单位剂量的坦索罗辛干粉或其药学上可接受的盐。在某些实施方案中,本发明还涉及治疗患有良性前列腺增生的患者的良性前列腺增生的方法,该方法包括(a)提供根据本发明的受控释放制剂,(b)将所述制剂分散在液体中以分散所述粉末,然后(c)口服摄取包含如此分散的粉末的液体。在某些实施方案中,本发明还涉及制备根据本发明的受控释放制剂的方法,包括(a)在受控释放基质中制备坦索罗辛或其药学上可接受的盐的干粉,和(b)将粉末称入小药囊。在某些实施方案中,本发明还涉及可饮用的组合物,其包含(a)液体和(b)坦索罗辛的干粉或其药学上可接受的盐,其中分散有受控释放基质。附图的简要说明现在将参考附图更详细地描述本发明,其中:图1是根据本发明的单个颗粒的示意图。图2a,2b和2c是一系列预言图,每个图描绘了目前仅以胶囊或片剂形式(正方形)批准的fda批准的剂型与根据本发明的一种剂型实施方案(三角形)的药物释放百分比随时间的变化。图2a比较了本发明剂型的释放特征(释放曲线)与第一颗粒组合(a)的释放特征(释放曲线)。图2b比较了释放特征(释放曲线),其中在本发明剂型中,第一组合颗粒有意补充有另外的颗粒(b)以形成第二颗粒组合(a+b)。图2c比较了释放特征(释放曲线),其中在本发明剂型中,第二颗粒组合有意地补充有另外的颗粒(c)以形成颗粒的第三组合(a+b+c)。图3比较了本发明实施方案与的体外溶出度。图4比较了本发明实施方案(“禁食”和“进食”)与(“禁食”和“进食”)的体内生物等效性。具体实施方式本发明的示例性实施方案涉及含有坦索罗辛或其药学上可接受的盐,特别是盐酸坦索罗辛或其它药学上可接受的盐的制剂,其设计用于提供可被患吞咽困难的患者容易吞咽的延长释放口服剂型。本发明的实施方案利用活性药物成分(api)和制剂组分的属性来实现这一点,针对所述活性药物成分和制剂组分对受控释放口服悬浮液剂型的贡献,对所述活性药物成分和制剂组分进行精心选择。已经用受控释放悬浮液代替受控释放胶囊或片剂剂型的本发明实施方案提供了吞咽困难患者可以吞咽它的优点。这允许对否则将不能摄取片剂或胶囊的患者的治疗。本发明的一个示例性实施方案的组合物包含单位剂量小药囊中的受控释放粉末,其含有0.4mg盐酸坦索罗辛,已知的bph治疗日剂量。本发明另一个示例性实施方案的组合物包含0.4mg盐酸坦索罗辛的延长释放悬浮液,其分散在合适的液体,特别是水的玻璃杯中。在一个实施方案中,本发明涉及药物剂型,其包含有效治疗良性前列腺增生的量的组合,所述组合是(a)坦索罗辛或其生理学上可接受的盐的至少一种混合物的多个颗粒在(b)受控释放基质中的组合,其中当被引入随后被患者空腹摄取的一定量的液体时,所述组合在患者中表现出随时间的受控释放特征(释放曲线),其实质上与在餐后30分钟摄取的((r)-5-(2-{[2-(2-乙氧基苯氧基)乙基]氨基}丙基)-2-甲氧基苯-1-磺酰胺盐酸盐)受控释放片剂或胶囊的受控释放特征(释放曲线)相同。如所讨论的,药物剂型可以是小药囊的形式,但也可以是可以溶解在液体中的胶囊形式,例如水溶性胶囊,或者可以破碎或分成两个或更多块以使粉末状内容物倒入液体中。还可以通过直接压缩颗粒组合来形成片剂。虽然这种片剂如果尺寸足够大将不能避免吞咽困难,但这种片剂会避免的食物效应。坦索罗辛或其药学上可接受的盐的量可以在很宽的范围内变化,但最优选是治疗bph的有效量,尤其是0.4mg的单位剂量。通常,患者将每天被施用0.4mg的单一单位剂量,但是如果需要可以施用第二单位剂量(每天总共0.8mg)。还考虑了盐酸坦索罗辛的剂量范围为0.1至1mg,优选0.4至0.8mg,用于本文所述的小药囊和其他剂型。“药学上可接受的盐”是指药学上化学和制剂中常规的所有这些盐。如本文所用,“药学上可接受的盐”是指所公开化合物的衍生物,其中通过制备其盐来修饰母体化合物。药学上可接受的盐包括存在于本文公开的化合物中的酸性(例如羧酸)或碱性基团(例如伯,仲或叔胺)的盐。药学上可接受的盐的实例包括但不限于碱性残基,例如胺的无机或有机酸盐;酸性残基,如羧酸的碱(碱金属)或有机盐;等等。药学上可接受的盐包括母体化合物的常规无毒盐或季铵盐,例如由无毒无机或有机酸形成。例如,这种常规的无毒盐包括衍生自无机酸的那些,所述无机酸例如盐酸,氢溴酸,硫酸,氨基磺酸,磷酸,硝酸等;和由有机酸制备的盐,所述有机酸如乙酸,丙酸,琥珀酸,乙醇酸,月桂酸,癸酸,肉豆蔻酸,棕榈酸,硬脂酸,油酸,亚油酸,乳酸,苹果酸,酒石酸,柠檬酸,抗坏血酸,双羟萘酸,马来酸,羟基马来酸,苯乙酸,谷氨酸,苯甲酸,水杨酸,对氨基苯磺酸,2-乙酰氧基苯甲酸,富马酸,甲苯磺酸,甲磺酸,乙烷二磺酸,草酸和羟乙磺酸。化合物的药学上可接受的盐可以由母体化合物(例如化合物的非质子化碱形式,通常称为化合物的“游离碱”)、其含有碱性或酸性部分、通过常规化学品方法合成。通常,这些盐可以通过使这些化合物的游离酸或碱形式与适当的碱或酸在水中或在有机溶剂中或在两者的混合物中反应来制备;通常,优选非水介质如乙醚,乙酸乙酯,乙醇,异丙醇或乙腈。合适的盐的列表可参见remington'spharmaceuticalsciences,20thed.,lippincottwilliams&wilkins,baltimore,md,2000,p.704,其公开内容通过引入并入本文。药学上可接受的盐也可以通过使游离酸或碱形式的化合物分别与适当的碱或酸在熔融过程中,任选地在其它药学上可接受的赋形剂(例如蜡)存在下反应来制备。如本文所用,术语“熔融过程”是指这样的过程,其中化合物的游离酸或碱形式溶解在一种或多种熔融形式的赋形剂中(即,在室温下为固体)以制备溶液,其中碱或酸分别与游离酸或碱形式的化合物相互作用,形成所需的药学上可接受的盐。本发明考虑的药物活性剂的其他盐是为了改变相对于母体药物化合物(例如,化合物的游离酸或游离碱形式)的溶解度和/或溶出度,包括,但是不限于,果胶酯酸盐,单宁酸,植酸盐,水杨酸盐,糖精酸盐,双氧噁噻嗪酸盐(acesulfamate),没食子酸盐和对苯二甲酸盐。坦索罗辛(和其他药物)的显著副作用是它在口中留下苦味,这在考虑吞咽液体制剂时是有问题的。因此,本发明的优选实施方案涉及配制坦索罗辛作为糖精酸盐(或其他糖盐)。我们考虑在本文另外描述的任何实施方案中特异性使用坦索罗辛糖精酸盐作为活性成分。例如,如果具体实例涉及盐酸坦索罗辛,那么通过本段的效果,同样阐述了其中用坦索罗辛糖精酸盐替代盐酸坦索罗辛的类似实施方案。作为使用糖盐的替代或补充,可以使用调味剂和/或甜味剂。可用于本发明的调味剂包括但不限于天然调味剂,天然水果调味剂,人造调味剂,人造水果调味剂,增味剂或其混合物。天然调味剂,人造调味剂或其混合物包括但不限于薄荷(例如,胡椒薄荷或留兰香(绿薄荷)),柠檬,酸橙(lime),橙,草莓,薄荷醇,肉桂,香草,人造香草,巧克力,人造巧克力或泡泡糖(bubblegum)。天然水果调味剂,人造水果调味剂或其混合物包括但不限于樱桃,葡萄,橙子,草莓或柠檬。增味剂包括但不限于柠檬酸。尽管调味剂通常作为次要组分提供并且其量有效地为液体药物组合物提供可口的风味,但优选添加至少一种调味剂;更优选地,可以使用多达两种调味剂。用于本发明制剂中的调味剂的范围为约0.01至约0.15克/100ml。调味剂通常以根据个体风味而变化的量使用,并且可以例如以小药囊的最终组合物的重量/重量的约0.01%至约10%的量计。甜味剂的实例包括甜味剂,人造甜味剂和基于二肽的甜味剂,例如单糖,二糖和多糖,例如木糖,核糖,葡萄糖,甘露糖,半乳糖,果糖,葡萄糖,蔗糖,糖,麦芽糖,部分水解的淀粉,或玉米糖浆固体和糖醇如山梨糖醇,木糖醇,甘露醇,糖精酸盐,即钠或钙糖精酸盐,环己基氨基磺酸盐,乙酰磺胺酸钾(双氧噁噻嗪酸钾)(acesulfam-k),甘草酸铵,甘草酸二钾和游离酸形式的糖精l-天冬氨酰苯丙氨酸甲酯(saccharinl-aspartylphenylalaninemethylester)和它们的混合物。通常,甜味剂的存在量相当于小药囊总组合物的约1至60%重量/重量,该量部分取决于是否存在其它甜味剂成分和所需的甜度水平。通常,当使用糖时,其含量为组合物的约10%至约50%w/v。应当理解,可以使用甜味剂的组合。当使用时,甜味剂也可以单独使用或彼此组合使用。当使用人造甜味增强剂时,其可以以小药囊的最终组合物的约0.05%至约15%重量/重量的量存在。其它添加剂包括表面活性剂,例如十二烷基硫酸钠(月桂基硫酸钠),脱水山梨糖醇月桂酸酯,聚丙烯酸酯及其盐,例如聚丙烯酸钠,二辛基磺基琥珀酸钠,羟基化钠(sodiumoxylate),酒石酸钠及其混合物,或其它对于本领域技术人员而言众所周知的表面活性剂;或稀释剂,例如羟丙基甲基纤维素,纤维素,滑石,淀粉,例如玉米淀粉,大米淀粉(ricestarch),木薯淀粉,小麦淀粉,马铃薯淀粉,预胶化淀粉,或淀粉羟乙酸钠,或明胶,及其混合物,或本领域技术人员熟知的其他稀释剂。在某些实施方案中,坦索罗辛或其药学上可接受的盐以干粉形式提供在受控释放制剂中,用于分散在合适的液体,例如水,果汁(橙汁,苹果汁,菠萝汁等)中。通常,调味剂,甜味剂,表面活性剂,稀释剂和其它常规赋形剂将作为粉末本身在受控释放制剂中组合。坦索罗辛或药学上可接受的盐在与api相容的受控释放基质中提供,其形式促进受控释放制剂在液体中的悬浮,并且一旦含有悬浮的受控释放制剂的液体由患者摄取,则缓慢释放api。在一个特别优选的实施方案中,受控释放制剂包含在小药囊中。将api和受控释放基质混合,优选均匀混合,并减少成干粉,优选通过挤出混合物作为挤出物,然后粉碎或优选微粉化。然后将所得干粉称重并如果需要包装成小药囊,以及调味剂,甜味剂,表面活性剂,稀释剂和其它常规赋形剂。选择重量使得例如每个小药囊提供0.4mg坦索罗辛或药学上可接受的盐的单位剂量。给予患者小药囊,遵照指示以每天将一个小药囊分散到合适的液体中以治疗bph(或其他疾病或障碍)。患者将小药囊分散到他们选择的合适液体、例如水或果汁中,然后摄取所得悬浮液。由于悬浮液对于患有吞咽困难的患者而言更容易容忍,因此患者依从性应该比目前用坦索罗辛胶囊观察到的程度增加。此外,由于本发明的悬浮液(或胶囊或片剂)的释放在接近餐服用时不会显著增加(如的情况),本发明的悬浮液(或胶囊或片剂)可以空腹服用,例如,在早晨或就寝时间,从而也增加了患者依从性和施用灵活性。实际上,食物对本发明剂型的影响与食物对的影响根本不同。虽然食物摄取减缓了本发明剂型中活性成分的释放,但与的情况形成鲜明对比,其中在没有食物的情况下释放速率增加,在本发明剂量的情况下释放速率减慢不构成安全问题。api的干粉和受控释放基质将包含多个选择和组合的颗粒,使得药物剂型当引入随后由患者摄取的一定量的液体时,表现出所需的随着时间推移的患者中api受控释放特征(释放曲线)。参考图1,显示了根据本发明的颗粒10的一个实施方案的图示。该颗粒包含受控释放基质12,其中分布有api11的分子,优选均匀分布。当这些颗粒与悬浮液体一起被摄入患者体内时,api分子11将从受控释放基质12中释放出来。影响药物从基质材料中释放的因素是本领域熟知的。例如,参考m.varma等人,“factorsaffectingmechanismandkineticsofdrugreleasefrommatrix-basedoralcontrolleddrugdeliverysystems,”am.j.drugdeliv.,2(1):43-57(2004),其内容通过引用并入本文。本发明利用以下事实:在由api+基质的相同混合物产生的不同颗粒之间或由api+基质的不同混合物产生的不同颗粒之间存在api释放速率的差异,以产生颗粒的复合分组,基于它们在api的释放上的差异而有意地单独选择,它们共同组合以以期望的受控api释放速率释放api。在一个优选的实施方案中,选择可以像分离异常小的颗粒和/或异常大的颗粒一样简单,例如借助于一个或多个网筛,直到获得颗粒的组合,所述颗粒的组合当与剂型的其它成分、例如甜味剂和其他赋形剂组合时,模仿患者体内的释放。在另一个优选的实施方案中,产生单个颗粒,单独测试颗粒的释放性质,并对颗粒与小药囊的预期共内容物(co-content)、和体内液体的组合进行测试,并记录和编目结果。如此编目的各种类型的单个颗粒被选择并组合,采用的方式被计算以得到所需释放特征(释放曲线),然后通过实际测试确认该计算。以这种方式选择和组合单个颗粒示于图2a-2c中。图2a显示了具有颗粒(a)的第一组合的本发明剂型的一个实施方案。该颗粒(a)的第一组合可以给出与在相同测试条件下由fda批准的剂型显示的释放特征(释放曲线)(正方形)显著不同的释放特征(释放曲线)(三角形)。本发明在该特定实施方案中考虑通过有意地补充额外的颗粒(b)以形成颗粒(a+b)的第二组合来改变颗粒(a)的第一组合,所述颗粒(a+b)的第二组合具有与通过fda批准的剂型所展示的(如图2b所示)更接近的释放特征(释放曲线)。可以以相同的方式进行进一步的改变,直到两个释放特征(释放曲线)实质上相同,如图2c所示。“实质上相同”是指两个释放特征(释放曲线)之间至少80%一致,优选至少90%一致,更优选至少95%一致,最优选至少99%一致。在一个实施方案中,根据本发明的药物剂型包含多个颗粒,选择所述颗粒的粒度分布使得当将药物剂型引入一定量的液体并由患者摄取时,实现所需的随时间的释放速率。在另一个实施方案中,根据本发明的药物剂型包含多个颗粒,所述颗粒选自api和受控释放基质的不同混合物,并且组合所述颗粒使得当将药物剂型引入一定量的液体并由患者摄取时实现期望的随时间的释放速率。在一个特别优选的实施方案中,药物剂型包含第一受控基质中的api颗粒以及第二受控基质中的api颗粒,其中第一受控基质和第二受控基质是不同的。在第二个特别优选的实施方案中,药物剂型包含第一受控基质中的第一浓度的api的颗粒以及第二受控基质中的第二浓度的api的颗粒,其中api的第一浓度不同于api的第二浓度,并且其中第一受控基质与所述第二受控基质相同或不同。根据本发明的药物制剂本身不包含任何溶解作为ph的函数的肠溶包衣。这些包衣是本领域技术人员公知的,允许剂型即使被摄取通过胃而不释放活性成分,然后活性成分仅在或主要在肠道中受到受控释放。这种肠溶包衣优选在5至7的ph下溶解。尽管本发明的剂型本身不含这种肠溶包衣,但将肠溶包衣材料组合在受控释放基质中可能是有利的。这些肠溶包衣材料优选选自虫胶,聚甲基丙烯酸/丙烯酸乙酯或甲基丙烯酸/丙烯酸甲酯/甲基丙烯酸甲酯共聚物,甲基丙烯酸/甲基丙烯酸甲酯共聚物,乙酸羟丙甲纤维素琥珀酸酯(hydroxypropylmethylcelluloseacetate-succinate),乙酸邻苯二甲酸纤维素,聚乙酸乙烯酯-邻苯二甲酸酯,羟丙基甲基纤维素邻苯二甲酸酯和/或乙酸纤维素-偏苯三酸酯。通过本领域技术人员已知的各种方法可以实现超过原位引起的释放的进一步延迟,并因此进一步改变活性成分的释放。可用于本发明的基质材料包括本领域技术人员已知的生理学相容的亲水性材料,例如,bauer等人,“berzogenearzneiformen”(“coatedpharmaceuticalforms”),wissenschaftlicheverlagsgesellschaftmbhstuttgart,1988,p.69参照以下,其通过引用并入本文,因此形成本公开的一部分。所用的亲水基质材料优选为聚合物,特别优选纤维素醚,纤维素酯和/或丙烯酸树脂,优选聚(甲基)丙烯酸酯。所用的基质材料非常特别优选乙基纤维素,羟丙基甲基纤维素,羟丙基纤维素,羟甲基纤维素,聚(甲基)丙烯酸和/或其衍生物,例如它们的盐,酰胺或酯。其他优选的基质材料包括疏水性材料,例如疏水性聚合物,蜡,脂肪,长链脂肪酸,脂肪醇或相应的酯或醚,或其混合物。所用的疏水性材料特别优选为c12-c30脂肪酸甘油单酯或甘油二酯和/或c12-c30脂肪醇和/或蜡,或其混合物。适用于缓释基质的材料还包括聚环氧烷,优选聚环氧乙烷;聚乙酸乙烯酯;和聚乙烯吡咯烷酮。所用的缓释基质材料也可以是所述亲水和疏水性材料的混合物。基质组分将构成本发明剂型的主体。对于含有优选的0.4mg坦索罗辛的单位剂型,基质组分的总量将为50-1000mg,优选75-500mg,最优选100-400mg。为了调节活性物质的释放速率,本发明的制剂除水不溶性聚合物外还可任选地含有非延迟的,优选水溶性的聚合物,其量最多为30wt%,如聚乙烯吡咯烷酮;或水溶性纤维素,优选羟丙基甲基纤维素或羟丙基纤维素;和/或亲水性成孔剂,如蔗糖,氯化钠或甘露醇;和/或本领域已知的增塑剂。为了进一步延缓活性成分的释放,根据本发明的药物制剂还可优选含有均匀分布在缓释基质中的活性成分。在优选的实施方案中,受控释放材料包含至少一种选自乙基纤维素,羟丙基甲基纤维素及其衍生物的材料。在更优选的实施方案中,受控释放材料包含(i)乙基纤维素或其衍生物,和(ii)羟丙基甲基纤维素或其衍生物的混合物。在最优选的实施方案中,乙基纤维素或其衍生物选自乙基纤维素,乙基纤维素和乙酸邻苯二甲酸纤维素的掺合物;羟丙基甲基纤维素或其衍生物选自羟丙基甲基纤维素,乙酸羟丙甲纤维素琥珀酸酯和羟丙基甲基纤维素邻苯二甲酸酯。粒度和基质组成是影响本发明制剂中活性成分释放特征(释放曲线)的因素。粒度范围为1-1000μm,尤其是1-500μm,优选<10-400μm,最优选<10-350μm。在这些范围内的较小尺寸的颗粒可以改善分散产品的美观性,例如,如美国专利no.5,149,541中所述,其涉及(欧车前果壳(psylliumhusk))。尺寸在100-600μm,优选125-500μm,最优选150-450μm的颗粒的组合提供特别好的结果,其中基质基于纤维素衍生物,特别是乙基纤维素和/或羟丙基甲基纤维素,和其酸酯。在最优选的制备实施方案中,本发明的剂型通过以下制备:(a)挤出挤出物,所述挤出物包含:(i)坦索罗辛或其盐,(ii)羟丙基甲基纤维素或其衍生物的混合物,和(iii)乙基纤维素或其衍生物,(b)将挤出物减少成粉末,(c)从所述各种尺寸的粉末颗粒中选择,以得到具有颗粒尺寸分布的所选颗粒的组合,使得当通过患者空腹摄取所选颗粒的组合以及一定量的液体和任选的赋形剂时,在所述患者中产生坦索罗辛或其盐在给定时间段过程内的体内释放特征(释放曲线),其实质上模拟在餐后30分钟由所述患者摄取的相同时间段内体内释放特征(释放曲线)。以下针对挤出产品示出了用于使任何合适的活性成分适应本发明的简要方案。简要方案:小药囊的颗粒组分的开发步骤:建立与盐酸坦索罗辛的基质相容性第一层替代基质组分50至80%va64(乙烯基吡咯烷酮-乙酸乙烯酯)2至10%peg1500(聚乙二醇500)/40至90%va64/10%至40%hpmc10%peg1500/40%va6410%tpgs(生育酚丙二醇琥珀酸酯)/40至10%tpgs/40%va64hpmcasmf(乙酸羟丙基甲基纤维素-琥珀酸酯)eudragitl30eudragitrspohpmct(乙酸羟丙基甲基纤维素-偏苯三酸酯)第二层替代基质组分甲基丙烯酸共聚物a型,nf甲基丙烯酸共聚物b型,nf甲基丙烯酸共聚物c型,nf聚环氧乙烷聚乙烯吡咯烷酮乙烯醋酸乙烯酯聚(l-乳酸-共-羟乙酸)polycaprolate微晶蜡黄原胶(xanthumgum)丙二醇乙基纤维素甘油二硬脂酸酯甘油三硬脂酸酯甘油二油酸酯甘油三油酸酯基质稀释剂多库酯钠黄原胶聚山梨醇酯80微晶纤维素聚乙二醇基质润滑剂硬脂酸月桂基硫酸钠淀粉预胶化淀粉滑石将基质组分的混合物挤出或滚圆。准备尺寸范围内的颗粒:产生单独的研磨颗粒级分;<10微米,10至20微米,20至30微米,30至40微米,40至50微米,50至60微米和60至70微米。混合粒度级分以建立合适的粒度混合物小药囊混合物的开发:添加表面活性剂,稀释剂粉末和甜味剂表面活性剂和/或分散剂十二烷基硫酸钠(月桂基硫酸钠)脱水山梨醇月桂酸酯聚丙烯酸酯聚丙烯酸钠二辛基磺基琥珀酸钠羟基化钠(sodiumoxylate)酒石酸钠稀释剂hpmc纤维素滑石玉米淀粉大米淀粉木薯小麦淀粉马铃薯淀粉预胶化淀粉淀粉乙醇酸钠明胶调味剂(gras代理商)甘露醇山梨糖醇草莓调味剂柠檬调味剂酸橙调味剂三氯蔗糖混合表面活性剂,稀释剂粉末和甜味剂表征在稀释剂中的小药囊以便饮用准备与小药囊组分兼容的各种浆料;在8盎司水中的浆料在8盎司果汁中的浆料橙子酸果蔓番茄葡萄柚在8盎司苹果酱中的浆料表征每个目标产品概况(见表1)效力药物释放特征(释放曲线)杂质表1:口服混悬液粉末的开发目标产品概况dtpp=开发目标产品概况药物释放方法使用用于盐酸坦索罗辛片剂的usp方法。在各种介质中比较实验制剂与ld第1阶段:在酸性缓冲液中2小时:在ph1.2的0.003%聚山梨醇酯80(使用hcl)使用装置2用于片剂(供参考),小药囊不需要腔室100rpm测试时间0,30分钟,1小时,1.5小时和2小时随后是第2阶段:磷酸盐缓冲液ph7.2中的溶出曲线将装置2用于片剂(供参考),小药囊不需要腔室100rpm测试0,1小时,2小时,3小时,4小时,5小时和6小时根据supac-mr指南要求,在ph1.2、4.5和6.8介质中产生额外的溶出曲线通过以下来确定等效性:实现多点溶出度相似性(f2)值>50,采用针对实验制剂和ld获得的结果与ld溶出曲线相比,最佳配方将显示>50的溶出度相似性(f2)值现在将参考以下非限制性实施例更详细地描述本发明。实施例实施例1:(预测):将以重量百分比表示的下列组分在混合器中均匀混合,例如辊式混合器,振荡混合器,剪切混合器或强制混合器:(给出的百分比是基于配方的总重量的重量。)将混合物通过双螺杆挤出机挤出,例如,可从leistritz(nurnberg)公司购得的zse18hp40d型双螺杆挤出机,优选用配有偏心螺杆端的螺杆。可以使用具有8个孔的可加热喷嘴板作为喷嘴,每个孔的直径为1.0mm。挤出参数可以设定为,例如以下值:螺杆速度:150rpm;通过量:2公斤/小时;产品温度:60℃至140℃,优选80℃至140℃,最优选110℃到140℃,采用相应的桶温。将棒状挤出物切割成预定长度,并在合适的设备中通过微粉化将切割的块减少成粉末。根据尺寸分离粉末颗粒,并且基于单个颗粒的释放特征(释放曲线)的知识,在此基础上选择所述颗粒并将其重新组合成预期具有所需的api释放特征(释放曲线)的新粉末。将该粉末称重并与调味剂,表面活性剂和甜味剂一起装入小药囊中,每个小药囊含有0.4mg盐酸坦索罗辛的单位剂量。实施例2:(预测):以与实施例1类似的方式,将以下所示重量百分比的组分在混合器中均匀混合,挤出,切割和微粉化:根据尺寸分离粉末颗粒,并且基于各个颗粒的释放特征(释放曲线)的知识,在此基础上选择所述颗粒并将其与实施例1中的所需颗粒一起重新组合成预期具有所需api释放特征(释放曲线)的新粉末。将该粉末称重并与调味剂,表面活性剂和甜味剂一起装入小药囊中,每个小药囊含有0.4mg盐酸坦索罗辛的单位剂量。实施例3:(预测):用载体和释放控制剂挤出结晶活性药物成分,研磨并包装在小药囊中。(给出的百分比是基于配方的总重量的重量。)实施例4:(实际):将以下成分组合为挤出前掺合物:盐酸坦索罗辛10wt.%ethocelstandard10premium45wt.%乙酸羟丙甲纤维素琥珀酸酯,45wt.%nf,as-mmp等级*所有“wt.%”基于挤出前掺合物的总重量将挤出前掺合物引入料斗并通过leistritz(nurnberg,germany)zse18hp40:1双螺杆挤出机的模头(die)挤出,该挤出机以100±10rpm的螺杆速度操作;通过量:0.5±0.2千克/小时;熔板温度:160℃±5℃;加热区温度:160℃±5℃;和模头加热器温度:160℃±5℃。将挤出物挤出到传送带上并冷却。使用具有0.033”穿孔筛网的fitzmill研磨冷却的挤出物,使叶片处于锤位置,直至获得粉末。将该粉末进行40目和100目筛网以除去细颗粒和粗颗粒,并使得到的粉末的粒度范围在150-425μm之间。然后将一部分该粉末与干燥赋形剂组合入具有以下配方的小药囊:实施例5:(预测):实施例4的制剂不是组合入小药囊,而是组合入水溶性的两部分胶囊。可以将内容物通过以下引入液体,例如水或果汁:简单地通过使整个胶囊落入液体中,或者通过分离胶囊的两个部分并将胶囊内容物倒入液体中。实施例6:(预测):实施例4的制剂不是组合入小药囊,而是在压片机上压制成圆形或其它形状的丸剂。可以像使用胶囊一样吞服丸剂。然而,因为本发明的胶囊具有不同的组成,它们应该没有胶囊食物效应。实施例7:(实际):溶出度研究目的该研究的主要目的是通过hplc建立实施例4的小药囊制剂的体外特征与根据usp<711>坦索罗辛溶出度测试要求(胶囊usp专论溶出度测试3)测量的胶囊的体外特征进行比较的相似性:总测试时间为8小时;酸阶段:2小时在0.003%聚山梨醇酯80ph1.2(500ml)中100rpm;缓冲剂阶段:总共6小时,磷酸盐缓冲液ph7.2;在3小时:65%至85%溶解;和在8小时:nlt溶解80%。方案:溶出参数酸阶段500ml酸阶段介质加入到每个溶出容器中,并使其平衡至37℃±0.5℃。将各制剂称重并将全部内容取出并小心洒入六(6)个溶出容器中的每个和启动计时器,在2小时,将单一的5毫升样品使用插管从每个容器中取出。从溶出介质表面和桨叶顶部之间的中间区域取样,距离容器壁和轴不小于1cm。使用0.45μm尼龙膜将样品直接过滤到玻璃hplc小瓶中。溶出500毫升预热的(至37℃)缓冲剂阶段浓缩液加入到含有酸阶段介质的六(6)个溶出容器的每个。然后总体积为1000ml和具有ph7.2。该计时器被允许继续。在第3小时和8小时,将单一的5毫升样品使用插管从每个容器中取出。从溶出介质表面和桨叶顶部之间的中间区域取样,距离容器壁和轴不小于1cm。使用0.45μm尼龙膜将样品直接过滤到玻璃hplc小瓶中。样品将在环境实验室条件下储存三(3)天后到期。hplc参数结果:将实施例4的制剂的溶溶出与胶囊进行比较。结果如图3所示。两条曲线非常相似,证明在体外和体内进一步研究是合理的。实施例8:(实际):临床研究目的该研究的主要目的是在禁食和进食条件下确定实施例4的制剂(下文中的“坦索罗辛drs”)与胶囊的生物等效性。次要目的包括评估食物对坦索罗辛drs药代动力学(pk)的影响,或缺乏食物对坦索罗辛drs药代动力学(pk)的影响;评估有和没有食物情况下的单剂量坦索罗辛drs的安全性和耐受性。该研究设计为两阶段研究。在第0天/第1期研究开始前30天内筛选潜在受试者的研究合格性。第1阶段:配方评估和施用方案第1阶段是开放标签,部分随机,三期交叉设计。受试者被随机分为两个序列之一。在禁食和进食条件下共有12名受试者参加了第1阶段。中途退出者没有被代替。施用方案如下:在第1阶段之后,进行样品的生物分析分析。第2阶段:生物等效性和食物效应第2阶段是一项开放标签,随机,单中心,单剂量,4期,交叉,生物利用度研究。在每个期,受试者接受单剂量的坦索罗辛drs(测试)或(参考)。在第2阶段共招募了36名受试者。中途退出者没有被代替,除非受试者入选率低于30名完成受试者。施用方案如下:入选标准除非另有说明,否则受试者必须满足以下所有入选标准才有资格参加本研究:1.健康的成年男性志愿者18至60岁(包括端点在内)。2.能够理解并提供签署的知情同意书。3.愿意遵守研究的要求。4.坐位血压收缩压介于100和140mmhg之间,舒张压介于60和90mmhg之间(含端点在内)。5.坐姿脉搏率在45到100次/分钟之间(含端点在内)。6.正常活动和另外被判断为健康状况良好,根据医疗史和体格检查。7.体重指数(bmi)19.0和32.0kg/m2。8.体重>55公斤。排除标准如果受试者符合以下任何排除标准,则他们未参加该研究:1.历史上任何临床显著心血管,肝,肾,肺,血液,胃肠,内分泌,免疫,皮肤或神经系统疾病。2.第1阶段1期和第2阶段4期报到前28天之内任何使用处方药物。目前服用用于治疗高血压的任何药物。目前服用用于治疗高胆固醇血症的任何药物。3.第1阶段1期和第2阶段4期报到前7天内任何使用非处方(otc)药物。4.对坦索罗辛或其他相关药物的过敏反应史。5.异常和临床相关的(如由主要研究者确定[pi])心电图(ecg)描图。6.在施用前28天内临床显著疾病或手术(包括流感,流感样症状,腹泻,呕吐)或研究前医疗评估或施用时的急性疾病。7.在研究药物的初始剂量之前28天内使用公知改变肝酶活性的任意的药物(例如,奥美拉唑或其它质子泵抑制剂[ppis])8.筛查时对乙型肝炎,丙型肝炎或人类免疫缺陷病毒(hiv)的阳性检测。9.由研究人员确定的在医学筛查期间发现的任何临床上显著的异常实验室测试结果。临床意义的定义将与进行测试的当地实验室的每个测试的正常水平相关。注意:高于或低于正常范围的测试值不一定表示该值是“临床显著的”。决定应由调查员自行决定,必要时与医疗监测员协商。10.在筛选或1期报到时丙氨酸氨基转移酶(alt)或天门冬氨酸转氨酶(ast)水平>正常上限的1.25倍。11.阳性药物筛选或连续筛查。12.研究开始(第一剂量)前3个月内使用任何尼古丁产品的历史。13.主要精神疾病的历史,这在研究者观点中,可能会影响受试者参与研究的能力。收容入院的受试者将没有资格参与。14.在研究开始(第一剂量)前30天内接触任何研究药剂。15.在研究开始(第一剂量)前30天内暴露于坦索罗辛。16.受试者在研究开始(第一剂量)前56天内捐献(标准捐赠量或更多)血液或血液制品(除了血浆,如下注释)。17.受试者在研究开始(第一剂量)前7天内曾进行血浆捐献。18.受试者具有研究者认为会干扰提供知情同意或遵守研究指导的能力,或可能混淆研究结果的解释或使受试者处于不适当风险中的病症。研究程序:筛选在在第1阶段第0天/第1期研究开始前30天内和第2阶段第42天/第4期的30天内,筛选潜在的受试者的研究资格。在到达以进行筛选访问之前,将指示受试者禁食至少8小时(为了实验室概况)。筛选评估将包括以下内容:6.签署知情同意书7.确定资格,纳入和排除标准(在筛选访问时可以确定的数量)8.医疗史9.之前(在过去30天内使用)和伴随施用(目前正在使用)10.身体检查,包括没有鞋子的身高和体重11.bmi确定12.坐下至少3分钟后的生命体征(血压,脉搏,呼吸和体温)13.十二导联心电描记法,仰卧至少3分钟14.实验室概况,包括血液学,化学小组和尿液分析15.hiv抗体,乙型肝炎表面抗原和抗丙型肝炎的抗体滴度16.滥施用物的毒理学筛查和酒精呼气测醉器试验。研究中继续的受试者的结果必须为阴性。17.将指导受试者遵守以下限制:a.在第1期(第1阶段)或第4期(第2阶段)报到前28天内停止使用处方药。b.在第1期(第1阶段)或第4期(第2阶段)报到前7天内停止使用非处方药(包括任何非甾体类抗炎药[nsaids]和含阿司匹林的药物)。c.在第1期(第1阶段)或第4期(第2阶段)报到前7天内停止使用维生素或草药补充剂。d.在每个研究期的报到前24小时内,戒除含有罂粟种子的食物。e.从报到前48小时到每个研究期的最后一次样本收集,不要食用含酒精的产品。f.从每个研究期的报到前24小时戒除含咖啡因、黄嘌呤衍生物或黄嘌呤相关化合物的食品或饮料,或能量饮料。g.从第1期(第1阶段)或第4期(第2阶段)报到前7天到整个研究中戒除含有葡萄柚和/或塞维利亚桔子的食物或饮料。h.从筛选和整个研究中戒除st.jon的麦芽汁。i.从筛查到整个研究过程中,不要使用烟草或其他含尼古丁的产品。j.从筛查到整个研究过程中,不要使用雄激素或促蛋白合成类固醇。报到(每期)受试者将在施用日前一天大约下午4:00进入机构报到(报到在第1天为第0天施用,报到在第6天为第7天施用,报到在第13天为第14天施用,报到在第41天为第42天施用,报到在第48天为第49天施用,报到在第55天为第56天施用,和报到在第62天为第63天施用)。1.将审查纳入/排除标准,并审查和评估持续资格。2.将在第-1天(第1阶段)和第41天(第2阶段)进行基线十二导联心电描记法。3.每次研究期报到时将进行药物筛选和酒精呼气测醉器测试。结果必须为阴性才能继续进行研究。4.自筛选访问以来受试者的医疗史的任何变化将被记录并添加到医疗史中。自第1期(第1阶段)或第4期(第2阶段)施用以来,医疗史的临床显著变化将被记录为不良事件(ae)。5.将收集临床化学和血液学并评估任何临床显著的异常值。6.将询问受试者自筛选访问或之前研究期间采取的任何药物以及遵守限制。7.标准晚餐将在下午5:00-7:00pm之间或在诊所正常时间供应给受试者。将在预定施用(第二天)前至少10.5至11小时提供可选的零食。除了方案定义的高脂肪餐外,在此零食之后将开始至少10小时的监督禁食。8.研究者将在每次研究药物施用之前审查所有上述信息和实验室结果并确定受试者的持续合格性。研究第1期(第1阶段)或第4期(第2阶段)1.根据以下评估受试者对研究的持续资格:纳入/排除标准和遵守禁食和处方和非处方药,草药和其他方案概述的食品限制的要求2.在第1期(第1阶段)或第4期(第2阶段)施用研究药物之前,将受试者随机分配至治疗序列在每个研究期3.在第0天,7天,14天,42天,49天,56天及63天早晨,受试者接受坦索罗辛drs或flomax(禁食或进食),这是在监督过夜禁食至少10小时后进行的。食品将被控制和标准化,这针对每个住房期以及针对所有受试者。在至少10小时过夜禁食之后,随机的受试者在药物施用前30分钟将接受标准化的高脂肪,高热量餐。一个实例餐将是两个鸡蛋炒黄油,两条咸肉,两片黄油吐司,4盎司土豆煎饼和八盎司全脂牛奶。可以替换此试验餐,只要制成的餐提供了类似的来自蛋白质、碳水化合物和脂肪的卡路里量,并且具有可比的餐的一致性。受试者必须在30分钟或更少时间内吃这餐的总含量。在每个研究期施用后大约4.5和9.5小时将提供标准化餐。晚间零食被允许在每个研究期施用后大约14小时。4.在研究药物的施用之前(施用之前在120分钟内),受试者将在评估之前坐至少3分钟后进行生命体征(血压,脉搏,体温,和呼吸)。5.用于pk分析的血液样本将在以下时间点进行:分析物时间(小时)坦索罗辛t01x1x2x3x4x5x6x8x10x12x18x24x30x36x48x‘在施用日施用之前120分钟内采样。基于5个小时的报道t1/2。6.临床化学和血液学评估的血液样本将在筛选,在每个研究报到和研究退出时收集。a.临床化学(血液化学)将包括alt,白蛋白,碱性磷酸酶,ast,血液尿素氮(bun),钙,二氧化碳(碳酸氢盐),氯化物,肌酸酐,葡萄糖,无机磷酸盐,镁,钾,钠,总胆红素,总蛋白质,尿酸b.血液学将包括血细胞比容(hct),血红蛋白(hgb),白细胞(wbc)计数和血小板计数和差别的7.尿液分析的尿样将在筛选和研究退出时收集。a.尿液分析将包括葡萄糖,胆红素,比重,血,ph值,蛋白质,尿胆红素,亚硝酸盐,白血球酯酶和显微镜下评定(如果认为有必要通过细胞异常的证据)。8.所有受试者在各施用天他们的计划的施用时间后禁食不少于4小时。在各施用天施用后4小时禁食后,标准餐将在约束期间在指定时间提供。餐计划将对于每个研究期相同。9.一旦受试者接受第一剂研究药物并且持续到最后一次剂量或提前终止后48小时,将开始监测治疗紧急不良事件(teae)。10.生命体征:坐血压和脉搏率将在以下测量:在第0天,7天,14天,42天,49天,56天及63天(评估之前坐至少3分钟后)施用后约12小时和24小时[±15分钟],并且在被视为医疗必要的任何其它时间。从诊所退出或提前终止受试者将被限制在研究中心,直到每期施用后至少48小时。在该期完成所有方案定义的程序后,让受试者离去。将提供关于清洗期(食物/药物限制)以及何时返回以进行下一研究期(在两次剂量之间至少清洗7天)的提醒。在最终抽血(第16天第1阶段和第65天第2阶段)或提前退出后,将进行以下评估:11.审查ae和伴随药物12.身体评估,包括出血迹象13.禁食实验室概况(不包括血清学和药物/酒精测试)14.体检,包括出血迹象15.十二导联心电描记法,仰卧至少3分钟然后,受试者将从研究中心离去。如果受试者早期退出研究,将尽一切努力使受试者在施用后至少48小时保持在诊所中。如果他们不能留在诊所,将尽一切努力让受试者在施用后约48小时返回诊所以进行上述评估。总研究持续时间约为第1阶段的17天和第2阶段的25天(不包括筛选期)。对临床地点的限制将是从每个研究期施用前一天的大约下午4:00到施用后48小时(每个施用期总共约72小时)。评估标准:药代动力学将根据血浆浓度-时间过程数据估计以下非房室pk参数:auct,auc∞,cmax,tmax,kel,cl/f,t1/2,和vd/f。针对在浓度-时间数据中未显示末端对数线性相的情况没有报告auc∞,kel,cl/f,和t1/2的数值。将估计单剂量pk参数。安全性安全性评估将包括筛查,施用前和施用后的生命体征,ecg,临床实验室检测(血液学,血液化学和尿液分析),ae的记录和体格检查。统计方法:药代动力学药代动力学参数(auct,auc∞,cmax,tmax,kel,cl/f,t1/2,和vd/f将在受试者接受坦索罗辛的天数在坦索罗辛的血浆浓度时间过程数据的非房室分析后被估计。将计算所有pk参数的算术平均值,标准偏差和变异系数。另外,将计算auct,auc∞,和cmax的几何平均值。方差分析(anova)将对变换的(in-transformed)pk参数auct,auc∞,和cmax进行。每个anova将包括以下的计算:最小二乘平均值(lsm),调整平均值之间的差异,以及与这些差异相关的标准误差。均值的比率将使用用于变换的(in-transformed)auct,auc∞,和cmax的lsm计算。使用变换的(in-transformed)浓度数据计算参数auct,auc∞,和cmax的各分析物差异的90%置信区间[ci]。ci将表示为相对于lsm的百分比。将进行以下比较:第1阶段第2阶段如果源自用于比较#9的变换的(in-transformed)pk参数、auct、auc∞、和cmax的分析(如上面概述的)几何最小二乘平均值的比率的90%ci是在可接受的范围内,则结论是没有食物与坦索罗辛drs的相互作用。结果坦索罗辛drs显示出惊人的结果,即坦索罗辛drs在禁食条件下与进食的相比提供了类似的pk曲线。结果如图4所示。包装标签剂量和给药说明书指出“它应该在每天同一餐后大约半小时施用”。这些说明是由于在禁食状态下施用时对的全身暴露要高得多。研究数据表明,进食与禁食的cmax差异为56%。同一研究中的坦索罗辛drs显示进食与禁食状态的cmax仅有25%的差异。总体而言,坦索罗辛drs禁食的pk曲线与进食曲线相似。尽管上文已经描述的内容特别涉及坦索罗辛,但是本领域技术人员将理解,本文使用的方法可以以类似的方式用于其他活性成分。应用于此类其他活性成分的本发明方法和所得剂型被认为是本发明的一部分。合适的其他活性剂列表可选自不应被压碎的口服剂型列表2015或不应被压碎的口服剂型列表2016,这两者均可在网站ismp.org上找到。这些列表通过引用并入本文。其它活性成分将以有效用于使用其它活性成分的治疗目的的量存在于所得剂型中。将这些其它活性成分与本发明的基质材料组合并如本文所述进行处理,以产生小药囊和本文所述的其他剂型,以提供对吞咽困难以及可能还有食物效应的缓解。坦索罗辛还可以在本文所述的实施方案中与具有对bph的补充作用的其他活性成分组合,例如,5α-还原酶抑制剂,尤其是(度他雄胺=(1s,3as,3bs,5ar,9ar,9bs,11as)-n-[2,5-双(三氟甲基)苯基]-9a,11a-二甲基-7-氧代-1,2,3,3a,3b,4,5,5a,6,9b,10,11-十二氢茚并[5,4-f]喹啉-1-甲酰胺)或其药学上可接受的盐。这种组合可能对雄激素性秃发有用。在这种组合中度他雄胺的剂量为0.5mg,或另一种有效治疗bph的量。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1