一种提高药物溶解性的环糊精-金属有机骨架组合物的制作方法
本发明涉及一种提高药物溶解性的环糊精-金属有机骨架组合物。
背景技术:
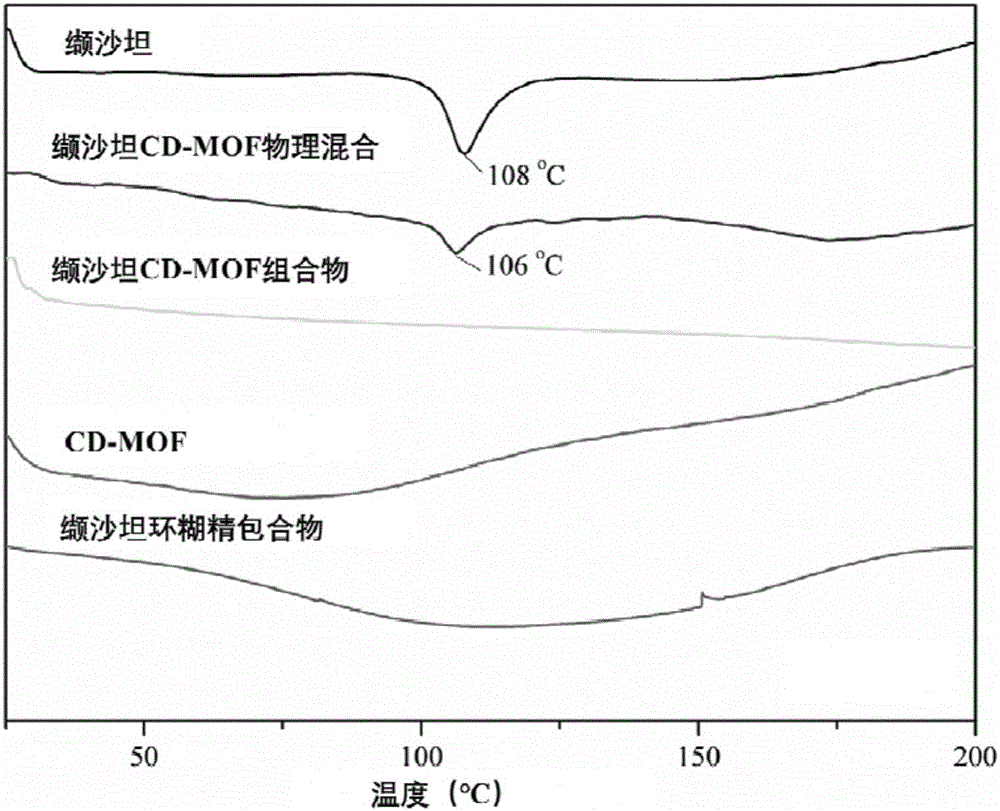
:高血压是最常见的心血管疾病之一,不仅直接危害健康,还加速动脉粥样硬化的过程,血压升高是中国人群脑卒中、冠心病、心力衰竭和肾脏疾病的主要危险因素。我国心、脑血管疾病的发生和死亡人数在不断上升,其中死亡人数占我国总死亡率已达40%,高血压已经成为危害健康的普遍问题。“沙坦”类药物即血管紧张素ⅱ受体拮抗剂(arb)类抗高血压药物,问世10年多来,因其降压作用明显,耐受性好,尤其临床试验和循证证据表明其具有对心血管疾病的独特疗效和保护作用,已成为临床最普遍的抗高血压药物之一,引起临床普遍关注,且长期应用后干咳、停药反跳及体位性低血压等不良反应发生率低,已被who多个治疗指南推荐为伴有心血管疾病和蛋白尿的高血压患者的一线降压药。沙坦类药物多为bcsⅱ类药物,在水中溶解性差,导致沙坦类药物口服生物利用度低。为了提高沙坦类药物口服生物利用度,必须提高沙坦类药物的溶解性。缬沙坦是血管紧张素ⅱ受体拮抗剂(arb)类抗高血压药物,问世10年多来,因其降压作用明显,耐受性好,尤其临床试验和循证证据表明其具有对心血管疾病的独特疗效和保护作用,已成为临床最普遍的抗高血压药物之一,已引起临床普遍关注,且长期应用后干咳、停药反跳及体位性低血压等不良反应发生率低,已被who多个治疗指南推荐为伴有心血管疾病和蛋白尿的高血压患者的一线降压药。缬沙坦为bcsⅱ类药物,在水中溶解性差,导致缬沙坦口服生物利用度低。为了提高缬沙坦口服生物利用度,必须提高缬沙坦药物的溶解性。技术实现要素:本发明的目的在于提供一种提高药物溶解性的环糊精-金属有机骨架组合物。在本发明的第一方面,提供了一种药物环糊精-金属有机骨架组合物,包含:(a)环糊精-金属有机骨架材料;以及(b)负载于所述骨架材料或与其混合的药物,其中,所述药物为沙坦类药物。在另一优选例中,所述沙坦类药物为奥美沙坦酯、奥美沙坦、厄贝沙坦、替米沙坦、缬沙坦、氯沙坦钾、氯沙坦、坎地沙坦酯、坎地沙坦、依普沙坦、阿齐沙坦或两种以上的组合物。在另一优选例中,所述沙坦类药物的分子量为350-650。在另一优选例中,所述沙坦类药物的分子量为400-500。本发明中,药物以两种方式负载于所述骨架材料:双环糊精分子对药物分子的包合;以及药物在环糊精-金属有机骨架中间大空腔中形成纳米团簇。在另一优选例中,所述环糊精-金属有机骨架材料中的金属离子选自下组:li+、na+、k+、rb+、cs+、mg2+、cd2+、sn2+、ag+、yb+、ba2+、sr2+、ca2+、pb2+、la3+。在另一优选例中,金属离子为ca2+、na+或k+,进一步优选k+。在另一优选例中,与金属离子成盐或碱的阴离子包括但不限于oh-、no3-、co32-、hco3-、ch3coo-、scn-、c6h5cooh=c6h5coo-、cl-、br-、i-、o2-、s2-、hs-、hso4-、clo-、clo3-、mno4-,优选oh-。在另一优选例中,所述环糊精-金属有机骨架材料为氢氧化钾环糊精-金属有机骨架材料、碳酸钾环糊精-金属有机骨架材料、氯化钾环糊精-金属有机骨架材料、乙酸钾环糊精-金属有机骨架材料,优选乙酸钾γ-环糊精-金属有机骨架材料。在另一优选例中,所述环糊精-金属有机骨架材料中的环糊精选自下组:α-环糊精、β-环糊精、γ-环糊精、羟丙基-β-环糊精、磺丁基-β-环糊精、甲基-β-环糊精、羧甲基-β-环糊精。在另一优选例中,环糊精选自下组:α-环糊精、β-环糊精、γ-环糊精,进一步优选为γ-环糊精。在另一优选例中,所述环糊精-金属有机骨架材料为碱性或中性环糊精-金属有机骨架材料。碱性环糊精-金属有机骨架材料分散到含有机酸的有机溶剂中孵育即得到中性环糊精-金属有机骨架材料。在另一优选例中,所述有机酸选自下组:甲酸、乙酸、柠檬酸、富马酸、酒石酸、偏酒石酸、苹果酸、己二酸或其混合溶剂,优选甲酸、乙酸或其混合溶剂,最优选乙酸。在另一优选例中,所述有机溶剂选自甲醇、乙醇、丙醇、异丙醇、正丁醇或其混合溶剂,优选甲醇、乙醇或其混合溶剂,最优选乙醇。在另一优选例中,所述的组合物中环糊精-金属有机骨架与药物的摩尔比例为1:0.2-1:2,优选为1:0.5-1:1.8,进一步优选为1:1.5。在另一优选例中,所述组合物还具有以下一个或多个特征:(1)所述环糊精-金属有机骨架材料的平均粒径为5纳米-1000微米;(2)所述组合物的载药量为0.1%-50%。在另一优选例中,所述环糊精-金属有机骨架材料的平均粒径为100-1000纳米(纳米级)或1-100微米(微米级)。在另一优选例中,所述组合物的载药量为10%-40%,进一步优选为20%-30%。本申请组合物对沙坦类药物具有增溶效果,如将缬沙坦在水中的溶解度提高了10-50倍,优选为10-40倍,进一步优选为30-40倍,再如将阿奇沙坦在水中的溶解度提高了10-200倍,再如将厄贝沙坦在水中的溶解度提高了10-1000倍。本发明的第二方面,提供第一方面所述的组合物的制备方法,所述制备方法包括将所述环糊精-金属有机骨架材料与药物溶液混合后得到所述组合物的步骤。在另一优选例中,所述混合的温度为25~65℃,优选30~50℃,最优选30~40℃。在另一优选例中,所述混合的时间为10分钟~3天,优选为1小时~12小时,最优选为30分钟~2小时。在另一优选例中,所述药物为沙坦类药物,选自:奥美沙坦酯、奥美沙坦、厄贝沙坦、替米沙坦、缬沙坦、氯沙坦钾、氯沙坦、坎地沙坦酯、坎地沙坦、依普沙坦、阿齐沙坦或两种以上的组合物。在另一优选例中,将药物溶解于选自下组的溶剂中制备所述药物溶液:甲醇、乙醇、丙醇、异丙醇、正丁醇、氯仿、二甲基甲酰胺或其混合溶剂,优选甲醇、乙醇或其混合溶剂,最优选乙醇。在另一优选例中,所述环糊精-金属有机骨架材料中的环糊精与药物的投料摩尔比为1:1.5-1:25。在另一优选例中,环糊精与药物的投料摩尔比为1:1-1:50,较佳为1:1-1:30、1:1-1:25或1:1-1:22、1:15-1:25。在另一优选例中,所述制备方法还包括后处理步骤:收集固体并进行干燥。本发明的第三方面,提供第一方面所述的组合物的制备方法,所述制备方法包括将所述药物与所述环糊精-金属有机骨架材料混合进行研磨得到所述组合物的步骤。在另一优选例中,所述药物为沙坦类药物,选自:奥美沙坦酯、奥美沙坦、厄贝沙坦、替米沙坦、缬沙坦、氯沙坦钾、氯沙坦、坎地沙坦酯、坎地沙坦、依普沙坦、阿齐沙坦或两种以上的组合物。在另一优选例中,所述环糊精-金属有机骨架材料与药物的投料比为1:0.2-1:2,较佳为1:0.4-1:1.8。在另一优选例中,所述药物为粉体形式或药物溶液形式。在另一优选例中,将药物溶解于选自下组的溶剂中制备所述药物溶液:甲醇、乙醇、丙醇、异丙醇、正丁醇、氯仿、二甲基甲酰胺或其混合溶剂,优选甲醇、乙醇或其混合溶剂,最优选乙醇。在另一优选例中,所述环糊精-金属有机骨架材料为粉体形式或药物溶液形式。在另一优选例中,将环糊精-金属有机骨架材料溶解于选自下组的溶剂中制备所述药物溶液:甲醇、乙醇、丙醇、异丙醇、正丁醇、氯仿、二甲基甲酰胺或其混合溶剂,优选水、甲醇、乙醇或其混合溶剂,最优选乙醇、甲醇。在另一优选例中,所述研磨是采用球磨机进行研磨。在另一优选例中,球磨机采用直径为6-10mm较佳为8mm的锆珠进行研磨。在另一优选例中,珠与(药物+环糊精-金属有机骨架材料)的重量比例15:1-1:1,较佳为10:1-3:1。在另一优选例中,研磨时间为1min-2h,较佳为5-80min。本发明的第四方面,提供第一方面所述的组合物的制备方法,包括以下步骤:(i)将环糊精、金属离子源溶解于水中制备成第一溶液,将所述药物与有机溶剂混合制成第二溶液;(ii)将第二溶液加入第一溶液中进行混合,得到混合物;(iii)对所述混合物进行固体收集和干燥,即得所述的组合物。在另一优选例中,所述金属离子源选自下组:氢氧化钾、碳酸钾、氯化钾。在另一优选例中,所述第一溶液中所述环糊精的浓度为0.01-200mm。在另一优选例中,所述环糊精与所述金属离子源中金属离子的摩尔比为1:1-1:10。在另一优选例中,所述药物为沙坦类药物,选自:奥美沙坦酯、奥美沙坦、厄贝沙坦、替米沙坦、缬沙坦、氯沙坦钾、氯沙坦、坎地沙坦酯、坎地沙坦、依普沙坦、阿齐沙坦或两种以上的组合物在另一优选例中,将药物溶解于选自下组的有机溶液中制备所述第二溶液:甲醇、乙醇、丙醇、异丙醇、正丁醇、氯仿、二甲基甲酰胺或其混合溶剂,优选甲醇、乙醇或其混合溶剂,最优选乙醇。在另一优选例中,在步骤ii)中,将第二溶液加入第一溶液中进行混合,可加入形态调节剂混合得到混合物。在另一优选例中,所述形态调节剂为聚乙二醇、聚维酮、聚山梨醇、失水山梨醇单月桂酸酯、聚氧乙烯月桂醇醚、乳化剂op、乳百灵a、普流罗尼、十二烷基硫酸钠、十二烷基苯磺酸钠、十六烷基三甲基溴化铵、或其组合。在另一优选例中,所述形态调节剂为聚乙二醇20000、聚乙二醇2000、聚乙二醇4000、聚乙二醇6000、聚乙二醇8000、聚乙二醇10000、聚乙二醇20000、或其组合。在另一优选例中,按1-20mg/ml上清液的比例加入所述形态调节剂,较佳为5-10mg/ml上清液。本发明的第五方面,提供一种药物组合物,包含:第一方面所述的组合物;和药学上可接受的载体。“药学上可接受的载体”指的是:一种或多种相容性固体或液体填料或凝胶物质,它们适合于人使用,而且必须有足够的纯度和足够低的毒性。“相容性”在此指的是组合物中各组份能和本发明的化合物以及它们之间相互掺和,而不明显降低化合物的药效。药学上可以接受的载体部分例子有纤维素及其衍生物(如羧甲基纤维素钠、乙基纤维素钠、纤维素乙酸酯等)、明胶、滑石、固体润滑剂(如硬脂酸、硬脂酸镁)、硫酸钙、植物油(如豆油、芝麻油、花生油、橄榄油等)、多元醇(如丙二醇、甘油、甘露醇、山梨醇等)、乳化剂(如)、润湿剂(如十二烷基硫酸钠)、着色剂、调味剂、稳定剂、抗氧化剂、防腐剂、无热原水等。在另一优选例中,所述的载体选自下组:稀释剂、赋形剂、填充剂、粘合剂、润湿剂、崩解剂、吸收促进剂、表面活性剂、吸附载体、润滑剂、或其组合。在另一优选例中,所述的药物组合物配制为固体剂型或液体剂型,较佳适用于口服给药。在另一优选例中,固体剂型包括胶囊剂、片剂、丸剂、散剂和颗粒剂。在另一优选例中,液体剂型包括药学上可接受的乳液、溶液、悬浮液、糖浆或酊剂。在另一优选例中,所述药物组合物为胶囊剂、片剂、颗粒剂。在另一优选例中,所述药物组合物还包含表面活性剂,选自下组:聚山梨坦-80、聚山梨坦-60、聚乙二醇甘油脂肪酸酯、脱水山梨糖醇脂肪酸酯及两种以上的混合物。应理解,在本发明范围内中,本发明的上述各技术特征和在下文(如实施例)中具体描述的各技术特征之间都可以互相组合,从而构成新的或优选的技术方案。限于篇幅,在此不再一一累述。附图说明图1为实施例13的差示扫描量热(dsc)图。图2为实施例13的气体吸附图。图3为实施例13的红外光谱图。图4为实施例13的粉末x-射线衍射(pxrd)图。图5为实施例14制备的载缬沙坦cd-mof组合物胶囊在四种不同介质中的溶出曲线。图6为实施例14中市售代文胶囊在四种不同介质中的溶出曲线。图7为实施例17中缬沙坦cd-mof组合物片剂与国产某市售片剂在水中的溶出曲线。图8为实施例18溶解速率图。图9为实施例20中阿齐沙坦cd-mof组合物表征图,(a)x-射线衍射、(b)红外、(c)气体吸附、(d)差式扫描量热。图10为实施例20中阿齐沙坦cd-mof组合物的x-射线小角散射图。图11为实施例27中cd-mof载阿齐沙坦的分子模拟图,a为环糊精分子对的包合,b为阿齐沙坦在空腔中形成纳米团簇。图12为实施例28中cd-mof载缬沙坦的分子模拟图。图13为实施例28中cd-mof载缬沙坦等的x-射线小角散射图。图14为实施例28中cd-mof载缬沙坦等的固态核磁图。具体实施方式本发明人经过广泛而深入地研究,首次研发出一种载沙坦类药物的环糊精-金属有机骨架复合物,将沙坦类药物负载于环糊精-金属有机骨架材料或与其混合,能显著提高沙坦类药物在水中的溶解度和溶解速率,解决沙坦类药物溶解性差、口服绝对生物利用度低的问题,制备组合物所用原料和溶剂廉价易得,步骤简单,利于工业化生产。环糊精环糊精是由直链淀粉经葡萄糖基转移酶作用下生成的一系列环状低聚糖的总称,通常含有6~12个d-吡喃葡萄糖单元。其中研究得较多并且具有重要实际意义的是含有6、7、8个葡萄糖单元的分子,分别称为α、β-和γ-环糊精。环糊精是迄今所发现的类似于酶的理想宿主分子,并且其本身就有酶模型的特性。金属有机骨架材料金属有机骨架材料(metal-organicframeworks,mofs)是由有机桥连配体通过配位键的方式将无机金属中心连接起来形成无限延伸的网络状结构的晶体材料。由于mofs超高的孔隙率和巨大的比表面积,以及其由无机和有机不同成分组成的结构使得其结构多样并可调节,促使mofs在许多方面如气体储存、催化、药物载体等领域具有潜在的应用价值。环糊精-金属有机骨架材料如本文所用,术语“基于环糊精的金属有机骨架材料”、“环糊精-金属有机骨架材料”、“环糊精-金属有机骨架化合物”可互换使用,是利用环糊精在水溶液中能与第一、二主族金属离子以一种有机配位的方式形成一种新的晶体,这种晶体具有多孔、表面积大、储存气体等特点。这种绿色、多孔材料能够吸附一些结构不稳定的药物,其巨大的空腔能够对药物起到保护作用,这使得其用于商业发展成为可能,尤其是由于环糊精-金属有机骨架为可食用衍生物,适于人类食用。将环糊精作为有机配体,金属离子作为无机金属中心,可形成新的、安全性较高、可药用的环糊精-金属有机骨架,即cd-mof。如本文所用,术语“碱性环糊精-金属有机骨架材料”是以碱金属和环糊精为原料制备的环糊精-金属有机骨架材料,呈碱性,将其溶解于水中制成10mg/ml的水溶液时,其ph约为11-13。如本文所用,术语“中性环糊精-金属有机骨架材料”、“经酸化的环糊精-金属有机骨架材料”可互换使用,是将碱性环糊精-金属有机骨架材料进行酸化处理得到的近中性的环糊精-金属有机骨架材料,将其溶解于水中制成10mg/ml的水溶液时,其ph约为5-8。一种优选的酸化处理的方法如下:称取一定量的环糊精-金属有机骨架置于乙醇中,加入一定量的冰醋酸,25℃下,振摇孵育一定时间后,所得固体用乙醇洗涤,即得近中性的环糊精-金属有机骨架。药物的环糊精-金属有机骨架复合物如本文所用,术语“载药物的环糊精-金属有机骨架组合物”、“载药物的环糊精-金属有机骨架复合物”、“载药物的环糊精-金属有机骨架”、“载药物的cd-mof复合物”、“载药物的cd-mof”、“cd-mof载药物”、“药物cd-mof组合物”、“cd-mof载药物”可互换使用,上述术语均代表将沙坦类药物负载于环糊精-金属有机骨架材料或将沙坦类药物与环糊精-金属有机骨架材料混合所得的样品。下面结合具体实施例,进一步阐述本发明。应理解,这些实施例仅用于说明本发明而不用于限制本发明的范围。下列实施例中未注明具体条件的实验方法,通常按照常规条件,或按照制造厂商所建议的条件。除非另外说明,否则百分比和份数按重量计算。通用方法沙坦类药物含量测定方法:采用紫外分光光度法,如,缬沙坦的紫外检测波长为250nm,阿齐沙坦的紫外检测波长为250nm,替米沙坦的紫外检测波长为290nm,厄贝沙坦的紫外检测波长为245nm。溶解度测定方法:称取过量的待测样品,置于水中,摇床振摇(25℃、200rpm)三天,离心过滤,按照上述“含量测定方法”测定溶液中药物的浓度。记为待测样品在25℃水中的溶解度。增溶倍数:待比较样品的溶解度与药物在水中(25℃)的溶解度的比值。载药量与载药摩尔比的测定与计算方法:精密称定5mg(m1)待测样品,置于100ml容量瓶中,用纯水溶解,过滤后,按照“含量测定条件”对药物含量进行测定,计算药物的质量m2。载药量(%)=m2/m1×100;载体与药物的摩尔比为:(m2/m药物):((m1-m2)/m载体),m载体为载体的分子量,m药物为药物的分子量。实施例1:缬沙坦微米级氢氧化钾cd-mof组合物-孵育量取3000ml纯水于烧杯中,加入97.3gγ-cd搅拌,再加入33.6gkoh,超声,使γ-cd和koh完全溶解,制得母液。将母液倒入反应釜中,设置转速为300rpm,温度50℃。再向反应釜中加入1800ml甲醇,溶解澄清后加入38.4gpeg20000。室温静置过夜后离心,去上清,沉淀分别使用100ml乙醇洗涤两遍,再用100ml甲醇洗涤两遍。沉淀于40℃真空干燥箱中干燥12h,即得微米级氢氧化钾-环糊精-金属有机骨架,粒径为1-10微米。称取200mg微米级氢氧化钾cd-mof于15ml离心管中,加入10ml缬沙坦乙醇溶液(环糊精-金属有机骨架材料中的环糊精与缬沙坦的摩尔比例为1:22),摇床振摇孵育(37℃,150rpm)20h,离心,下层沉淀物60℃真空干燥4h,即得载缬沙坦的微米级氢氧化钾cd-mof组合物。所得组合物中载药量为29.4%(w/w),cd-mof与缬沙坦的摩尔比为1:1.4,将缬沙坦在水中的溶解度提高了34.5倍(缬沙坦原料在25℃的水中溶解度为0.08mg/ml)。实施例2:缬沙坦微米级乙酸钾cd-mof组合物-孵育在实施例1中的基础上进一步制备微米级乙酸钾cd-mof:向下层沉淀物中加入0.4l无水乙醇分散,进行离心洗涤,再向沉淀中加入0.4l无水乙醇和20ml冰醋酸,搅拌中和,离心去除上清。最后向沉淀中加入0.4l乙醇,离心(4000rpm,5min)洗涤两遍。将洗涤后的沉淀置于60℃真空干燥箱中干燥4h。即得微米级乙酸钾环糊精-金属有机骨架,粒径为1-10微米。按照实施例1中所述的载药方法(环糊精-金属有机骨架材料中的环糊精与缬沙坦的摩尔比例为1:20),制备缬沙坦cd-mof组合物,所得组合物中载药量为30.7%(w/w),cd-mof与缬沙坦的摩尔比为1:1.6,将缬沙坦在水中的溶解度提高33.7倍。实施例3:缬沙坦微米级氢氧化钾cd-mof组合物-搅拌称取200mg实施例1中制备的微米级氢氧化钾cd-mof于20ml西林瓶中,加入10ml缬沙坦乙醇溶液(环糊精-金属有机骨架材料中的环糊精与缬沙坦的摩尔比例为1:20),40℃加热磁力搅拌1h,离心,下层沉淀物真空干燥,即得载缬沙坦微米级氢氧化钾cd-mof组合物。所得组合物中载药量为29.7%,组合物中cd-mof与缬沙坦的摩尔比为1:1.5,增溶倍数为35.6。实施例4:缬沙坦微米级乙酸钾cd-mof组合物-搅拌称取40g实施例2中的微米级乙酸钾cd-mof于烧杯中,加入1l浓度为0.25g/ml缬沙坦乙醇溶液(γ-cd与缬沙坦的投料摩尔比为1:22),按照实施例3方法进行载药。作为对照,同时以γ-cd代替cd-mof,用相同的方法制备缬沙坦γ-cd组合物。缬沙坦cd-mof物理混合物的制备:按照载药比例分别称取缬沙坦与微米级乙酸钾cd-mof,置于烧杯中,用玻璃棒进行搅拌混合,得物理混合物。结果:所得缬沙坦cd-mof组合物中载药量为31.1%,组合物中cd-mof与缬沙坦的摩尔比为1:1.5,增溶倍数为39.5,显著高于缬沙坦γ-cd组合物(增溶倍数为4.6)。溶出测定:称取相当于一个剂量(80mg)的缬沙坦cd-mof组合物,平行6次,进行溶出实验。转速100rpm,温度为37.0±0.5℃,介质为900ml去离子水,不同时间点取样5ml,在250nm处用紫外测吸光度,计算溶出量。同时对比测定缬沙坦原料、缬沙坦与cd-mof的物理混合物。结果:表1,缬沙坦微米级乙酸钾cd-mof组合物和物理混合物均显著高于缬沙坦原料,而组合物又显著高于物理混合物。表1缬沙坦cd-mof组合物、物理混合物及原料的溶出实施例5:缬沙坦纳米级氢氧化钾cd-mof组合物取2000ml纯水于5000ml大烧杯中,称取64.9gγ-cd于大烧杯中,搅拌,再向烧杯中加入22.4gkoh,超声,使γ-cd和koh完全溶解,制得母液。将母液倒入反应釜中,转速300rpm,温度50℃时,向反应釜中加入1200ml甲醇,再加入peg20000(8mg/ml)。冷水浴中静置过夜。离心去上清,沉淀分别使用150ml乙醇洗涤两遍,于40℃真空干燥箱中干燥12h。粒径为100-500纳米。按照实施例3方法进行载药(环糊精-金属有机骨架材料中的环糊精与缬沙坦的摩尔比例为1:20),得缬沙坦纳米级氢氧化钾cd-mof组合物。结果:所得组合物中载药量为31.7%,cd-mof与缬沙坦的摩尔比为1:1.5,增溶倍数为36.2。实施例6:缬沙坦纳米级乙酸钾cd-mof组合物在实施例5的基础上按照实施例2的方法进行改进制备纳米级乙酸钾cd-mof,粒径为100-500纳米。按照实施例3方法进行载药(环糊精-金属有机骨架材料中的环糊精与缬沙坦的摩尔比例为1:20),得缬沙坦纳米级氢氧化钾cd-mof组合物。结果:所得组合物中载药量为32.8%,cd-mof与缬沙坦的摩尔比为1:1.7,增溶倍数为36.3。实施例7:缬沙坦碳酸钾cd-mof组合物取2500ml纯水于烧杯中,加入81.1gγ-cd和34.5g碳酸钾,制得母液。将母液倒入反应釜中,加入2500ml甲醇,反应澄清后加入40.0gpeg20000使完全溶解后,将反应液室温静置过夜后离心去上清,沉淀分别使用100ml乙醇洗涤两遍,再用100ml甲醇洗涤两遍。于40℃真空干燥箱中干燥12h,即得碳酸钾微米级cd-mof,粒径为300-700微米。按照实施例3的方法进行载药(环糊精-金属有机骨架材料中的环糊精与缬沙坦的摩尔比例为1:20),得缬沙坦碳酸钾cd-mof组合物。结果:所得组合物中载药量为24.6%,cd-mof与缬沙坦的摩尔比为1:1.1,增溶倍数为30.5。实施例8:缬沙坦氯化钾cd-mof的制备将γ-cd、氯化钾溶于5ml水中,使其浓度分别为0.125m、0.5m,超声10min使其充分溶解。然后预加0.5ml甲醇至γ-cd与氯化钾混合溶液内,在密闭容器中50℃条件下加热甲醇,反应6h后,用乙醇和甲醇各洗涤2次,将所得晶体50℃真空干燥12小时,即得氯化钾cd-mof,粒径为1-10微米。按照实施例3的方法进行载药(环糊精-金属有机骨架材料中的环糊精与缬沙坦的摩尔比例为1:20),得缬沙坦氯化钾cd-mof组合物。结果:所得组合物中载药量为22.3%,cd-mof与缬沙坦的摩尔比为1:1.3,增溶倍数为34.8。实施例9:共结晶法将163.0mgγ-cd与56.0mgkoh溶解于5ml水中,超声溶解。再加入5ml缬沙坦甲醇溶液(γ-cd与缬沙坦的投料摩尔比为1:22),再按8mg/ml上清液的比例加入peg20000作为形态调节剂,静置半小时后,离心,弃去上清液,下层固体用异丙醇洗涤,将所得晶体50℃条件下真空干燥过夜,即得缬沙坦环糊精-金属有机骨架共结晶组合物。结果:所得组合物中载药量为33.9%,cd-mof与缬沙坦的摩尔比为1:1.8,增溶倍数为38.9。实施例10:缬沙坦乙酸钾β-cd-mof组合物称取β-环糊精200g、氢氧化钾78.9g加2l纯水至烧杯中溶解后,加入至5l反应釜中,室温25℃条件下,转速为100rpm,加入2l甲醇,搅拌16h,将混悬的反应液离心,沉淀即为氢氧化钾β-cd-mof。将40ml冰醋酸加入至氢氧化钾β-cd-mof的乙醇混悬体系中,搅拌5min使之分散均匀,用滤纸进行抽滤,滤饼继续用无水乙醇洗涤2次,滤饼干燥后即为乙酸钾β-cd-mof。称取200mg乙酸钾β-cd-mof于20ml西林瓶中,加入10ml缬沙坦乙醇溶液(环糊精-金属有机骨架材料中的环糊精与缬沙坦的摩尔比例为1:22),37℃加热搅拌1h,离心,下层沉淀物真空干燥,即得缬沙坦乙酸钾β-cd-mof组合物。结果:所得组合物中载药量为14.7%,cd-mof与缬沙坦的摩尔比为1:0.5,增溶倍数为15.2。实施例11:缬沙坦微米级乙酸钾cd-mof组合物称取20g实施例1中制备的微米级乙酸钾cd-mof于1l烧杯中,加入300ml浓度为0.1g/ml缬沙坦乙醇溶液(环糊精-金属有机骨架材料中的环糊精与缬沙坦的摩尔比例为1:5),40℃加热磁力搅拌1h,离心,下层沉淀物真空干燥,即得载缬沙坦微米级氢氧化钾cd-mof组合物。所得组合物中载药量为12.4%,组合物中cd-mof与缬沙坦的摩尔比为1:0.5,增溶倍数为35.2。实施例12:缬沙坦微米级乙酸钾cd-mof组合物称取20g实施例1中制备的微米级乙酸钾cd-mof于1l烧杯中,加入500ml浓度为0.18g/ml缬沙坦乙醇溶液(环糊精-金属有机骨架材料中的环糊精与缬沙坦的摩尔比例为1:16),40℃加热磁力搅拌1h,离心,下层沉淀物真空干燥,即得载缬沙坦微米级氢氧化钾cd-mof组合物。所得组合物中载药量为22.82%,组合物中cd-mof与缬沙坦的摩尔比为1:1.0,增溶倍数为34.9。实施例13:缬沙坦/cd-mof组合物的表征作为对比,制备缬沙坦/γ-环糊精包合物:称取1297mgγ-环糊精(1mmol)溶解于30ml水中,称取435mg缬沙坦(1mmol)溶解于15ml乙醇中,将缬沙坦溶液滴加到搅拌状态下的γ-环糊精水溶液中,维持温度在40℃,500rpm下继续搅拌3h,除去乙醇。余下液体冷冻干燥,即得缬沙坦/γ-环糊精包合物。所得环糊精包合物中缬沙坦的含量(w/w)为15.5%,γ-环糊精与缬沙坦的摩尔比为1:0.64,包合物在水中的溶解度为0.38mg/ml,将缬沙坦在水中的溶解度提高了4.75倍。图1:缬沙坦在108℃可见一个宽的融化吸热峰,而环糊精包合物和实施例4中的缬沙坦cd-mof组合物该峰完全消失,物理混合物中该吸收峰重新出现,表明缬沙坦被环糊精包合,组合物中缬沙坦被cd-mof负载。图2:气体吸附结果显示,乙酸钾cd-mof的比表面积为996m2/g,载药摩尔比为1:0.5的缬沙坦cd-mof组合物(实施例11)的langmuir比表面积将至307m2/g,而载药摩尔比为1:1.5的缬沙坦cd-mof组合物(实施例4)的langmuir比表面积几乎为零,表明缬沙坦被cd-mof负载。图3:红外光谱图显示载缬沙坦中性微米级cd-mof组合物有别于缬沙坦原料药和中性微米级cd-mof以及其两者的物理混合物。对比cd-mof、val/cd-mof和cd-mof与val的物理混合物的图谱,三个样品在2926cm-1均有较宽的吸收带,可以归属为cd-mof中-ch-的伸缩振动(νch)。val由于自身酰胺键和羰基的存在,在1731cm-1以及1600cm-1处有强吸收(碳氧双键特征吸收)以及1445cm-1处ν(c=c)芳环振动,cd-mof均不具有以上特征吸收;val/cd-mof在1731cm-1(酰胺羧基)以及1445cm-1ν(c=c)振动在强度上明显减弱或消失,而物理混合物依然存在。val的特征峰被掩盖表明缬沙坦被cd-mof负载。图4:粉末x-射线衍射结果表明缬沙坦在13-15o与20-22o有两个宽衍射峰;cd-mof具有典型的晶体结构。载药后缬沙坦cd-mof组合物中相应的cd-mof衍射峰仍然存在,表明载药后的缬沙坦cd-mof组合物仍为晶体结构。实施例14:缬沙坦微米级乙酸钾cd-mof组合物胶囊剂的制备制备工艺:称取实施例4制备的载缬沙坦微米级乙酸钾cd-mof组合物10g,加入10ml浓度为0.05mg/ml的聚山梨坦-80乙醇溶液均匀润湿,用高纯氮气吹30min,过30目筛制粒,干燥,整粒,填充胶囊。并与市售代文胶囊在四种不同ph介质中进行溶出度与溶出速率比较。溶出度测定:采用转篮法,转速100rpm,温度37℃,四种不同介质:ph6.8磷酸盐缓冲液、ph4.5醋酸盐缓冲液、ph1.2盐酸溶液和去离子水各1000ml,在3,5,10,15,30,60,90,120min取点,每次取5ml,在250nm处用紫外测吸光度。结果表明,载缬沙坦微米乙酸钾cd-mof组合物胶囊(图5)在各介质中的溶出差异低于市售胶囊(图6),显著提高了市售胶囊在ph1.2和水中的溶出速率,有助于改善缬沙坦的生物利用度。实施例15:大鼠体内生物利用度12只健康雄性sd大鼠购自上海实验动物研究中心,体重约为200±20g,随机分为2组,每组6只,分别灌胃给药给予缬沙坦cd-mof组合物(实施例6制备)、原研代文胶囊15mg/kg,于0、0.08、0.25、0.5、0.75、1、1.5、2、3、4、8、12与24h分别眼眶取血0.3ml。将载缬沙坦中性微米级cd-mof组合物与原研代文胶囊进行生物利用度对比并评价。采用高效液相色谱串联质谱(lc-ms/ms)分析方法测定大鼠血浆样品中缬沙坦浓度。采用das2.0软件计算12只健康雄性sd大鼠的药动学参数,结果见表2,与代文组相比,缬沙坦/cd-mof组的平均相对生物利用度为149.3%,表明cd-mof载缬沙坦后,显著提高了缬沙坦在大鼠体内的生物利用度。同时采用spss17.0软件对药动学参数(药时曲线下面积auc0-24h、达峰浓度cmax、达峰时间tmax)进行t检验统计分析,结果显示,两组的auc0-24h、cmax及tmax均具有显著性差异,进一步说明了缬沙坦被cd-mof载药后生物利用度得到显著提高。表2.大鼠体内药动学参数药动学参数缬沙坦cd-mof组合物代文胶囊auc0-24h(μg/ml·h)77.71±13.12*52.05±23.02tmax(h)0.72±0.25**2.22±1.08cmax(μg/ml)16.88±4.85**7.90±3.70注:与代文组比较*p<0.05;**p<0.01。实施例16:比格犬体内生物利用度12只健康成年比格犬购自上海交大农学院教学实验实习场,体重约为8~12kg,分成3组,每组4只(雌雄各半),采用三周期三交叉实验设计,第一周期三组分别口服缬沙坦/cd-mof组合物胶囊(a药,按照实施例4制备,加入吐温-80乙醇溶液制粒)、代文胶囊(b药)、缬沙坦/γ-cd包合物胶囊(c药);第二周期三组分别口服b药、c药、a药;第三周期三组分别口服c药、a药、b药,每两周期间的清洗期为2周。实验前比格犬禁食12h,自由饮水,服药后4h内禁止饮食,受试期间进食统一餐。给药时将胶囊直接塞入会咽部,使比格犬自动吞咽并注入约50ml清水送下,于0、0.25、0.5、1、1.5、2、2.5、3、3.5、4、6、8、10、12、24、36、48h分别前肢内侧皮下静脉取血2ml。采用lc-ms/ms分析方法测定比格犬血浆样品中缬沙坦浓度。比格犬药动学结果显示(表3),缬沙坦/cd-mof组合物胶囊与缬沙坦γ-cd包合物胶囊相比,平均auc0-48h提高了47.3%、平均cmax提高了42.2%;缬沙坦/cd-mof组合物胶囊与代文胶囊相比,平均auc0-48h提高了59.8%、平均cmax提高了119.1%。以上结果表明,cd-mof载缬沙坦后,与其他两组相比,可提高缬沙坦在比格犬体内的生物利用度。表3比格犬体内药动学参数药动学参数缬沙坦/cd-mof代文缬沙坦/γ-cdauc0-48h(μg/ml·h)10.52±6.106.58±2.457.14±3.69tmax(h)1.08±0.561.75±0.991.08±0.52cmax(μg/ml)4.38±2.09*2.00±0.773.08±1.73注:与代文组比较*p<0.05。实施例17:缬沙坦/cd-mof组合物片剂制备工艺:称取下述处方中的载缬沙坦中性微米级cd-mof组合物与各辅料至混合机中均匀混合,选用12mm冲头进行粉末直压法压片。处方:溶出测定:采用桨法,在转速100rpm,温度为37.0±0.5℃,介质为900ml去离子水,在0.5,3,5,10,15,30,60min取点,每次取5ml,用0.45μm滤膜过滤,稀释适当倍数,在250nm处用紫外测吸光度(n=6)。结果如图7所示,缬沙坦微米级乙酸钾cd-mof组合物片剂30min溶出91%,而市售某缬沙坦片剂30min的溶出量为58%。实施例18:缬沙坦微米级乙酸钾cd-mof组合物-研磨法称取7.0g实施例1中制备的微米级乙酸钾cd-mof与3.0g缬沙坦(环糊精-金属有机骨架材料中的环糊精与缬沙坦的摩尔比例为1:1.5),球磨机(锆珠与粉体的重量比例为3:1)分别研磨5、10和60min,得到缬沙坦微米级乙酸钾cd-mof组合物。其增溶倍数依次为32.2、32.5、38.9倍,显著高于研磨法(环糊精与缬沙坦的摩尔比例为1:1.5,锆珠与粉体的重量比例为3:1,研磨60min)制备的缬沙坦γ-cd组合物(增溶倍数为4.5倍)。检测研磨5min获得的组合物的溶解速率,结果如图8所示,10min溶出87.1%。以相同的方法研磨单纯的药物缬沙坦(锆珠与粉体的重量比例为3:1)60min,其增溶倍数为1.4倍。溶解速率如图8所示,10min溶出4.8%。称取7.0g微米级乙酸钾cd-mof与3.0g缬沙坦(环糊精-金属有机骨架材料中的环糊精与缬沙坦的摩尔比例为1:1.5),对其进行简单混合,得到缬沙坦微米级乙酸钾cd-mof组合物。其增溶倍数为23.3倍。溶解速率如图8所示,10min溶出19.6%,相比于研磨的组合物,释放速率较慢。将7.0g缬沙坦和伽马环糊精进行简单物理混合(环糊精与缬沙坦的摩尔比例为1:1.5),制备缬沙坦和伽马环糊精组合物,缬沙坦和伽马环糊精组合物对缬沙坦的增溶倍数为4.5倍。溶解速率如图8所示,10min溶出24.8%。实施例19:替米沙坦cd-mof组合物称取1.944gγ-cd(1.5mmol)、0.672gkoh(12mmol)(nγ-cd:nkoh=1:8)和1543.85mg(3mmol)替米沙坦于100ml锥形瓶中,加入40ml纯水使其溶解,50℃水浴加热搅拌(300rpm)1h,加入24ml无水甲醇,继续搅拌5min,室温静置2h。离心(4000rpm,5min),沉淀用甲醇洗涤3遍后,置70℃鼓风干燥箱干燥。测得载药量为9.8%,实验测得替米沙坦在水中的饱和溶解度(25℃)为0.117mg/ml,而cd-mof载替米沙坦后在水中的饱和溶解度为11.425mg/ml,相比替米沙坦水中的溶解度提高了98倍。实施例20:阿齐沙坦乙酸钾纳米cd-mof组合物称取520mg阿齐沙坦于50ml具塞锥形瓶中,加入40ml无水乙醇,置60℃水浴加热搅拌使药物完全溶解,得到阿齐沙坦无水乙醇溶液(13mg/ml)。称取1.2g乙酸钾纳米cd-mof置50ml具塞锥形瓶,60℃,400rpm,水浴加热搅拌4h。离心(4000rpm,5min),弃去上清,取沉淀置烘箱干燥。阿齐沙坦/γ-cd包合物:称取8.45gγ-cd(10ml纯水溶解)和1.5g阿齐沙坦(10ml无水乙醇溶解),投料摩尔比cd:阿齐沙坦=2:1,采用行星式球磨机,加入一半体积的直径8mm锆珠研磨,研磨1h后,混悬液旋蒸后烘箱干燥。结果:阿齐沙坦/cd-mof的载药量为22.77%,载药摩尔比(azl:cd)为1:1。得到的环糊精/阿齐沙坦包合物含药量为13.14%,载药摩尔比(azl:cd)=0.49:1。阿齐沙坦环糊精包合物将阿齐沙坦在水中的溶解度提高了9.4倍,阿齐沙坦/cd-mof将阿齐沙坦在水中的溶解度提高了339.7倍,cd-mof的增溶能力显著高于环糊精包合(36.1倍)。阿齐沙坦cd-mof物理混合物(pm)的制备:按照载药比例分别称取阿齐沙坦与乙酸钾纳米级cd-mof,置于烧杯中,用玻璃棒进行搅拌混合,得物理混合物。阿齐沙坦/cd-mof组合物的表征:xrd结果(图9中a)表明阿齐沙坦与cd-mof均有明显特征衍射峰,载药后xrd结果无阿齐沙坦衍射峰,说明药物在cd-mof中的存在形式为无定型。同样,γ-cd包合阿齐沙坦后阿齐沙坦的存在形式也为无定型。ft-ir结果(图9中b)表明阿齐沙坦在1775cm-1和1693cm-1处有酰胺键的振动,分别为酰胺羧基和酰胺羰基。cd-mof载药后,阿齐沙坦/cd-mof的酰胺羧基发生位移至1770cm-1而酰胺羰基发生位移至1646cm-1,说明了cd-mof与阿齐沙坦的强相互作用为阿齐沙坦的羧基或羰基与cd-mof上的羟基发生分子间氢键。然而物理混合物(pm,实施例)则没有发生位移。而γ-cd包合阿齐沙坦后阿齐沙坦上1693cm-1处酰胺羰基发生位移至1640cm-1,说明了γ-cd与阿齐沙坦的相互作用位置主要为酰胺羰基与γ-cd上的羟基。气体吸附(图9中c)表明cd-mof的langmuir比表面积为996m2/g,且具有一定的孔径分布,而载药后阿齐沙坦/cd-mof中则由于药物的负载比表面积下降。dsc结果(图9中d)表明阿齐沙坦在205℃存在熔融吸热峰,而载药后其熔融吸热峰消失而物理混合物依然存在,说明阿齐沙坦在cd-mof中的存在状态发生改变,包合物中阿齐沙坦也同样发生了相同的变化。saxs结果(图10)显示当cd-mof载阿齐沙坦后,由于阿齐沙坦在cd-mof中的负载,尤其是在cd-mof大空腔位置的占据,1.6nm处散射信号显著减弱,各散射峰均向高场位移,推测阿齐沙坦被cd-mof负载,且在cd-mof大空腔处占据,原位形成纳米团簇。实施例21:阿齐沙坦大鼠体内生物利用度实验将8.45gγ环糊精溶解在10ml纯水中,1.5g阿齐沙坦溶解在10ml无水乙醇溶液中,投料摩尔比cd:azl2:1,两种溶剂混合后加入到行星式球磨机内,加入一半体积的直径8mm镐珠研磨1h,混悬液旋蒸后烘箱干燥,即得包合物。得到的环糊精/阿齐沙坦包合物含药量为13.14%,摩尔比(azl:cd)=0.49:1。与阿齐沙坦原料药相比增溶了9.4倍。取健康雄性sd大鼠18只,随机分为3组,分别为阿齐沙坦原料药组、阿齐沙坦/cd-mof组、阿齐沙坦/γ-cd包合物组,每组6只。实验前大鼠禁食不禁水12h,称重,分别灌胃给药给予阿齐沙坦原料药组、实施例20制备的阿齐沙坦/cd-mof组、实施例20制备的阿齐沙坦/γ-cd包合物组5ml/kg(阿齐沙坦给药剂量为1mg/kg),于给药前及给药后5min、15min、30min、1h、1.5h、2h、4h、8h、12h、24h分别眼眶取血约0.3ml,血样置肝素化的ep管中,离心(4000rpm,5min),分离血浆,置-80℃冰箱中保存,实验过程中允许大鼠自由饮水。结果:阿奇沙坦/cd-mof的吸收明显高于和快于阿齐沙坦/γ-cd包合物和原料药。阿齐沙坦/cd-mof与阿齐沙坦原料药相比auc提高了9.7倍,与阿奇沙坦/γ-cd包合物比较auc提高了1.4倍。表4阿齐沙坦大鼠体内药动学参数药动学参数阿齐沙坦/cd-mof阿奇沙坦/γ-cd包合阿齐沙坦原料药auc(0-12h)(mg/l*h)30.65±14.2421.80±7.703.16±0.42tmax(h)1.50±1.300.90±0.426.67±2.07cmax(mg/l)2.63±1.022.06±0.410.19±0.01实施例22:载缬沙坦不同温度考察微米级乙酸钾cd-mof选自实施例2,参考实施例3的制备方法分别在20、60和70℃条件下对缬沙坦进行载药。水浴加热搅拌转速为400r/min,载药时间为2h,载药混悬液离心取沉淀,40℃真空干燥,制备组合物。结果:20、60和70℃条件下的载药量分别为22.9%、28.8%、29.3%,组合物中cd-mof与缬沙坦的摩尔比为1:1.0、1:1.4、1:1.4。实施例23:载缬沙坦不同溶剂考察参考实施例3的制备方法,选择甲醇、乙醇:甲醇1:1混合溶剂、乙酸乙酯作为载药溶剂。微米级乙酸钾cd-mof选自实施例2,载药条件为40℃,缬沙坦乙醇溶液浓度为250mg/ml,水浴加热搅拌转速为400r/min,载药时间为2h,载药混悬液离心取沉淀,40℃真空干燥,制备组合物。结果:甲醇、甲醇:乙醇1:1混合溶剂、以及乙酸乙酯中的载药量分别为26.4%、24.5%、22.37%,组合物中cd-mof与缬沙坦的摩尔比分别为1:1.2、1:1.1、1:1.0。实施例24:不同投料摩尔比-微米级乙酸钾cd-mof参考实施例3的制备方法,设置缬沙坦乙醇溶液浓度为100mg/ml,150mg/ml和200mg/ml,微米级乙酸钾cd-mof选自实施例2。水浴加热搅拌转速为400r/min,载药时间为2h,载药混悬液离心取沉淀,40℃真空干燥,制备组合物。结果:微米级乙酸钾cd-mof与缬沙坦投料摩尔比为1:10、1:15和1:20的载药量分别为23.7%、26.6%、28.7%,组合物中cd-mof与缬沙坦的摩尔比分别为1:1.1、1:1.2、1:1.4。实施例25:不同投料摩尔比-微米级氢氧化钾cd-mof参考实施例3的制备方法,设置缬沙坦乙醇溶液浓度为100mg/ml和150mg/ml,微米级氢氧化钾cd-mof选自实施例1。水浴加热搅拌转速为400r/min,载药时间为2h,载药混悬液离心取沉淀,40℃真空干燥,制备组合物。结果:微米级氢氧化钾cd-mof与缬沙坦投料摩尔比为1:10和1:15的载药量分别为22.4%、25.9%,组合物中cd-mof与缬沙坦的摩尔比分别为1:1.0、1:1.2。实施例26:厄贝沙坦称量1g厄贝沙坦原料于100ml锥形瓶中,加入50ml无水乙醇(浓度为20mg/ml),置50℃恒温水浴磁力搅拌加热,药物完全溶解后,加入1.16g实施例2中的微米级乙酸钾cd-mof(药物与cd-mof的投料摩尔比3:1),加热搅拌(300rpm)4h。离心去上清,沉淀置真空干燥箱干燥(50℃,5h)。称取载药cd-mof5mg于10ml容量瓶中,用纯水溶解并定容至刻度。离心(4000rpm,5min)后取上清用紫外分光光度法在药物含量。结果显示,载药量为11.19%,组合物中cd-mof与厄贝沙坦的摩尔比分别为1:0.4,增溶倍数为339倍。实施例27:γcd-mof载阿齐沙坦的分子模拟阿齐沙坦在γcd-mof中的分布形式主要为两种状态:一部分为环糊精分子对对阿齐沙坦的包合(图11中a);一部分为阿齐沙坦在γcd-mof的大空腔中形成纳米团簇(图11中b)。具体为:阿奇沙坦分子首先倾向于蜷缩在γcd-mof中双环糊精结构产生的疏水空腔中(对接自由能为-10.1kcal/mol),阿齐沙坦中的疏水部分正好陷入环糊精的疏水空腔中,而亲水部分则在双环糊精对的缝隙中存在。利用了阿齐沙坦分子中疏水部位的特性,继续对接的阿齐沙坦分子以不同的体系能量对接到γcd-mof的大空腔中。6个阿齐沙坦分子被6个环糊精分子对包合,3个阿齐沙坦分子负载于大空腔中,组合物中γcd-mof与缬沙坦的理论载药摩尔比为1:1。实施例28:γcd-mof载缬沙坦的分子模拟及表征缬沙坦在γcd-mof中的分布形式主要为两种状态:一部分为环糊精分子对对缬沙坦的包合;一部分为缬沙坦在γcd-mof的大空腔中形成纳米团簇。具体为:第一个缬沙坦分子更倾向于蜷缩在γcd-mof中双环糊精结构产生的疏水空腔中(对接自由能-8.5kcal/mol),从而空出亲水空腔接纳更多的缬沙坦分子。缬沙坦中的羧基与γcd-mof晶体缝隙中的氢氧根以及环糊精中的羟基发生静电相互作用,产生氢键。利用缬沙坦分子中亲水部位和疏水部位的特性,继续对接的缬沙坦分子以不同的体系能量对接到γcd-mof的大空腔中。图12所示,6个缬沙坦分子被6个环糊精分子对包合,5个缬沙坦分子负载于大空腔中,组合物中γcd-mof与缬沙坦的理论载药摩尔比为1:1.3。图13:x-射线小角散射(saxs)结果表明:cd-mof为典型的体心立方晶体,长程有序,具有较好的周期性。载药后(实施例4的缬沙坦/cd-mof1:1.5和实施例11的缬沙坦/cd-mof1:0.5),缬沙坦/cd-mof的布拉格长周期(2.2nm)均与cd-mof保持一致,但由于药物分子进入cd-mof的骨架导致半高宽均增加。当缬沙坦/cd-mof为1:0.5时,cd-mof在各个峰的位置没有发明显位移,当缬沙坦/cd-mof为1:1.5时,(200)晶面上1.6nm空腔位置由于被药物分子占据信号显著减弱。图14:固态核磁结果显示:cd-mof载药后(实施例4的缬沙坦/cd-mof1:1.5和实施例11的缬沙坦/cd-mof1:0.5)整体信号线宽显著增加,尤其对于高载药量的val/cd-mof(1:1.5),缬沙坦苯环(c12-23)的裂分信号消失,cd-mof上糖苷键(c4)线宽明显增加,说明药物与cd-mof载体之间存在较强相互作用。缬沙坦分子处于一个受限环境中并很可能进入双γ-cd分子对中。载药后cd-mof羟基(c6)向高场偏移,val/cd-mof1:0.5和1:1.5在57.5ppm处分别偏移1.9ppm和1.7ppm;相应地,val/cd-mof1:0.5和1:1.5中缬沙坦在170-180ppm处的羧基(c10)由裂分峰变为一个宽峰,表明载药过程中缬沙坦的羧基和cd-mof的羟基可能通过形成氢键发生相互作用。载药后对于缬沙坦的烷基链(8-36ppm),val/cd-mof1:0.5在此处的线型和强度均发生显著变化,而val/cd-mof1:1.5中信号与缬沙坦的线型相似,表明缬沙坦分子部分聚集在cd-mof中间的大空腔内形成纳米团簇。实施例29:厄贝沙坦量取3000ml纯水于烧杯中,加入97.3gγ-cd搅拌,再加入33.6gkoh,超声,使γ-cd和koh完全溶解,制得母液。将母液倒入反应釜中,设置转速为300rpm,温度50℃。再向反应釜中加入1800ml甲醇,溶解澄清后加入38.4gpeg20000。室温静置过夜后离心,去上清,向下层沉淀物中加入0.4l无水乙醇分散,进行离心洗涤,再向沉淀中加入0.4l无水乙醇和20ml冰醋酸,搅拌中和,离心去除上清。最后向沉淀中加入0.4l乙醇,离心(4000rpm,5min)洗涤两遍。将洗涤后的沉淀置于60℃真空干燥箱中干燥4h。即得微米级乙酸钾环糊精-金属有机骨架,粒径为1-10微米。称量4.29g厄贝沙坦原料和14.93g微米级乙酸钾cd-mof(环糊精-金属有机骨架材料中的环糊精与厄贝沙坦的摩尔比例为1:1),充分混匀,以摩尔比1:1的比例投料研磨,使用球磨机(锆珠与粉体的重量比例为3:1),分别研磨5、15、20min。分别称取研磨后的样品20mg(过量)于10ml离心管中,加入5ml纯水,立即涡旋(1000rpm)1min,0.22μm滤膜过滤,用紫外分光光度法测饱和溶解度。结果显示,厄贝沙坦原料在纯水中的饱和溶解度为8.4μg/ml,厄贝沙坦与cd-mof共研磨5、15、20min后饱和溶解度分别为145.2、153.4、193.8μg/ml,分别增溶了17.3、18.3、23.1倍。实施例30阿齐沙坦称取7.0g实施例29所制备的微米级乙酸钾cd-mof与3.0g阿齐沙坦(环糊精-金属有机骨架材料中的环糊精与阿齐沙坦的摩尔比例为1:1),使用球磨机(锆珠与粉体的重量比例为3:1)研磨5min,得到阿齐沙坦微米级乙酸钾cd-mof组合物。其增溶倍数为25.4倍。实施例31缬沙坦与环糊精的混合物称取7.0g伽马环糊精与3.0g缬沙坦(伽马环糊精与缬沙坦的摩尔比例为1:1.5),使用球磨机(锆珠与粉体的重量比例为3:1)分别研磨60min、5min,得到缬沙坦和伽马环糊精组合物,增溶倍数分别为5.7倍、4.8倍。实施例32称取7.0g实施例29所制备的微米级乙酸钾cd-mof与3.0g缬沙坦(环糊精-金属有机骨架材料中的环糊精与缬沙坦的摩尔比例分别为1:1.5、1:0.5),使用球磨机(锆珠与粉体的重量比例分别为10:1、3:1)研磨60min,得到缬沙坦微米级乙酸钾cd-mof组合物,增溶倍数分别为为35.1倍、23.0倍。实施例33称取648mgγ-cd和224mgkoh,加入20ml纯水,搅拌至完全溶解;倒入50ml塑料离心管中,加入12ml无水甲醇;放入恒温水箱中50℃加热20min。加入8mg/mlpeg20000震荡摇匀;静置1h后以3500rpm离心5min,倒掉上清液。用12ml无水乙醇洗涤沉淀3次后真空干燥制备得到氢氧化钾cd-mof。称取7.0g微米级氢氧化钾cd-mof与3.0g缬沙坦(环糊精-金属有机骨架材料中的环糊精与缬沙坦的摩尔比例为1:1),使用球磨机(锆珠与粉体的重量比例为3:1)研磨5min,得到缬沙坦微米级乙酸钾cd-mof组合物。其增溶倍数为53.9倍。实施例34取2000ml纯水于5000ml大烧杯中,称取64.9gγ-cd于大烧杯中,搅拌,再向烧杯中加入22.4gkoh,超声,使γ-cd和koh完全溶解,制得母液。将母液倒入反应釜中,转速300rpm,温度50℃时,向反应釜中加入1200ml甲醇,再加入peg20000(8mg/ml)。冷水浴中静置过夜。离心去上清,沉淀分别使用150ml乙醇洗涤两遍,于40℃真空干燥箱中干燥12h。粒径为100-500纳米。称取7.0g所制备的纳米级乙酸钾cd-mof与3.0g缬沙坦(环糊精-金属有机骨架材料中的环糊精与缬沙坦的摩尔比例为1:1.5),使用球磨机(锆珠与粉体的重量比例为3:1)研磨5min,得到缬沙坦纳米级乙酸钾cd-mof组合物。其增溶倍数为27.3倍。实施例35量取3000ml纯水于烧杯中,加入97.3gγ-cd搅拌,再加入33.6g氢氧化钠,超声,使γ-cd和氢氧化钠完全溶解,制得母液。将母液倒入反应釜中,设置转速为300rpm,温度50℃。再向反应釜中加入1800ml甲醇,溶解澄清后加入38.4gpeg20000。室温静置过夜后离心,去上清,沉淀分别使用100ml乙醇洗涤两遍,再用100ml甲醇洗涤两遍。沉淀于40℃真空干燥箱中干燥12h,即得微米级氢氧化钠环糊精金属有机骨架。称取7.0g微米级氢氧化钠环糊精金属有机骨架与3.0g缬沙坦(环糊精-金属有机骨架材料中的环糊精与缬沙坦的摩尔比例为1:1),使用球磨机(锆珠与粉体的重量比例为3:1)研磨5min,得到缬沙坦微米级氢氧化钠环糊精金属有机骨架组合物。实施例36量取3000ml纯水于烧杯中,加入97.3gγ-cd搅拌,再加入16.8g氢氧化钙,超声,使γ-cd和氢氧化钙完全溶解,制得母液。将母液倒入反应釜中,设置转速为300rpm,温度50℃。再向反应釜中加入1800ml乙醇,溶解澄清后加入38.4gpeg4000。室温静置过夜后离心,去上清,沉淀分别使用100ml乙醇洗涤两遍,再用100ml乙醇洗涤两遍。沉淀于40℃真空干燥箱中干燥12h,即得微米级氢氧化钙环糊精金属有机骨架。称取7.0g微米级氢氧化钙环糊精金属有机骨架与3.0g缬沙坦(环糊精-金属有机骨架材料中的环糊精与缬沙坦的摩尔比例为1:1),使用球磨机(锆珠与粉体的重量比例为3:1)研磨5min,得到缬沙坦微米级氢氧化钙环糊精金属有机骨架组合物。在本发明提及的所有文献都在本申请中引用作为参考,就如同每一篇文献被单独引用作为参考那样。此外应理解,在阅读了本发明的上述讲授内容之后,本领域技术人员可以对本发明作各种改动或修改,这些等价形式同样落于本申请所附权利要求书所限定的范围。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1