苯并异硒唑衍生物与抗代谢药物联合用于制备治疗肿瘤药物中的应用的制作方法
本发明属于肿瘤治疗技术领域,具体涉及一种苯并异硒唑衍生物与抗代谢药物联合用于制备治疗肿瘤药物中的应用。
背景技术:
原发性肝细胞癌(primarylivercancer,下称肝癌)是人类最常见的恶性肿瘤之一。据报道,肝癌发病率位居全球所有恶性肿瘤发病率的第六位,而死亡率则居第三位。我国每年约有38.3万人死于肝癌,占全球肝癌死亡病例数的51%,严重威胁人类的生命健康。
由于肝癌细胞增殖速度快,早期难以诊断,往往当肝癌被诊断出来的时候,大多数病人已经发展到了肝癌晚期,且经常伴随肝内或肝外的转移,这些因素是造成肝癌高死亡率的主要原因。因此,寻找能够有效抑制肝癌细胞增殖的药物是治疗肝癌的关键。
抗代谢药物,其能与体内代谢物发生特异性结合,从而影响或拮抗代谢功能的药物,通常它们的化学结构与体内的核酸或蛋白质代谢物相似。其中氟尿嘧啶(5-fu)作为典型的抗代谢药物,是一种多种癌症的一线抗癌药物,其适用于肝癌、卵巢癌、肺癌等多种癌症,能够通过多种机制发挥广谱的抗癌作用。然而,抗代谢药物一般于用药后第二周即可出现骨髓毒性,常表现为白细胞及血小板减少,因此,当抗代谢类药物单独作为抗癌药物使用时,其较高的毒副作用,导致抗癌效果较差。
技术实现要素:
本发明提供苯并异硒唑衍生物与尿嘧啶衍生物联合用于制备治疗肿瘤药物中的应用。
根据本发明,所述苯并异硒唑衍生物具有如式a所示化合物的结构,其选自式a所示化合物、其前体、活性代谢产物、立体异构体、药学上可接受的盐、前药和溶剂合物中的至少一种,
其中,r1、r2相同或不同,彼此独立的选自h、或下列基团:c1-12烷基、c3-20环烷基;优选地,选自h、或下列基团:c1-6烷基、c3-10环烷基;示例性地,r1、r2选自h。
其中,r选自c1-12亚烷基、亚苯基、亚联苯基、亚三苯基,或者
优选地,所述式a所示化合物中,r1、r2相同或不同,彼此独立的选自h、-ch3、-ch2ch3、-ch(ch3)2、-c(ch3)3、-ch(ch2)4、或-ch(ch2)5;r选自-ch2-、-c2h4-、-c4h8-、亚苯基-c6h4-。
根据本发明示例性的技术方案,所述苯并异硒唑衍生物选自1,2-二[2-(1,2-苯并异硒唑-3(2h)-酮)]-丁烷,其结构如下所示:
根据本发明,所述抗代谢药物可以选自嘧啶拮抗剂、嘌呤拮抗剂和叶酸拮抗剂中的至少一种。
例如,所述嘧啶拮抗剂可以选自尿嘧啶衍生剂和/或胞嘧啶衍生剂。优选地,所述嘧啶拮抗剂可以选自氟尿嘧啶(5-fluorouracil,简写为5-fu)、替加氟(ftorafur)、卡莫氟(carmofur)、5-乙炔尿嘧啶(eniluracil)、去氧氟尿苷和盐酸阿糖胞苷等中的至少一种;例如,所述嘧啶拮抗剂选自氟尿嘧啶、替加氟或者卡莫氟;示例性地,所述嘧啶拮抗剂选自氟尿嘧啶(5-fu)。例如,所述嘌呤拮抗剂可以选自次黄嘌呤与鸟嘌呤的衍生物;优选地,所述嘌呤拮抗剂可以选自巯嘌呤。例如,所述叶酸拮抗剂可以选自甲氨喋呤。
根据本发明,所述苯并异硒唑衍生物与抗代谢药物的摩尔比为(1~10):(1~10);例如摩尔比为(1~8):(1~8),(1~7):(1~7),(1~6):(1~6),(1~5):(1~5),(1~4):(1~4);示例性地,摩尔比为1:1、2:1、1:2、1:3。
根据本发明,所述肿瘤包括实体瘤和非实体瘤,可以是良性的和恶性的。例如,所述肿瘤包括但不限于:肝癌、肺癌、乳腺癌、结肠癌、鼻咽癌、胃癌、皮肤癌、膀胱癌、卵巢癌、前列腺癌、骨癌、脑癌、直肠癌、食管癌、舌癌、淋巴癌、口腔上皮癌、上皮宫颈癌或慢性髓系白血病。优选地,所述肿瘤为耐药的肿瘤,所述耐药是指对作用于分裂和增殖状态的肿瘤细胞的抗肿瘤药物耐药。
进一步的,本发明提供苯并异硒唑衍生物与抗代谢药物联合用于抑制肿瘤细胞增殖中的应用。例如,苯并异硒唑衍生物与抗代谢药物联合用于抑制肿瘤细胞体外或体内增殖中的应用;优选地,用于抑制肿瘤细胞体外增殖中的应用。
例如,所述肿瘤细胞包括但不限于:人肝癌细胞hepg2、人肝癌细胞be17402、人肝癌细胞huh7721、人结直肠癌细胞lovo、人结直肠癌细胞rko、人结直肠癌细胞sw480、人肺癌细胞a549、人肺癌细胞h1299、人肺癌细胞spca-1、人上皮宫颈癌细胞hela、人乳腺癌细胞mcf-7、人慢性髓性白血病细胞k562、人食管癌细胞kyse150、人食管癌细胞kyse450或人食管癌细胞kyse510。根据本发明示例性的技术方案,所述肿瘤细胞选用人肝癌细胞hepg2。
根据本发明示例性的技术方案,所述苯并异硒唑衍生物与抗代谢药物联合用于抑制人肝癌细胞hepg2的增殖。例如,用于抑制人肝癌细胞hepg2的体外增殖;优选地,造成hepg2细胞s期(dna合成期)阻滞,以及抑制hepg2细胞内trxr(硫氧还蛋白氧化还原酶)活性。
进一步的,本发明提供苯并异硒唑衍生物与抗代谢药物联合抑制肿瘤细胞增殖的方法,将苯并异硒唑衍生物与抗代谢药物按一定配比作用于肿瘤细胞。
优选地,所述一定配比为:苯并异硒唑衍生物与抗代谢药物的摩尔比为(1~10):(1~10);例如摩尔比为(1~8):(1~8),(1~7):(1~7),(1~6):(1~6),(1~5):(1~5),(1~4):(1~4);示例性地,摩尔比为1:1、2:1、1:2、1:3。本建议为达到体内组织部位的要求浓度,由于要求体内代谢可能出现的差异另行按照药物代谢特征折算。
优选地,所述苯并异硒唑衍生物与抗代谢药物的浓度为1~100μm,例如5~60μm、10~50μm;示例性地,浓度为15、20、30、40μm。
优选地,以肿瘤细胞重量计,所述苯并异硒唑衍生物的给药量为0.04~1mg/g;例如,给药量为0.045~0.8mg/g、0.0.06~0.0.1mg/g、0.15~0.5mg/g;示例性地,给药量为0.18mg/g、0.045mg/g、0.09mg/g。
优选地,以肿瘤细胞重量计,所述抗代谢药物的给药量为0.005~0.05mg/g;例如,给药量为0.008~0.04mg/g、0.01~0.03mg/g;示例性地,给药量为0.01mg/g、0.013mg/g、0.026mg/g、0.039mg/g。
优选地,所述作用的时间为10~120h,例如15~105h,24~96h;示例性地,作用时间为24h、48h、72h。
进一步的,本发明还提供一种药物组合物,所述药物组合物包括苯并异硒唑衍生物与抗代谢药物。优选地,所述苯并异硒唑衍生物与抗代谢药物具有如上文所述的含义和浓度配比。
优选地,所述药物组合物还可任选地包含至少一种药学上可接受的辅料。
优选地,所述药学上可接受的辅料是制药领域中常用或已知的各种辅料,包括但不限于:稀释剂、粘合剂、抗氧化剂、ph调节剂、防腐剂、润滑剂、崩解剂等。
例如,所述稀释剂选自乳糖、淀粉、纤维素衍生物、无机钙盐、山梨醇等。所述粘合剂例如:淀粉、明胶、羧甲基纤维素钠、聚乙烯吡咯烷酮等。例如,所述抗氧化剂选自维生素e、亚硫酸氢钠、亚硫酸钠、丁羟基茴香醚等。例如,所述ph调节剂选自盐酸、氢氧化钠、柠檬酸、酒石酸、tris、乙酸、磷酸二氢钠、磷酸氢二钠等。例如,所述防腐剂选自对羟基苯甲酸甲酯、对羟基苯甲酸乙酯、间甲酚、苯扎氯铵等。例如,所述润滑剂选自硬脂酸镁、微粉硅胶、滑石粉等。所述崩解剂例如:淀粉、甲基纤维素、黄原胶、交联羧甲基纤维素钠等。
所述药物组合物的剂型可以是口服剂的形式,例如片剂、胶囊、丸剂、粉剂、颗粒剂、悬浮剂、糖浆剂等;也可以是注射给药的剂型,例如注射液、粉针剂等,通过静脉内、腹膜内、皮下或肌肉内的途径。所有使用的剂型形式都是药学领域普通技术人员所熟知的。
进一步的,本发明还提供上述药物组合物治疗肿瘤的方法,将治疗有效量的所述药物组合物施用于有此需求的个体。
优选地,所述个体可以为哺乳动物,如人类。
优选地,所述肿瘤具有如上文所述的含义,优选为肝癌。
本发明的有益效果是:
本发明出人意料地发现,bs与抗代谢药物(尤其是5-fu)联合应用对肿瘤细胞增殖具有优异的协同抑制效果,尤其是对于肝癌细胞增殖抑制效果明显,对于肿瘤的治疗具有重大价值。二者联用,可造成hepg2细胞s期(dna合成期)阻滞,并且抑制了hepg2细胞内trxr(硫氧还蛋白氧化还原酶)活性。
具体来讲,bs的使用,有效弥补了5-fu在早期用药时对肝癌细胞无明显生长抑制作用和易产生耐药性的缺陷,且bs也很好地发挥了其抗肿瘤活性和较为彻底地细胞杀伤力。二者联用,较5-fu单药使用时,毒性较大的5-fu用量明显减少,提升抗癌用药的安全性。
bs和5-fu联合抑制肿瘤的机制:
5-fu作为典型的抗代谢药物,在体内先转变为氟尿嘧啶核苷(5-fur)及氟尿嘧啶脱氧核苷(5-fudr),它们进一步转变为相应的磷酸核苷和脱氧核苷(5-fudrp)。5-fudrp可抑制胸腺嘧啶核苷酸合成酶(thmidylatesynthetase,ts)活力,从而阻断dump甲基化形成dtmp的过程,使细胞增殖阻滞于s期(dna合成期)而死亡,但是毒副作用明显。bs具有的硫氧还蛋白还原酶抑制作用,可以通过凋亡诱导促进肿瘤细胞死亡,因此,与5-fu联合应用可以发挥二者不同作用机制,产生协同作用,抗肿瘤效果优异。
术语定义和说明
除非另有说明,本申请说明书和权利要求书中记载的基团和术语定义,包括其作为实例的定义、示例性的定义、优选的定义、表格中记载的定义、实施例中具体化合物的定义等,可以彼此之间任意组合和结合。这样的组合和结合后的基团定义及化合物结构,应当属于本申请说明书记载的范围内。
术语“c1-12烷基”应理解为优选表示具有1~12个碳原子的直链或支链饱和一价烃基,优选为c1-6烷基。“c1-6烷基”应理解为优选表示具有1、2、3、4、5、6个碳原子的直链或支链饱和一价烃基。所述烷基是例如甲基、乙基、丙基、丁基、戊基、己基、异丙基、异丁基、仲丁基、叔丁基、异戊基、2-甲基丁基、1-甲基丁基、1-乙基丙基、1,2-二甲基丙基、新戊基、1,1-二甲基丙基、4-甲基戊基、3-甲基戊基、2-甲基戊基、1-甲基戊基、2-乙基丁基、1-乙基丁基、3,3-二甲基丁基、2,2-二甲基丁基、1,1-二甲基丁基、2,3-二甲基丁基、1,3-二甲基丁基或1,2-二甲基丁基等或它们的异构体。特别地,所述基团具有1、2、3、4、5、6个碳原子(“c1-6烷基”),例如甲基、乙基、丙基、丁基、异丙基、异丁基、仲丁基、叔丁基,更特别地,所述基团具有1、2、3或4个碳原子(“c1-4烷基”),例如甲基、乙基、正丙基、异丙基或丁基。
术语“c3-20环烷基”应理解为表示饱和的一价单环或双环烃环,其具有3~20个碳原子,优选“c3-10环烷基”。术语“c3-10环烷基”应理解为表示饱和的一价单环或双环烃环,其具有3、4、5、6、7、8、9或10个碳原子。所述c3-10环烷基可以是单环烃基,如环丙基、环丁基、环戊基、环己基、环庚基、环辛基、环壬基或环癸基,或者是双环烃基如十氢化萘环。
术语“c1-12亚烷基”应理解为优选表示丢失两个氢原子的具有1~12个碳原子的直链或支链饱和一价烃基,优选为c1-4亚烷基。“c1-4亚烷基”应理解为优选表示丢失两个氢原子的具有1、2、3、4个碳原子的直链或支链饱和一价烃基。所述亚烷基是例如亚甲基、亚乙基、亚丙基、亚丁基。
术语“有效量”或者“治疗有效量”是指足以实现预期应用(包括但不限于如下定义的疾病治疗)的本发明所述化合物的量。治疗有效量可以取决于以下因素而改变:预期应用(体外或者体内),或者所治疗的受试者和疾病病症如受试者的重量和年龄、疾病病症的严重性和给药方式等,其可以由本领域普通技术人员容易地确定。具体剂量将取决于以下因素而改变:所选择的特定化合物、所依据的给药方案、是否与其它化合物组合给药、给药的时间安排、所给药的组织和所承载的物理递送系统。
附图说明
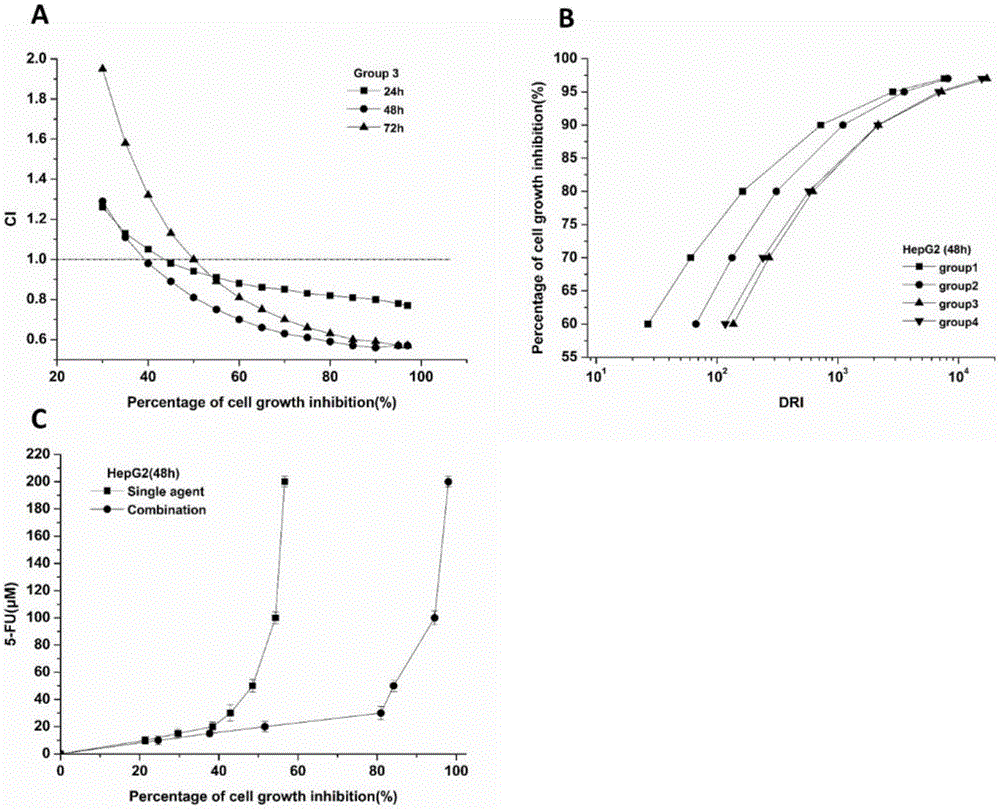
图1是实施例3中bs和5-fu联合给药作用于hepg2细胞的相互作用评价图;
其中,(a)为实施例3处理hepg2细胞24小时、48小时、72小时,ci随细胞增殖抑制率的变化曲线;(b)为实施例1-4处理hepg2细胞48小时,细胞增殖抑制率随dri的变化曲线;(c)为实施例3和5-fu单药分别处理hepg2细胞48小时,5-fu用药量随细胞增殖抑制率的变化曲线。数据值为平均值±标准差,n=3。
图2是15μm5-fu、15μmbs以及两者联合(15μm5-fu:15μmbs=1:2)作用于hepg2细胞48h后的细胞周期分布图。
其中,a:对照组;b:15μm5-fu;c:15μmbs;d:15μm5-fu:15μmbs=1:2。
图3是15μm5-fu、15μmbs以及两者联合(15μm5-fu:15μmbs=1:2)作用48h对hepg2细胞周期的影响。数据值为平均值±标准差,n≥3。
图4是15μm5-fu、15μmbs及两者联合(15μm5-fu:15μmbs=1:2)作用于hepg2细胞48h后蛋白变化图;
其中,a:细胞内trxr、trx的蛋白表达水平;b:trxr的相对蛋白表达量;c:trx的相对蛋白表达量。数据均为平均值±标准差,n≥3。*p<0.05表示与对照相比具有显著性差异;#p<0.05表示与5-fu组相比具有显著性差异;※p<0.05表示与单药组相比均具有显著性差异。
图5是bs和5-fu抑制hepg2细胞增殖线;其中,图(a)为5-fu作用于hepg2细胞;图(b)为bs作用于hepg2细胞。
具体实施方式
下文将结合具体实施例对本发明的技术方案做更进一步的详细说明。下列实施例仅为示例性地说明和解释本发明,而不应被解释为对本发明保护范围的限制。凡基于本发明上述内容所实现的技术均涵盖在本发明旨在保护的范围内。
除非另有说明,以下实施例中使用的原料和试剂均为市售商品,或者可以通过已知方法制备。
实验方法:
1.试验药品及细胞:
bs:化学名1,2-二[2-(1,2-苯并异硒唑-3(2h)-酮)]-丁烷,参考中国专利第02158917.8号申请。
5-fu:购自北京大学第三医院。
hepg2细胞:购自协和医科大学基础医学院细胞中心。
2.试验方法:
2.1细胞增殖抑制率检测法(srb法):
(1)将贴壁培养的细胞消化为单细胞悬液,调整细胞浓度为2×104个/ml,均匀接种于96孔板(corning),每孔180μl;
(2)将96孔板置于培养箱中24h,待细胞贴壁后按照指定浓度加入化合物20μl/孔;
(3)药物作用终点,每孔加入100μl4℃预冷的10%(v/v)三氯乙酸,将96孔板于4℃固定1h;
(4)小心弃去固定液,用去离子水从96孔板一侧缓缓冲洗4遍,用电热吹风吹干或者室温自然干燥(干燥后的96孔板可以室温长期保存);
(5)每孔中加入100μl0.057%(w/v)的srb染色液,室温染色30min;
(6)小心弃去染液,每孔用150μl1%(v/v)乙酸洗涤4次,用电热吹风吹干或者室温自然干燥(干燥后的96孔板可以室温长期保存);
(7)每孔中精确加入200μl10mm的tris脱色液,室温静置使之自然溶解或置于摇床上使之溶解;
(8)用酶标仪(thermofisherscientific)检测样品在492nm处的吸光值。依据以下公式计算细胞增殖抑制率:
其中,对照孔接种细胞混悬液180μl,以同等体积培养基(20μl)代替化合物;空白孔接种时加入180μl培养基,给药时加入20μl培养基。每个药物浓度设置三个复孔。
2.2westernblot法:
提取组织总蛋白并测定蛋白浓度(30~50μg)加入5×上样缓冲液,以ripa裂解液补足体积,混匀后95℃变性处理10min。充分混匀后,加入洁净的1.5mm制胶板至适当位置正丁醇进行水封。加入1ml1×分离胶缓冲液,室温放置过夜;插入1.5mm加样梳,室温静置2h等待胶凝。
拔去加样梳,常法上样;80v恒压电泳,至溴酚蓝前沿进入分离胶,将电压提高至160v继续恒压电泳,至溴酚蓝迁移接近分离胶末端时停止电泳。
精确剪取与分离胶同样大小的6张滤纸和一张0.2μmpvdf膜,将pvdf膜于甲醇中浸泡10s,去离子水中浸泡3min,电转缓冲液中浸泡3min;
电转三明治由正极到负极的顺序依次安放:一张海绵、三张滤纸、pvdf膜、分离胶、三张滤纸、一张海绵(全部预先用电转缓冲液浸泡),每层之间用玻璃棒赶出气泡,夹好电转夹连同冰盒一起放入电转槽;
将电转槽置于冰水浴中,250ma恒流电转2h。
电转完毕后,将pvdf膜取出,置于5%封闭;tbst洗膜3次,将pvdf膜与稀释适当比例的二抗溶液一同封闭于杂交袋中,室温孵育1h,tbst洗膜3次,显影;
打开凝胶成像系统,将ecl发光液的a液和b液等体积混匀后均匀分布在pvdf膜上,进行曝光分析。
2.3dtnb还原法:
1.于96孔板中加入50μg蛋白样品,用0.1m磷酸钠缓冲液补足体积至80μl,37℃孵育30min;
2.各样品孔加入20μl5mmnadph溶液,对照孔加入等体积的0.1m磷酸钠缓冲液;
3.各孔用排枪加入100μl10mmdtnb溶液后,立即用酶标仪检测405nm处吸光值,第一次读数前震荡10s,每15s测定一次,测定30次。取最大反应速率为酶活性指标。实验独立重复三次。
2.4流式细胞术检测法:
1)取对数生长期的细胞接种于六孔板(corning)中,5×104个/孔,每孔含细胞悬液2ml,将培养皿置于培养箱中培养。待24h细胞贴壁后,弃去培养基,向其中加入相应浓度的药液2ml,对照组加入等体积的培养基;
2)药物作用24h后,回收培养基于5ml离心管中,1200rpm离心2min,弃上清。培养皿中的贴壁细胞用4℃预冷的pbs润洗一遍,加入300μl胰酶后置于培养箱中消化3min;
3)用1ml培养基将细胞吹打成单细胞,用1mlpbs重悬离心管中的沉淀细胞,与相应组别培养皿中的贴壁细胞合并,400rpm离心2min,弃上清。用1mlpbs重悬细胞,过300目细胞筛;
4)用190μl结合液重悬沉淀细胞,加入10μlannexinv-fitc,室温避光孵育10min,4000rpm离心2min,弃上清;
5)用195μl结合液重悬沉淀细胞,加入5μlpi溶液,立即于流式细胞仪(bdfacscalibur)上检测。
2.5药液配制
bs药物母液:称取bs,加入dmso将其溶解,制备成10mm的储备液,于-20℃保存备用。
cddp母液:称取cddp,加入pbs将其溶解,制备成10mm的储备液,于室温保存备用。
用前根据研究需要,用不含血清的培养基将母液稀释到所需浓度(15、20、30、40μm)的工作液。
实施例1
以hepg2细胞为作用对象,bs与5-fu按照摩尔比2:1同时给药。
实施例2
以hepg2细胞为作用对象,bs与5-fu按照摩尔比1:1同时给药。
实施例3
以hepg2细胞为作用对象,bs与5-fu按照摩尔比1:2同时给药。
实施例4
以hepg2细胞为作用对象,bs与5-fu按照摩尔比1:3同时给药。
实施例5bs与氟尿嘧啶联合应用抑制hepg2细胞增殖的体外实验研究
5.1bs与5-fu联合应用对hepg2细胞的增殖抑制作用
采用实施例1-4的药物联合方案分别作用于对数生长期的hepg2细胞24、48、72h,利用srb法测定细胞生长抑制率。表1联合列的结果显示,两药联合对hepg2的生长抑制也呈现出了一定的剂量、时间依赖性。实施例1-4四种联合方案在3个时间点的ic50均低于5-fu单药组,mi均高于5-fu单药组。其中实施例3、实施例4的ic50均低于bs单药组,尤以48、72h最为明显。从ic50和mi分析,四种联合方案中实施例3效果最好,即5-fu与bs浓度比为1:2。此条件下,药物作用24、48、72h后ic50分别为24.27±0.36、10.32±0.22、6.84±0.50μm,均明显低于其他实施例相同药物作用时间的ic50。三个时间点的mi分别为81.45%、94.55%、97.58%,也均高于其他实施例相同药物作用时间的mi,三个时间点以72h最为明显。
表1.单药和联合用药处理hepg2细胞后的ic50值和mi值
5.2bs与5-fu联合应用抑制hepg2细胞增殖的中位效应分析
为了正确合理地研究bs和5-fu的联合作用,采用chou-talalay的中位效应分析法[回2]来分析两者的相互作用方式。理论上,联合效应指数ci<1代表两种化合物之间具有协同作用。拥有协同作用的联合目的主要是为了降低所用药物的剂量,由此降低毒性并维持药效,尤其是像5-fu一样的细胞毒药物。对于癌症治疗,在高效果水平(90%,95%,99%有效剂量)的协同作用比在低效果水平(30%,50%有效剂量)拥有更明显的治疗相关性[回2]。因此重点分析细胞抑制率为60~97%对应的ci和剂量减少指数dri的结果。实施例1-4四种联合给药方案对hepg2细胞24、48、72h的中位效应分析结果见表2。
表2.bs与5-fu联合用药对hepg2细胞的中位效应分析结果表
5.3ci联合指数分析
表2的结果表明,实施例3在三个时间点其ci值均小于0.9,表现出明显的协同效果,即5-fu与bs比例为1:2。如图1a所示,在实施例3中药物作用的各个时间点,随着细胞生长抑制率增加,ci值逐渐降低。当细胞生长抑制率达到40%后,联合给药作用48h开始出现协同作用(ci<1);并且该曲线均在联合给药作用24、72h下方,可见实施例3最明显的协同作用出现在联合给药24h后。
5.4dri剂量减少指数分析
联合用药方案广泛应用于许多恶性疾病,如aids和癌症。采用联合用药方案的主要目的在于提高疗效,降低药物的毒性,延缓或者最小化药物耐受的发生。我们进一步分析了5-fu与bs联合作用于hepg2细胞24、48、72h后的剂量减少指数dri值。该概念最早是由choujh和周廷潮先生于1988年提出的,它是用来衡量联合时每种药的剂量相对于这种药单独作用时剂量减少的倍数。dri值越大,给药量减少的越多。鉴于药物联用的目的是降低5-fu的剂量,减轻细胞毒性,我们在此主要考察了5-fu的dri值(达到相同细胞生长抑制率所需5-fu单药浓度与联合给药中5-fu浓度的比)。结果如表3所示,对于5-fu而言,任一联合方案,任一作用时长,任一细胞增殖抑制率,其dri值都远大于1,尤以48h最为突出。我们继续比较了在此作用时间下不同联合方案的dri值,由表1b的细胞增殖抑制率-dri曲线可见,随着5-fu在联合给药所占比例的下降,该组对应曲线右移,即达到相同细胞生长抑制率所需5-fu剂量随其在联合方案中所占比例的减少而减少,实施例3对5-fu的减剂量效应最强,随后实施例4的曲线左移,减剂量效应减弱。为了进一步说明联合后5-fu所需剂量减少的程度,将实施例3与单药组所需5-fu剂量对应细胞生长抑制率作图(图1c),实施例3曲线在细胞生长抑制率达到40%以后相对于5-fu单药曲线明显下移,即明显降低5-fu用量,并且随着药物浓度的升高,减量作用愈加显著;同时,细胞生长抑制率随药物浓度的升高进入平台期,达到mi值。联合给药和单药mi值分别为94.55%、54.33%,这也与表1结果相吻合。
表3.bs与5-fu联合用药对hepg2细胞的dri值分析结果表
综合细胞生长抑制实验和中位效应的分析可得出:5-fu与bs在给药摩尔量1:2,作用48h时,表现出最佳的抗肿瘤作用。即在此条件下既可以提高或维持对hepg2细胞的增值抑制作用,又可以最大限度的降低5-fu的用量,同时起到增效减毒的作用。故我们采用这一最佳的药物比例和作用时间来进行后续的细胞实验,以探讨5-fu和bs联合作用对hepg2细胞周期、凋亡的影响。
5.55-fu和bs联合应用导致hepg2细胞周期阻滞
采用流式细胞术检测了15μm5-fu、15μmbs及两者联合(15μm5-fu:15μmbs=1:2)作用于hepg2细胞48h后的细胞周期分布。图2为检测结果典型图。表4为不同给药组作用24h对hepg2细胞周期的影响结果。
由图2和表4可见,与对照相比,15μm5-fu表现出了强烈的s期阻滞作用,由对照组的33.2%增加至53.0%,g2/m期的细胞比例为0。由于s期是dna合成期,这与5-fu抗代谢,抑制dna、rna合成的周期特异性作用机理相符。10μmbs单独作用48h,出现明显的g2/m期阻滞,由对照组的7.69%增加至16.5%。两药联合作用48h,则表现为s期阻滞。这些结果表明,在联合作用中5-fu的抗代谢作用占了主导地位,bs对g2/m期的阻滞作用消失,最终表现为s期阻滞。s期为dna合成期,此周期的阻滞更能有效的抑制细胞增殖。
表4.不同给药组作用24h对hepg2细胞周期的影响结果
数据值为平均值±标准差,n≥3;
*p<0.01,表示与对照组相比具有统计学差异;
#p<0.01,表示与对照组、bs组相比具有统计学差异。
5.65-fu和bs联合应用对hepg2细胞trxr的影响
5.6.15-fu和bs联合应用对hepg2细胞trxr活性的影响
bs是一种靶向trxr的新型有机硒化合物,何劼等已经证实其在体外可以抑制大鼠和多种肿瘤细胞系的trxr活性。前面的研究我们已经发现bs可以剂量依赖性的抑制hepg2细胞内trxr活性,为了考察bs与5-fu联用有无增强此抑制作用,我们将15μm5-fu、15μmbs及两者联合(15μm5-fu:15μmbs=1:2)作用于hepg2细胞48h后,提取细胞总蛋白,dtnb还原法测定细胞内trxr活性,结果如表所示,无论bs单药还是联合给药都会使hepg2细胞内trxr活性显著降低(p<0.05),抑制率分别为20.33%和30.91%,可见两者联合抑制作用增强,但不显著。此外,5-fu也显示出了轻微的trxr活性抑制作用,但与对照无显著性差异(p>0.05)。
表5.bs与5-fu联合应用对hepg2细胞trxr活性的影响
数据值为平均值±标准差,n=6;
*p<0.05(单项方差分析测试),表示与对照组相比具有统计学差异。
5.6.25-fu和bs联合应用对hepg2细胞trxr含量的影响
将上述提取的细胞总蛋白变性,进行westernblot实验(免疫印迹试验,蛋白上样量为30μg)。结果显示(见图4,以β-actin肌动蛋白作为内参)无论bs单药还是联合给药,细胞中trxr含量均较对照组升高,显示出与活性相反的变化趋势。而其催化底物trx的含量降低,其中联合给药组最低,与trxr蛋白表达趋势相反。这与我们前面得到的bs在抑制hepg2细胞中trxr活性的同时,会使细胞代偿性产生更多trxr的结论一致。
对比例1bs、5-fu单药对hepg2细胞的增殖抑制作用
将处于对数生长期的hepg2细胞接种于96孔板中,贴壁后与不同浓度的bs或5-fu分别作用24、48、72h,利用srb法检测细胞增殖抑制率。数据用origin7.5软件处理,通过hill方程拟合剂量反应曲线。结果如图2所示,bs和5-fu均可抑制hepg2细胞的增殖,并呈现出了一定的剂量、时间依赖性。
选取两药对hepg2细胞作用24、48、72h的半数抑制浓度(ic50)和最大生长抑制率(mi)。如表1单药ic50列所示:随着作用时间的延长,两种药物的ic50值均减小。5-fu单独作用24、48、72h的ic50值分别为773.81±0.82、65.02±0.38、42.56±0.47μm,bs单独作用24、48、72h的ic50值分别为25.49±0.59、16.25±0.30、10.26±0.45μm。结果表明,在相同浓度和作用时间下,bs对hepg2细胞生长抑制作用远超过5-fu,尤以24h最为突出。在此作用时间下,bs组ic50值为25.49±0.59μm,显示出一定的药物活性;5-fu组ic50值大于500μm,基本没有药物活性。
同时,两种药物对hepg2作用24、48、72h的mi见表1单药mi列。结果显示:随着作用时间的延长,两种药物的mi值均增大。值得注意的是在相同作用时间下,bs组的mi值均远高于5-fu组。当药物作用72h时,bs组的mi值接近100%,而5-fu组仅为58.73%。
以上结果表明,5-fu早期对hepg2肝癌细胞无明显的生长抑制作用,并且只对部分细胞显示出其药物活性,这与5-fu周期特异性的药物作用特点有关,而且在治疗过程中肿瘤细胞易对其产生耐药性,这也是5-fu在临床应用中存在的问题。而bs在早期则表现出一定的抗肿瘤活性和比较彻底的细胞杀伤力,可以弥补5-fu的不足,因此联合两种药物具有一定的可行性。
以上,对本发明的实施方式进行了说明。但是,本发明不限定于上述实施方式。凡在本发明的精神和原则之内,所做的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!