突变率测量方法与流程
本发明涉及一种突变率测量方法,该测量方法包括制造用于下一代测序(nextgenerationsequencing;ngs)的文库的步骤。
背景技术:
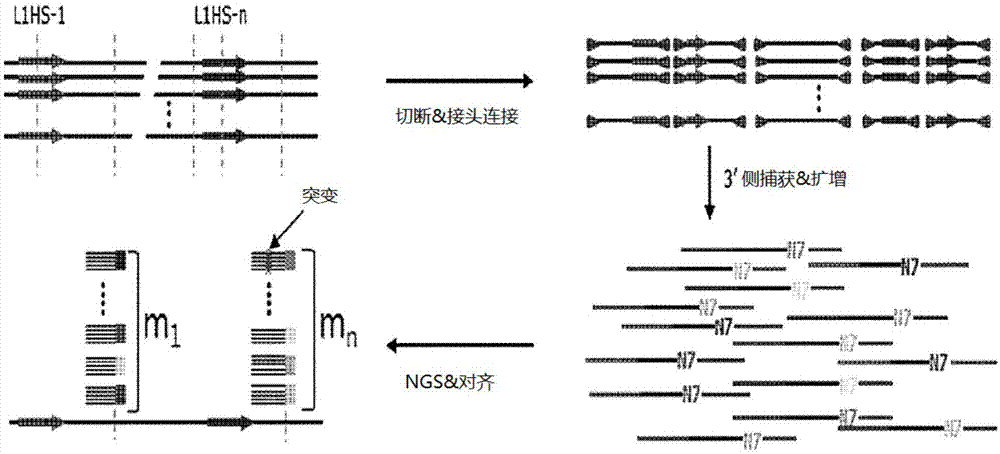
:突变为在疾病预测领域等多种生命科学领域中最受瞩目的部分中的一种。特别是,随着全球急剧进入高龄化社会,处于对健康的关注不断提高的趋势。因此,为了提高用于改善生活质量的疾病预测可能性,活跃进行关于突变率测量的研究。另外,作为用于检测突变的一种技术,在韩国公开专利第2015-0143025中公开了能够使用pna(peptidenucleicacid,肽核酸)来检测丝聚蛋白基因的突变。此外,正在持续报道在对表皮生长因子受体(egfr)的突变检测中利用pna的研究(tubercrespirdis(结核和呼吸疾病)2010;69:271-278)。但是,由于无法自然生成pna且只能利用合成来制造pna,因此存在费用非常昂贵且难以用于测量大规模突变率的限制。此外,利用pna的突变检测方法只能检测特定基因的突变,在检测随机发生的突变方面存在局限性。因此,目前仍然需要进行用于测量突变率的多种研究。技术实现要素:技术问题为此,本发明人为了开发不仅能够测量大规模突变率而且准确度还优异的突变率测量方法而深入研究,由此开发出了如下的突变率测量方法并且完成了本发明,其中,该测量方法包括通过对导入有随机序列和接头的目的基因进行扩增来分析碱基序列的步骤,能够提高准确度且广泛测量随机发生的突变的发生率。此外,由于本发明能够通过改变所应用的基因物质来测量包含在该基因物质中的变异体,并且根据发明的特性而去除由在实验过程中引入的各种噪音所导致的失真,因此本发明能提供更准确的测量值。例如,本发明可应用于在从线粒体dna中测量异质性(heteroplasmy)的内容和程度或者从微生物基因组中测量微生物的生态结构。技术方案本发明的主要目的是提供一种测量基因组突变率的方法,包括:(1)第一步骤,制造用于下一代测序(nextgenerationsequencing;ngs)的文库且包括下述(a)至(c)步骤:(a)利用限制酶来分别切断从个体提取出且具有界标的基因组dna,并且在切断出的各个基因组dna的两个末端连接包含彼此不同的随机序列的接头,从而制造dna-接头连接体;(b)将由所述(a)步骤制造的dna-接头连接体作为模板,并且利用在所述模板中与界标的5'末端结合的第一引物和与接头的3'末端结合的第二引物来执行pcr,从而获取扩增产物;和(c)将由所述(b)步骤制造的扩增产物作为模板,并且利用与所述模板的两个末端结合的引物对来执行pcr;(2)第二步骤,通过ngs来确定包含在所述文库中的各基因组断片的序列;(3)第三步骤,以标准基因组序列上的n个界标为基准,对齐由所述(c)步骤制造的扩增产物,从而将所述扩增产物分为n个组;(4)第四步骤,在按随机序列将构成组的扩增产物分为子组之后,为各子组选择一个基因组断片,从而为各组选择mi个基因组断片(所述mi为选自第i个界标中的基因组断片的数量);(5)第五步骤,通过比较mi个基因组断片的碱基序列,为各组确定排除突变后的一个代表碱基序列;(6)第六步骤,在n个组的mi个基因组断片中将具有与组的代表碱基序列不同的碱基序列的情况判断为突变,从而确定突变的总数量(m);以及(7)第七步骤,利用下述数学式1来计算突变率(amr)。发明效果本发明能够通过以区分模板的方式扩增从分析对象样品中选择性地捕获的基因组断片且按各模板比较碱基序列来准确地测量进行分析的基因组断片的规模和突变率。由于能够通过这种方法来测量环境变化即药物、放射线、基因结构、老化及个体经受的各种压力等给对象样品的突变发生带来的效果,因此本发明能够有效地应用到毒性实验、医学试验及与健康维持等相关联的试验、诊断、管理以及评价中。附图说明图1是示出用于从样品中测量突变率的方法的示意图。图2是示出用于构建包含随机序列和接头的基因组断片的dna序列文库的模板及引物的种类的图。图3a是示出在扩增基因组断片之后用2%琼脂糖凝胶进行电泳的结果的照片。图3b是示出用2%琼脂糖凝胶对基因组断片的dna序列文库进行电泳后的结果的照片。图4是示出通过ngs获得的关于碱基序列的分析结果的照片。图5是示出在基因组断片的碱基序列分析结果中一个具有彼此不同的碱基序列的基因组断片的照片。图6是示对28个样品的突变率测量结果的图表。具体实施方式为了实现上述目的,本发明的一方面提供一种测量基因组突变率的方法,包括:(1)第一步骤,制造用于下一代测序(nextgenerationsequencing;ngs)的文库且包括下述(a)至(c)步骤:(a)利用限制酶来分别切断从个体提取出且具有界标的基因组dna,并且在切断出的各个基因组dna的两个末端连接包含彼此不同的随机序列的接头,从而制造dna-接头连接体;(b)将由所述(a)步骤制造的dna-接头连接体作为模板,并且利用在所述模板中与界标的3'末端结合的第一引物和与接头的5'末端结合的第二引物来执行pcr,从而获取扩增产物;和(c)将由所述(b)步骤制造的扩增产物作为模板,并且利用与所述模板的两个末端结合的引物对来执行pcr;(2)第二步骤,通过ngs来确定包含在所述文库中的各基因组断片的序列;(3)第三步骤,以标准基因组序列上的n个界标为基准,对齐由所述(c)步骤制造的扩增产物,从而将所述扩增产物分为n个组;(4)在按随机序列将构成组的扩增产物分为子组之后,为各子组选择一个基因组断片,从而为各组选择mi个基因组断片(所述mi为选自第i个界标中的基因组断片的数量);(5)第五步骤,通过比较mi个基因组断片的碱基序列,为各组确定排除突变后的一个代表碱基序列;(6)通过在n个组的mi个基因组断片中将具有与各组的代表碱基序列不同的碱基序列的情况判断为突变,从而确定突变的总数量(m);以及第七步骤,利用下述数学式1来计算突变率(amr)。[数学式1](其中,amr是指突变率(accumulatedmutationrate);m是指突变的总数量;mi是指选自第i个界标中的基因组断片的数量;li是指在第i个界标的基因组断片中确定序列并进行分析的碱基数量。)第一步骤提供制造用于下一代测序(nextgenerationsequencing;ngs)的文库的步骤,其包括(a)步骤至(c)步骤。在本发明中,术语“下一代测序(nextgenerationsequencing;ngs)”是指针对基因组碱基序列的高速分析方法,能够与高通量测序技术(high-throughputsequencing)、大规模平行测序(massiveparallelsequencing)或第二代测序(secondgenerationsequencing)混合使用。在本发明中,术语“文库”是指利用限制酶等进行切断而获得的基因断片的集合,可以是通过将基因断片导入到载体而成的集合,但并不限于此。具体而言,在本发明中,所述文库可通过下述(a)至(c)步骤来制造,所述文库可用于测量突变率。所述(a)步骤提供以下步骤:利用限制酶来分别切断从个体提取出且具有界标的基因组dna,并且在切断出的各基因组dna的两个末端连接包含彼此不同的随机序列的接头,从而制造dna-接头连接体。在本发明中,术语“个体”可指包含需要测量突变率的人类在内的所有动物。从个体提取所述基因组dna的方法可以无限制地使用本领域中使用的方法。在本发明中,术语“界标”是指用于与基因组dna上的其他碱基序列区分的特定碱基序列。作为一例,界标可以是在基因组dna内重复呈现的特定碱基序列,具体而言,可以是line(longinterspersednuclearelement,长散在重复序列)或sine(shortinterspersednuclearelement,短散在重复序列)系列的重复碱基序列或者如特定限制酶识别部位那样在基因组内重复呈现的碱基序列,但并不限于此,只要是能够通过与其他碱基序列区分而测量特定部位的突变率的碱基序列则并不受限。在本发明的一实施例中,作为用于测量突变率的界标使用line系列的重复序列即l1hs碱基序列。在本发明中,术语“接头”是指为了获取包含全部或部分界标和限制酶切断部位的碱基序列的扩增产物而使用的部分双螺旋结构的碱基序列,所述接头可结合到利用限制酶切断的基因组dna的两个末端。具体而言,所述接头可包含随机序列。所述接头的一末端可包含与利用限制酶切断的基因组dna部位互补结合的序列。此外,所述接头可包含有在用于测量突变率的扩增产物的制造步骤中执行pcr时能够附接引物的碱基序列。在本发明的一实施例中,使用dpnⅱ限制酶来切断人类白血球细胞的基因组dna,并且将能够结合到所述限制酶的切断部位且包含随机序列的dpnⅱ接头附接到切断出的基因组dna中。另外,本发明的所述接头可以是为了全部捕获所述界标dna的互补链而在5'位置上结合有磷酸基的接头。此外,为了区分所述互补dna链,在接头的互补结合部位上可包含一个以上的错配核苷酸(mismatchnucleotide)。即,虽然在只捕获所述界标dna的一侧链的情况下,难以区分因在文库构建步骤等的反应中发生的化学变异引起的假阳性(falsepositive),但由于难以在dna的互补链的相同位置上同时发生这种化学变异,因此可通过使用能够全部捕获界标nda的两侧链的接头,去除因化学变异引起的噪音,并且进一步准确地测量突变率。此外,为了防止接头间的结合,可使用生成non-palindromoverhang(非回文突出)的限制酶。所述限制酶可以是bstni或avaii限制酶,但并不限于此。在本发明中,术语“随机序列”是指为了区分从个体提取出的基因组dna的起源而使用的任意5至11个核苷酸,可包含特定碱基序列。所述随机序列可结合到利用限制酶切断后的基因组dna的两个末端,并且按基因组dna的起源呈现不同的碱基序列,因此在从扩增产物测量突变率时,容易按基因组dna的起源测量突变率。所述随机序列可位于所述接头的部分双螺旋结构中的单一链上,但并不限于此。在本发明的一实施例中,将任意七个核苷酸作为随机序列使用。在本发明中,术语“dna-接头连接体”是指利用所述限制酶切断后的基因组dna和接头连接而成的结构体,该dna-接头连接体用作为用于测量突变率的扩增模板。具体而言,所述连接体可包含随机序列,例如所述随机序列可位于基因组dna与接头之间。所述(b)步骤提供以下步骤(b-1):将由所述(a)步骤制造的dna-接头连接体作为模板,并且利用在所述模板中与界标的3'末端结合的第一引物及与接头的5'末端结合的第二引物来执行pcr,从而获取扩增产物。所述第一引物为与界标的3'结合的引物,所述第二引物为与接头的5'末端结合的引物,在使用所述引物来执行pcr时发挥捕获界标3'部位的碱基序列的作用。在本发明中,术语“扩增产物”是指利用第一引物及第二引物执行的pcr的结果物,可包含界标、随机序列、基因组断片及接头。具体而言,所述扩增产物可包含界标的全部或部分序列,并且可包含接头的全部或部分序列,但并不限于此。在本发明中,术语“基因组断片”为包含作为突变率的测量对象的基因组dna的基因组,为了与其他基因组dna区分而可结合到界标及随机序列。具体而言,所述基因组断片可包含一个以上的碱基,并且可以全部或部分包含有在所述(a)步骤中利用限制酶进行的基因组dna的切断部位。在本发明的一实施例中,将包含由限制酶切断的基因组dna和随机序列及接头的dna-接头连接体作为模板,并且使用与包含在所述模板中的l1hs界标的3’末端结合的第一引物和与接头的5’末端结合的第二引物来执行pcr,从而获取扩增产物。所述步骤(b)可进一步包含以下步骤(b-2):将制造出的所述扩增产物作为模板,并且利用与所述界标的全部或部分碱基序列结合的正向引物和与除随机序列以外的全部或部分接头结合的逆向引物来执行巢式pcr(nestedpcr)。在本发明中,术语“巢式pcr”是指通过将一次pcr扩增产物用作为模板来去除不需要的扩增产物并且只特异性地筛选所需的扩增产物的pcr。由于所述巢式pcr能去除不需要的扩增产物,因此在本发明中测量突变率时可呈现出能提高准确度的效果。为了执行用于去除所述不需要的扩增产物的nesedpcr,可使用与捕获基因组断片的界标和接头分别结合的引物,具体而言,所述引物中的一种可以与界标的全部或部分碱基序列结合,所述引物中的另一种可以与接头的全部或部分碱基序列结合。此外,所述引物可以是附加适合下一代测序的碱基序列而成的形态的引物,但并不限于此。所述(c)步骤提供以下步骤:将由所述(b)步骤制造的扩增产物作为模板,并且利用与所述模板的两个末端结合的引物对来执行pcr。所述(c)步骤的引物对可以与由所述(b)步骤制造的扩增产物的两个末端结合。所述引物对由第一引物及包含按模板不同的指标的第二引物组成,因此容易区分所述扩增产物的文库。具体而言,所述按模板不同的指标一般可以是下一代测序中使用的通常指标。此外,所述引物对可以是附加适合下一代测序的碱基序列而成的形态的引物对。在本发明的一实施例中,使用包含适合下一代测序的碱基序列的引物对来制造ngs用文库(图2)。第二步骤提供以下步骤:通过ngs来确定构成所述文库的各基因组断片的序列。在本发明中,术语“基因组断片”及“ngs”与如上所述的说明相同。可利用在下一代测序中使用的碱基序列分析装置来分析所述基因组断片的序列,只要所述碱基序列分析装置为下一代测序中通常使用的装置则能够无限制地使用。所述文库可以是通过所述(c)步骤而附加适合下一代测序的碱基序列而成的形态,容易用于下一代测序中。第三步骤提供以下步骤:以标准基因组序列上的n个界标为基准,对齐由所述(c)步骤制造的扩增产物,从而将所述扩增产物分为n个组。具体而言,所述第三步骤为将包含从个体分离出的基因组dna且由所述(c)步骤制造的扩增产物对齐在标准基因组序列上的步骤,扩增产物的对齐可以以一个以上的界标为基准进行对齐并分为与界标相同的数量的组。在本发明中,术语“标准基因组序列”为特征个体的一般或平均基因组序列,是指当分别比较基因组序列的各种基因等时成为其基准的基因组的碱基序列。在本发明中,所述标准基因组序列使用与作为突变率的测量对象的基因组断片相同的个体的碱基序列,由于所述标准基因组序列与基因组断片之间的界标相同,因此容易按界标测量突变率。第四步骤提供以下步骤:在按随机序列将构成组的扩增产物分为子组之后,为各子组选择一个基因组断片,从而为各组筛选mi个断片,所述mi是指选自第i个界标中的基因组断片的数量。具体而言,所述第四步骤可通过在由所述第三步骤分组的各个组内按随机序列将扩增产物分为子组,并且为各子组选择一个基因组断片,从而为各组筛选mi个基因组断片。在为各子组选择一个基因组断片时,可以按各随机序列选择具有与分为子组后的扩增产物的一致碱基序列相同的序列的基因组断片,当在相同的随机序列中由五个以上的扩增产物支持一个基因组断片时,可将选择出的所述基因组断片视为有效的断片。第五步骤提供以下步骤:通过比较所述mi个基因组断片的相互间碱基序列,从而为各组确定排除突变后的一个代表碱基序列,第六步骤提供以下步骤:在n个组的mi个基因组断片中将具有与各组的代表碱基序列不同的碱基序列的情况判断为突变,从而确定突变的总数量(m)。具体而言,可通过排列属于相同的界标的同时由彼此不同的随机序列区分的mi个基因组断片的碱基序列而确定排除突变后的代表碱基序列,并且比较所述代表碱基序列和基因组断片的序列。当排列到相同的界标上且具有彼此不同的随机序列的基因组断片为10个以上时,如果存在一个具有彼此不同的碱基的基因组断片,则将该情况分类为突变,如果存在两个以上的具有彼此不同的碱基的基因组断片,则分类为多态性,从而确定突变的总数量。第七步骤提供以下步骤:利用下述数学式1来计算突变率(amr)。[数学式1]在上述数学式1中,amr是指突变率(accumulatedmutationrate),m是指突变的总数量,mi是指选自第i个界标中的基因组断片的数量,以及li是指在第i个界标的基因组断片中确定序列并进行分析的碱基数量。在本发明的一实施例中,通过所述第一步骤至第六步骤从人类白血球细胞的28个样品中确定突变的总数量,并且利用上述数学式1来计算突变率,其结果确认:在所述28个样品中每10万个碱基呈现出0.2至2.1个突变,平均发生0.9个突变(图6)。实施例下面,通过实施例对本发明的构成及效果进行更详细说明。这些实施例只是用于举例说明本发明,本发明的范围并非由这些实施例来限定。实施例1:构建基因组断片的dna序列文库转授保藏于韩国韩医学研究院kdc的人类白血球细胞的基因组dna的8个样品,并且按与图1相同的方法构建基因组断片的dna序列文库。具体而言,按样品使用dpnii限制酶在37℃下进行两小时的200ng基因组dna的切断之后,利用pcr提纯试剂盒(pcrpurificationkit)进行提纯,然后使其溶解于30μl缓冲液(elutionbuffer)。将切断出的各基因组dna50ng以及作为与包含随机序列的序列号1(5'-3':gagcaggtgactctggcttcctacacgacgctcttccgatctnnnnnnncacccacacttgacc,)和序列号2(5'-3':aattggtcaagtgtgggtg)的互补接合体的接头(adaptor)16pmole混合到包含dna连接酶(solgent)400u和1x缓冲剂的水溶液中,并且使其在室温下反应一小时,其中,所述序列号2通过互补结合到序列号1的3'侧而形成能够与dpnii切断部位结合的突出(overhang)。在通过上述反应将所述接头附接到限制酶的切断部位之后,利用pcr提纯试剂盒进行提纯并将其溶解于30μl缓冲液。为了将附接有接头的所述dna用作为pcr反应的模板,在分别提取1μl、2μl及4μl所述dna之后,使用与l1hs的3'部位结合的序列号3的l1_c引物和以接头为基准与5'部位结合的序列号4的a_c引物来以下述表1的条件执行pcr反应。此时,68℃、10分钟的3'部位的延长反应为用于填满接头的单链部位的步骤,是为了在连接的pcr反应中将两个链均用作为扩增反应的模板,可通过使用不同的dna聚合酶(dnapolymerase)来按独立反应实施。另外,所述引物如下述表2所示。[表1][表2]引物种类碱基序列(5'-3')l1_c引物gggagatatacctaatgctagatgacac(序列号3)a_c引物gagcaggtgactctggctt(序列号4)为了将作为所述pcr反应结果获得的基因组断片的扩增产物用作为连接的巢式pcr的模板,提取1μl该扩增产物,并且使用与扩增产物的5'末端结合的序列号5的l1_n引物和与3'末端结合的序列号6的a_n引物来以与上述表1相同的条件执行巢式pcr。提取作为所述巢式pcr结果获得的扩增产物0.1μl,并且通过使用序列号7的ngs_f引物和序列号8的ngs_r引物来以与上述表1相同的条件在所述扩增产物的两个末端附接ngs(nextgenerationsequencing)所需的碱基序列,从而构建ngs用序列文库(图2)。此时,为了区分扩增产物的文库,为各文库使用ngs_f引物指标(index)彼此不同的引物。另外,所述引物如表3所示。利用2%琼脂糖凝胶对所述扩增产物和构建的文库进行电泳,其结果确认呈现出相似的图案,并且确认能够通过上述方法构建用于正常测量突变率的文库(图3a及图3b)。[表3]实施例2:通过ngs数据分析来测量突变率使用碱基序列分析装置(hiseq2000,illumina)来确定由上述实施例1构建的文库的扩增产物的碱基序列。在所述扩增产物中不存在不清楚的碱基序列,在筛选出附接有ngs所需的碱基序列的扩增产物之后,通过将这些扩增产物摆列在标准基因组序列上且分别排列在l1hs界标上而进行分组。提取作为排列在各界标上的扩增产物的分子指标(molecularindex)的随机序列并按它们所来源的模板分为子组。然后,确定分为子组后的基因组断片所起源的一致碱基序列,并且选择具有所述碱基序列的基因组断片。此时,当在一个随机序列中存在五个以上的扩增产物时,视为一致碱基序列有效。作为分为子组后的结果确认到,作为一例,将f28样品的dna作为模板的扩增产物为总计43559个,并且具有6122个随机序列,所述扩增产物中的分组到第27个l1hs界标的扩增产物(cl_27)排列在从第一个染色体的14584433起至14584967的535bp大小的+链上,与2222bp大小的l1hs相隔-17bp。另外,确认到在0至20个随机序列中用0表示的第一个随机序列由“caaaaag”序列组成,用所述随机序列分为子组的扩增产物为20个(read_0至read_19),用1表示的第二个随机序列由“tgagaat”序列组成,用所述随机序列分为子组的扩增产物为19个(read_0至read18)(图4)。另外,在通过排列属于相同的界标的同时由彼此不同的随机序列定义的基因组断片的碱基序列而确定排除突变后的代表碱基序列之后,将该代表碱基序列与选择出的所述基因组断片的碱基序列进行比较。具体而言,当排列到相同的界标上且具有彼此不同的随机序列的基因组断片为10个以上时,如果存在具有彼此不同的碱基的一个基因组断片,则将该基因组断片分类为突变,如果存在具有彼此不同的碱基的两个以上基因组断片,则分类为多态性。其结果确认到,作为一例,分到第1484个l1hs的界标上的扩增产物(cl_1484)排列在从第11个染色体的49814618起至49814732的115bp大小的-链上,在所述扩增产物的分为子组后的基因组断片中的特定基因组断片中发生突变,具体而言能够识别发生突变的特定碱基的位置。此外,确认在总计8905个扩增产物的558026个碱基中的六个碱基中发生突变(图5)。另外,作为分组到所述第1484个l1hs的界标上的扩增产物的具体分析结果,确认由24个随机序列区分的基因组断片的碱基序列中的15个碱基序列与代表碱基序列一致(con_15/24),并且确认在由六个扩增产物构成的第八个随机序列中发生突变(mut_8_(6)),确认虽然剩余扩增产物的数量未达到五个,但由于扩增产物中的一部分与代表碱基序列不一致,因此在分析过程中去除该扩增产物中的一部分(图5)。按进行分析的界标来计算具有彼此不同的随机序列的基因组断片的数量、包含在碱基序列中的碱基的总数量以及突变事件的总数量,并且使用[数学式1]来计算突变率。[数学式1](其中,amr是指突变率(accumulatedmutationrate);m是指突变的总数量;mi是指选自第i个界标中的基因组断片的数量;li是指在从第i个界标捕获的基因组断片中确定序列并进行分析的碱基数量。)其结果确认到,在28个样品中呈现出每10万个碱基的突变率为0.2至2.1个的分布,具有平均0.9个突变(图6)。因此,确认能够通过本发明的突变率测量方法准确地识别进行分析的dna规模和突变数量。实施例3:确认利用变形的接头来测量突变率时的准确性提高利用avaii限制酶在37℃下进行两小时的实施例1中使用的基因组dna的切断之后,以与实施例1相同的方式进行提纯并使之溶解于缓冲液。由于在接头的互补结合部位上包含一个失配碱基对,并且在5'末端结合有磷酸基,因此能够通过ligation(连接)反应将该接头附接到捕获dna,且通过与实施例1相同的条件和步骤将使用互补结合序列号1的碱基序列和序列号9(5'-3':gtcggtcaagtgtgggtg)而成的接头附接到切断出的基因组dna并进行提纯而使之溶解于缓冲液以形成avaii限制酶的切断部位(图7)。通过与实施例1相同的条件和步骤对附接有接头的所述dna进行pcr扩增并制作ngs用文库(library)。通过与实施例2相同的过程和步骤对制作出的文库实施碱基序列确定及碱基序列分析,并且按各界标排列随机序列相同的碱基序列,通过确认接头的碱基序列1和2的失配部位而确认双螺旋的互补关系。因此,如上所述通过利用变形的接头来只将处于彼此互补关系的捕获dna的碱基序列彼此一致的变异选择为有效的突变,确认能够在实验过程中区分只在双螺旋的一侧链上发生的各种假变异,其结果能够去除因实验过程中发生的各种噪音导致的失真而提供更准确的测量值。本发明所属
技术领域:
的技术人员能够从上述说明中理解在不变更本发明的技术思想或必要特征的情况下可以以其他具体方式实施本发明。与此相关联地,应理解为以上所述的实施例在所有方面是示例性的而不是限定性的。本发明的范围由在上述详细说明之后叙述的权利要求书的范围来呈现,应解释为从权利要求书的含义及范围以及其均等概念导出的所有变更或变更后的形式包含在本发明的范围内。<110>韩国韩医学研究院<120>突变率测量方法<130>opa17106<150>kr10-2016-0075211<151>2016-06-16<150>kr10-2017-0061225<151>2017-05-17<160>9<170>kopatentin3.0<210>1<211>64<212>dna<213>人造序列<220><223>接头<400>1gagcaggtgactctggcttcctacacgacgctcttccgatctnnnnnnncacccacactt60gacc64<210>2<211>19<212>dna<213>人造序列<220><223>接头<400>2aattggtcaagtgtgggtg19<210>3<211>28<212>dna<213>人造序列<220><223>l1_c引物<400>3gggagatatacctaatgctagatgacac28<210>4<211>19<212>dna<213>人造序列<220><223>a_c引物<400>4gagcaggtgactctggctt19<210>5<211>57<212>dna<213>人造序列<220><223>l1_n引物<400>5gtgactggagttcagacgtgtgctcttccgatcttgcacatgtaccctaaaacttag57<210>6<211>20<212>dna<213>人造序列<220><223>a_n引物<400>6ctacacgacgctcttccgat20<210>7<211>52<212>dna<213>人造序列<220><223>ngs_f引物<400>7caagcagaagacggcatacgagatcgtgatgtgactggagttcagacgtgtg52<210>8<211>58<212>dna<213>人造序列<220><223>ngs_r引物<400>8aatgatacggcgaccaccgagatctacactctttccctacacgacgctcttccgatct58<210>9<211>18<212>dna<213>人造序列<220><223>接头<400>9gtcggtcaagtgtgggtg18当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1