2‑取代‑4‑甲基‑5‑(4,5‑二氢噻唑‑2‑基)‑1,3‑噻唑类化合物及其制备方法和生物活性评价与流程
本发明涉及有机合成技术领域,具体涉及2-取代-4-甲基-5-(4,5-二氢噻唑-2-基)-1,3-噻唑类化合物及其制备方法和生物活性。
背景技术:
海洋生物一直是新颖结构天然产物和活性天然产物的重要来源,尤其是海洋微生物已成为重要的海洋药物资源,越来越受到重视。在已发现的海洋天然产物中,噻唑(啉)环是许多有重要生理活性化合物的基本结构单位,药理研究结果表明这些天然产物具有广泛的抗肿瘤、抗病毒(shen,k.,peng,j.m.,li,w.,yan,x.w.,hu,a.x.,chin.j.struct.chem.2014,6,855)抗菌活性(mislin,g.l.,burger,a.,abdallah,m.a.,tetrohedron.2004,60,12139),甚至还表现出结合蛋白质、dna、rna(akaji,k.,kiso,y.,totalsynthesisofthiangazole.tetrahedron1999,55,10685)等性质,这些独特的生理活性与噻唑(啉)环结构有关(yingy.c.,taori,k.,kim,h.,j.am.chem.soc.,2008,130.8455.)。
除了从自然界发现外,人们致力于天然产物及其类似物的全合成和独特结构片段衍生物的合成研究(karegoudar,p.,karthikeyan,m.s.,prasad,d.j.,mahalinga,m.,holla,b.s.,kumarin.s.,eur.j.med.chem.2008,43,261)。因此对含噻唑或噻唑啉独特片段的化合物的合成具有一定的意义。噻唑或噻唑啉的传统合成方法是经α-卤代铜为原料与硫化试剂反应,其中经典的方法是hantzsch法((a)hantzsch,a.,weber,j.h.,ber,dtsch.,chem.ges.1887,20,3118(b)aoyama,t.,murata,s.,arai,i.,araki,n.,takido,t.,suzuki,y.,kodomari,m.,tetrahedron,2006,14,3201(c)han,f.s.,tokuyama,h.,tohru,f.,chemcommun,2007,3444(d)busacca,c.a.,dong,y.,spinelli,e.m.,tetrahedron.lett,1996,17,2935(d)lambert,s.g.,taylor,jo-a.m.,wegener,k.l.,woodhouse,s.l.,lincoln,s.f.,warda.d.,newj.chem.2000,24,541(e)liu,l.,lam,y.w.,wong,w.y.,j.organomet.chem.2006,691,1092(f)nasveschuk,c.g.,ungermannova,d.,liu,x.d.,phillips,a.j.org.lett.2008,16,3595)。文献(tangl.j.,qianb.h.,mag.z.,liuw.w.,chinesejstrucchem,2015,4,645-647.)合成路线采用酯经脱羧、酰氯化后再酰胺化的常规方法,本发明改进了合成路线直接由酯和胺酰胺化合成目标化合物,简化了反应路线,获得环保效应,适合工业化生产。
综上,本发明以天然海洋代谢产物largozole为模板,进行了含噻唑-噻唑啉骈合骨架的杂环化合物的设计与合成,并对化合物进行了生物活性测试,期望得到结构新颖且具有一定生物活性的化合物。
技术实现要素:
本发明的目的是提供2-取代-4-甲基-5-(4,5-二氢噻唑-2-基)-1,3-噻唑类化合物和其制备方法及对脲酶的抑制活性和对大肠杆菌(escherichiacoli)、枯草芽孢杆菌(bacillussubtilis)、金黄色葡萄球菌(staphylococcusaureus)的抑制作用。
本发明采用的技术方案是:
4-甲基-2-取代-5-(2-噻唑啉基)噻唑类化合物的合成,其特征是有如下通式:
式中
r=3a:c6h5;3b:3-ch3c6h4;3c:4-fc6h4;3d:4-clc6h4;3e:4-brc6h4;
3f:4-ch3oc6h4;3g:3-ch3oc6h4;3h:furanyl-2-yl;3i:thiophen-2-yl
一类2-取代4-甲基-5-(2-噻唑啉基)-1,3噻唑-类化合物的制备方法,步骤包括:
a、将腈类化合物与17%硫化胺水溶液在有机溶剂中反应,向反应液中加入冰水,析晶得到硫代酰胺类化合物
b、上述制得的硫代酰胺类化合物与2-氯乙酰乙酸乙酯在乙醇溶剂中回流,tlc跟踪,减压除去溶剂,冰乙酸和水(v/v,1∶3)重结晶得到2-取代-4-甲基噻唑-5-甲酸乙酯。
c、2-取代-4-甲基噻唑-5-甲酸乙酯与乙醇胺反应制备n-(2-羟乙基)-4-甲基-2-取代噻唑-5-甲酰胺。
d、n-(2-羟乙基)-4-甲基-2-取代噻唑-5-甲酰胺与五硫化二磷在甲苯溶剂中成环反应,不需柱层析,二氯甲烷和丙酮重结晶得到目标化合物。
所属步骤b中后处理方法为冰乙酸和水重结晶,采取简单方法重结晶进行产品的纯化,操作简单,降低了实验成本,适合工业化。
所属步骤c中2-取代-4-甲基噻唑-5-甲酸乙酯与过量乙醇胺直接酰胺化反应制备n-(2-羟乙基)-4-甲基-2-取代噻唑-5-甲酰胺,避开酰氯化反应,此法环保、操作简单、降低生产成本。
所属步骤d中反应的纯化步骤由二氯甲烷和丙酮(v/v,4∶1)重结晶取代柱层析
本发明有益效果:
1、本发明提供了由腈类化合物经硫化、环合、酰胺化后再环合反应合成噻唑-噻唑啉化合物的合成方法,避开酰氯化反应,且后处理全部以以重结晶方法纯化产物,制备反应操作简便,产率较高,对环境友好,适合工业化生产。
2、本发明对目标化合物进行了抑制脲酶活性,抑制大肠杆菌(escherichiacoli)、枯草芽孢杆菌(bacillussubtilis)和金黄色葡萄球菌(staphylococcusaureus)活性的生物活性评价,具有一定的生物活性,因而具有较好的医药、农药等行业应用前景。
附图说明
以下是目标化合物谱图附图说明
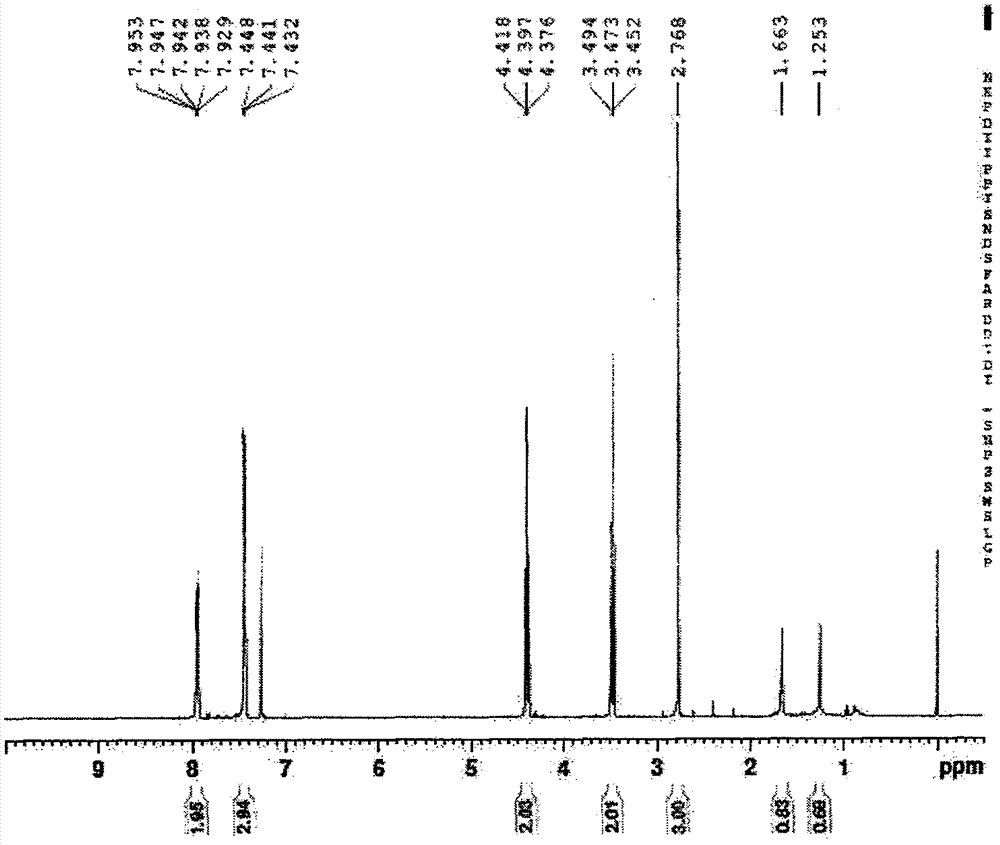
图1是4-甲基-2-苯基-5-(4,5-二氢噻唑-2-基)-1,3-噻唑氢核磁谱图
图2是4-甲基-2-苯基-5-(4,5-二氢噻唑-2-基)-1,3-噻唑碳核磁谱图
图3是4-甲基-2-(3-甲基苯基)-5-(4,5-二氢噻唑-2-基)-1,3-噻唑氢核磁谱图
图4是4-甲基-2-(3-甲基苯基)-5-(4,5-二氢噻唑-2-基)-1,3-噻唑碳核磁谱图
图5是4-甲基-2-(4-氟苯基)-5-(4,5-二氢噻唑-2-基)-1,3-噻唑备氢核磁谱图
图6是4-甲基-2-(4-氟苯基)-5-(4,5-二氢噻唑-2-基)-1,3-噻唑备碳核磁谱图
图7是目标产物4-甲基-2-(4-氯苯基)-5-(4,5-二氢噻唑-2-基)-1,3-噻唑氢核磁谱图
图8是目标产物4-甲基-2-(4-氯苯基)-5-(4,5-二氢噻唑-2-基)-1,3-噻唑碳核磁谱图
图9是4-甲基-2-(4-溴苯基)-5-(4,5-二氢噻唑-2-基)-1,3-噻唑氢核磁谱图
图10是4-甲基-2-(4-溴苯基)-5-(4,5-二氢噻唑-2-基)-1,3-噻唑碳核磁谱图
图11是4-甲基-2-(4-甲氧基苯基)-5-(4,5-二氢噻唑-2-基)-1,3-噻唑氢核磁谱图
图12是4-甲基-2-(4-甲氧基苯基)-5-(4,5-二氢噻唑-2-基)-1,3-噻唑碳核磁谱图
图13是4-甲基-2-(呋喃-2-基)-5-(4,5-二氢噻唑-2-基)-1,3-噻唑氢核磁谱图
图14是4-甲基-2-(呋喃-2-基)-5-(4,5-二氢噻唑-2-基)-1,3-噻唑碳氢核磁谱图
具体实施方式
实施例一:芳基(杂环基)硫代酰胺类化合物的制备
室温搅拌条件下,向干燥圆底烧瓶中加入15mmol芳腈、20ml无水甲醇,滴加17%(27.5mmol)的硫化铵溶液,充分混合反应液,于55℃反应,tlc监测反应进程。反应结束后,向反应液慢慢加入100ml冰水,析出黄色固体,抽滤,用冰水洗涤,干燥。粗产品可用乙醇和水进行重结晶。
实施例二:4-甲基-2-芳基(杂环基)-1,3-噻唑-5-甲酸乙酯的制备
8mmol取代硫代苯甲酰胺、10mmol2-氯乙酰乙酸乙酯、30ml无水乙醇,搅拌回流反应,tlc监测反应进程。冰水浴析晶或减压浓缩,残留物用冰乙酸重结晶得目标产物。将1g粗品溶于20ml冰乙酸,待完全溶解后,慢慢加入50ml冰水,白色晶体析出,抽滤,干燥。
实施例三:n-(2-羟乙基)-4-甲基-2-芳基(杂环基)-1,3-噻唑-5-甲酰胺的制备
4-甲基-2-芳基(杂环基)-1,3-噻唑-5-甲酸乙酯2mmol,乙醇胺15ml于一定温度下反应,tlc监测反应进程,反应结束后加30ml(15ml×2)乙酸乙酯,20ml水萃取两次,有机层用无水硫酸镁干燥减压浓缩既得产品。
[0019]方法中用各种温度尝试,所试验的温度包括25℃、45℃、65℃、85℃、105℃,tlc监测进程如表1。
表1以n-(2-羟乙基)-4-甲基-2-(3-甲苯基)-1,3-噻唑-5-甲酰胺为例在不同温度下的制备时间和收率
实施例四:目标产物2-取代-4-甲基-5-(2-噻唑啉基)-1,3-噻唑的制备
用15ml甲苯溶解[0019]中的酰胺产物,加入三乙胺9.6mmol、p2s54mmol,回流反应,tlc跟踪,待反应结束,冷却到室温,倾倒出甲苯层,水洗涤2-3次,有机相用无水硫酸钠干燥后浓缩,粗产物用二氯甲烷和丙酮重结晶。
实施例五:4-甲基-2-(呋喃-2-基)-1,3-噻唑-5-甲酸乙酯的制备
结构式
8mmol2-呋喃硫代苯甲酰胺、10mmol2-氯乙酰乙酸乙酯、30ml无水乙醇,搅拌回流反应,tlc监测反应进程,反应体系置于冰水浴析晶,抽滤、干燥。将1g粗品溶于20ml冰乙酸,待完全溶解后,慢慢加入60ml冰水,白色晶体析出,抽滤,得白色针状晶体,收率95.8%,m.p.66~67℃。ir(kbr,cm-1)v:3072.63,2025.91,1709.6,1618.32,1490.04,1318.07.1hnmr(cdcl3,300mhz)δ:7.58~6.58(m,3h,呋喃氢),4.38(q,2h,j=5.0hz,酯亚甲基-cooch2),2.79(s,3h,thiazole-ch3),1.41(t,3h,j=5.0hz,methyl,-o-ch2-ch3)
实施例六:4-甲基-2-(噻吩-2-基)-1,3-噻唑-5-甲酸乙酯的制备
结构式
制备方法同实施例五,以2-噻吩硫代苯甲酰胺代替2-呋喃硫代苯甲酰胺,得化合物4-甲基-2-(噻吩-2-基)-1,3-噻唑-5-甲酸乙酯。白色固体,m.p.57~59℃。ir(kbr,cm-1)v:3072.63,2025.98,1709.85,1618.32,1541.13,1277.51.1hnmr(cdcl3,300mhz)δ:7.60~7.11(m,3h,噻吩氢),4.37(q,2h,j=5.0hz,酯亚甲基-cooch2),2.77(s,3h,thiazole-ch3),1.40(t,3h,j=5.0hz,methyl,-o-ch2-ch3).
实施例七:4-甲基-2-(4-甲氧苯基)-1,3-噻唑-5-甲酸乙酯的制备
结构式
制备方法同实施例五,以4-甲氧基硫代苯甲酰胺代替2-呋喃硫代苯甲酰胺,白色针状晶体,收率82.6%,m.p.45~47℃。ir(kbr,cm-1)v:2922.59,1716.00,1618.12,1490.04,1101.26.1hnmr(cdcl3,300mhz)δ:7.85~7.31(m,4h,苯环氢),4.40(q,2h,j=5.0hz,酯亚甲基-cooch2),2.83(s,3h,ch3o-phenyl),2.46(s,3h,thiazole-ch3),1.43(t,3h,j=5.0hz,methyl,-o-ch2-ch3).
实施例八:4-甲基-2-(4-溴苯基)-1,3-噻唑-5-甲酸乙酯的制备
结构式
制备方法同实施例五,以4-溴苯硫代苯甲酰胺代替2-呋喃硫代苯甲酰胺,白色固体,收率80.5%,m.p.74~75℃.ir(kbr,cm-1)v:2922.59,1716.00,1618.12,1396.74,1101.26.1hnmr(cdcl3,300mhz)δ:8.15~7.39(m,4h,苯环氢),4.48(q,2h,j=5.0hz,酯亚甲基-cooch2),2.89(s,3h,thiazole-ch3),1.51(t,3h,j=5.0hz,methyl,-o-ch2-ch3).
实施例九n-(2-羟乙基)-4-甲基-2-(呋喃-2-基)-1,3-噻唑-5-甲酰胺的制备
结构式
4-甲基-2-(呋喃-2-基)-1,3-噻唑-5-甲酸乙酯2mmol,乙醇胺15ml于85℃反应50min,加水20ml稀释,用乙酸乙酯(15ml×2)萃取两次,有机相反复用水洗涤后加无水硫酸镁干燥,减压浓缩得产品。淡黄色固体,收率72.8%,m.p.106~108℃.ir(kbr,cm-1)v:3416.58,3131.75,2940.59,1647.2,1541.29,1492.90,1369.75,1303.75,1283.85,1223.25。1hnmr(cd3od,300mhz)δ:7.79~6.72(m,3h,呋喃氢),2.73(s,3h,thiazole-ch3),3.80~3.39(m,2个亚甲基4h,-nh--ch2-ch2-oh)
实施例十n-(2-羟乙基)-4-甲基-2-(噻吩-2-基)-1,3-噻唑-5-甲酰胺的制备
结构式
制备方法同实施例九,以4-甲基-2-(噻吩-2-基)-1,3-噻唑-5-甲酸乙酯代替4-甲基-2-(呋喃-2-基)-1,3-噻唑-5-甲酸乙酯。产品为淡黄色固体,收率89.4%,m.p.67-68℃.ir(kbr,cm-1)v:3423.69,3131.75,2940.59,1647.2,1541.29,1492.90,1369.75,1303.75,1283.85,1223.25。1hnmr(cdcl3,300mhz)δ:7.54~7.09(m,3h,噻吩氢),3.62~3.42(dd,2个亚甲基4h,-nh-ch2-ch2-oh),2.72(s,3h,thiazole-ch3),2.03(s,1h,-oh)
实施例十一n-(2-羟乙基)-4-甲基-2-(3-甲苯基)-1,3-噻唑-5-甲酰胺的制备
结构式
制备方法同实施例九,以4-甲基-2-(4-甲苯基)-1,3-噻唑-5-甲酸乙酯代替4-甲基-2-(呋喃-2-基)-1,3-噻唑-5-甲酸乙酯。产品为淡黄色固体,收率75%,m.p.141-142℃.ir(kbr,cm-1)v:3431.43,3300.70,2928.23,2880.92,1632.41,1555.36,1413.66,1312.38。1hnmr(cdcl3,300mhz)δ:8.26(s,1h,-nh-),7.8~7.10(m,4h,苯环氢),3.66~3.46(dd,2个亚甲基4h,-nh-ch2-ch2-oh),2.71(s,3h,thiazole-ch3),2.40(s,3h,ch3-phenyl)1.75(s,1h,-oh)
实施例十二目标产物4-甲基-2-苯基-5-(4,5-二氢噻唑-2-基)-1,3-噻唑(3a)的制备(3a)
结构式
n-(2-羟乙基)-4-甲基-2-苯基-1,3-噻唑-5-甲酰胺2mmol,溶于15ml甲苯中,加入三乙胺5ml和p2s55mmol,回流反应,tlc监测反应,反应结束冷却至室温,将上层甲苯层倾出,水洗2次,有机相用无水硫酸钠干燥后减压浓缩,用二氯甲烷和丙酮(v/v,5∶1)重结晶,收率62.8%。白色晶体,m.p.62-63℃。ir(kbr,cm-1)v:2842.81,1620.29,1576.69,1430.45,1332.14。。1hnmr(cdcl3,300mhz)δ:7.95~7.43(m,5h,phh),4.39(t,2h,j=6.0hz,thiazolineringn-ch2),3.47(t,2h,j=6.0hz,thiazolinerings-ch2),2.76(s,3h,thiazole-ch3)。13cnmr(cdcl3,300mhz)δ:168.19(s,thiazolec,s-c=n),159.66(s,thiazolinec,s-c=n),154.80(s,thiazolec,n-c),133.10~126.45(m,phenylc+thiazole5),64.17(s,thiazoline4,n-c),35.06(s,thiazoline5),21.49(s,c6h5-ch3)。(es+)m/z:338(m+h+,100)
实施例十三目标产物4-甲基-2-(3-甲基苯基)-5-(4,5-二氢噻唑-2-基)-1,3-噻唑(3b)的制备
结构式
制备方法同实施例十二,收率48.8%,白色晶体,m.p.68-69℃。ir(kbr,cm-1)v:2848.28,1600.14,1578.61,1458.74,1332.64。1hnmr(cdcl3,500mhz)δ:7.96~6.96(m,4h,苯环h),4.41(t,2h,j=5.0hz,噻唑啉n-ch2),3.48(t,2h,j=5.0hz,噻唑啉s-ch2),2.78(s,3h,噻唑ch3),2.43(s,3h,ch3ph).13cnmr(cdcl3,300mhz)δ:168.44(s,噻唑,s-c=n),159.63(s,噻唑啉,s-c=n),154.71(s,噻唑,n-c),138.83~114.35(m,苯环+噻唑c=c-s),64.21(s,噻唑啉n-c),35.08(s,噻唑啉c-c-s),21.35(s,ch3ph),17.82(s,噻唑ch3).(es+)m/z:291(m+h+,100).(es+)m/z:275(m+h+,100)
实施例十四目标产物4-甲基-2-(4-氟苯基)-5-(4,5-二氢噻唑-2-基)-1,3-噻唑(3c)的制备
结构式
制备方法同实施例十二,收率53.5%。白色晶体,m.p.130-131℃。ir(kbr,cm-1)v:2800,1618.84,1596.42,1405.63,1213.7。1hnmr(cdcl3,300mhz)δ:7.94~7.10(m,4h,苯环h),4.39(t,2h,j=6.0hz,噻唑啉n-ch2),3.46(t,2h,j=6.0hz,噻唑啉s-ch2),2.75(s,3h,噻唑ch3).13cnmr(cdcl3,300mhz)δ:166.92(s,噻唑,s-c=n),165.48(s,噻唑啉,s-c=n),162.98(s,苯环c-f),159.57(s,噻唑,n-c),129.70~116.03(m,苯环+噻唑c=c-s),64.16(s,噻唑啉n-c),35.10(s,噻唑啉c-c-s),17.75(s,噻唑ch3).19fnmr(cdcl3,300mhz):-109.33.(es+)m/z:279(m+h+,100)
实施例十五目标产物4-甲基-2-(4-氯苯基)-5-(4,5-二氢噻唑-2-基)-1,3-噻唑(3d)的制备
结构式
制备方法同实施例十二,收率51.2%,白色晶体,m.p.139-140℃。ir(kbr,cm-1)v:2850.24,1617.94,1593.30,1398.36,1230.95。1hnmr(cdcl3,300mhz)δ:7.88~7.40(m,4h,苯环h),4.39(t,2h,j=6.0hz,噻唑啉n-ch2),3.48(t,2h,j=6.0hz,噻唑啉s-ch2),2.76(s,3h,噻唑ch3).13cnmr(cdcl3,300mhz)δ:166.72(s,噻唑,s-c=n),159.53(s,噻唑啉,s-c=n),154.89(s,噻唑,n-c),136.64~126.78(m,苯环+噻唑c=c-s),64.18(s,噻唑啉n-c),35.12(s,噻唑啉c-c-s),17.77(s,噻唑ch3).(es+)m/z:295(m+h+,100)
实施例十六目标产物4-甲基-2-(4-溴苯基)-5-(4,5-二氢噻唑-2-基)-1,3-噻唑(3e)的制备
结构式
制备方法同实施例十二,收率40.5%,淡黄色晶体,m.p.136-137℃。ir(kbr,cm-1)v:2849.36,1617.94,1586.36,1497.79,1231.67。1hnmr(cdcl3,500mhz)δ:7.84~7.31(m,4h,苯环h),4.42(t,2h,j=5.0hz,噻唑啉n-ch2),3.51(t,2h,j=5.0hz,噻唑啉s-ch2),2.80(s,3h,噻唑ch3).13cnmr(cdcl3,300mhz)δ:166.80(s,噻唑,s-c=n),159.00(s,噻唑啉,s-c=n),154.94(s,噻唑,n-c),133.23~125.01(m,苯环+噻唑c=c-s),64.18(s,噻唑啉n-c),35.13(s,噻唑啉c-c-s),17.77(s,噻唑ch3).(es+)m/z:338(m+h+,100)
实施例十七目标产物4-甲基-2-(4-甲氧基苯基)-5-(4,5-二氢噻唑-2-基)-1,3-噻唑(3f)的制备
结构式
制备方法同实施例十二,收率42.6%,白色晶体,m.p.133-134℃。ir(kbr,cm-1)v:2849.24,1617.94,1586.36,1497.79,1231.67。1hnmr(cdcl3,500mhz)δ:8.06~7.72(m,4h,苯环h),4.34(t,2h,j=5.0hz,噻唑啉n-ch2),3.66(s,3h,ch3oph),3.46(t,2h,j=5.0hz,噻唑啉s-ch2),2.75(s,3h,噻唑ch3).13cnmr(cdcl3,300mhz)δ:168.11(s,噻唑,s-c=n),159.63(s,噻唑啉,s-c=n),154.65(s,噻唑,n-c),128.23~114.35(m,苯环+噻唑c=c-s),64.12(s,噻唑啉n-c),55.44(s,ch3oph),35.05(s,噻唑啉c-c-s),17.81(s,噻唑ch3).(es+)m/z:291(m+h+,100).
实施例十八目标产物4-甲基-2-(3-甲氧基苯基)-5-(4,5-二氢噻唑-2-基)-1,3-噻唑(3g)的制备
结构式
制备方法同实施例十二,收率64.6%,白色晶体,m.p.80-81℃。ir(kbr,cm-1)v:2850.62,1677.32,1581.36,1437.81,1255.17。1hnmr(cdcl3,500mhz)δ:7.89~6.84(m,4h,苯环h),4.41(t,2h,j=5.0hz,噻唑啉n-ch2),3.90(s,3h,ch3oph)3.49(t,2h,j=5.0hz,噻唑啉s-ch2),2.78(s,3h,噻唑ch3).13cnmr(cdcl3,300mhz)δ:168.01(s,噻唑,s-c=n),159.62(s,噻唑啉,s-c=n),154.72(s,噻唑,n-c),134.33~111.15(m,苯环+噻唑c=c-s),64.17(s,噻唑啉n-c),55.45(s,ch3oph),35.09(s,噻唑啉c-c-s),17.82(s,噻唑ch3).(es+)m/z:291(m+h+,100).
实施例十九目标产物4-甲基-2-(呋喃-2-基)-5-(4,5-二氢噻唑-2-基)-1,3-噻唑(3h)的制备
结构式
制备方法同实施例十二,收率48.5%,褐色晶体,m.p.99-100℃。ir(kbr,cm-1)v:3100.31,2848.28,1618.15,1582.27,1496.02,1258.62。1hnmr(cdcl3,500mhz)δ:7.75~6.37(m,3h,呋喃环h),4.42(t,2h,j=5.0hz,噻唑啉n-ch2),3.48(t,2h,j=5.0hz,噻唑啉s-ch2),2.72(s,3h,噻唑ch3).13cnmr(cdcl3,300mhz)δ:159.40(s,噻唑啉,s-c=n),157.90(s,噻唑,n-c),154.67(s,噻唑,s-c=n),148.48(s,呋喃2位碳),144.30(s,呋喃5位碳),125.72(s,噻唑c=c-s),112.41(s,呋喃3位碳),110.36(s,呋喃4位碳),64.15(s,噻唑啉n-c),35.10(s,噻唑啉c-c-s),17.72(s,噻唑ch3).(es+)m/z:291(m+h+,100).(es+)m/z:251(m+h+,100)
实施例二十目标产物4-甲基-2-(噻吩-2-基)-5-(4,5-二氢噻唑-2-基)-1,3-噻唑(3i)的制备
结构式
制备方法同实施例十二,收率48.5%,黄褐色晶体,m.p.112-113℃。ir(kbr,cm-1)v:3100.31,2848.28,1620.30,1587.59,1461.93,131.16。1hnmr(cdcl3,500mhz)δ:7.72~6.93(m,3h,呋喃环h),4.40(t,2h,j=5.0hz,噻唑啉n-ch2),3.49(t,2h,j=5.0hz,噻唑啉s-ch2),2.75(s,3h,噻唑ch3).13cnmr(cdcl3,300mhz)δ:161.88(s,噻唑啉,s-c=n),159.44(s,噻唑,s-c=n),154.50(s,噻唑,n-c),136.81(s,噻吩2位碳),128.68~125.73(m,呋喃3,4,5位碳+噻唑c=c-s),64.04(s,噻唑啉n-c),35.12(s,噻唑啉c-c-s),17.68(s,噻唑ch3).(es+)m/z:267(m+h+,100).
目标产物对脲酶活性的抑制作用的测定
采用sunrise全自动酶标仪进行目标产物对脲酶活性(1500,20ku)的初步筛选和ic50值的测定.
酶液
将称取好的24.000mg尿素酶放入经高锰酸钾溶液洗过的棕色瓶中,用移液枪加入6mlph6.8的磷酸盐缓冲液,均匀摇荡使尿素酶充分溶解以备用。
底物溶液
称取尿素3.000g、苯酚红2.000mg于容量瓶中,用ph为6.8的缓冲液将其定容至100ml,配成含0.002%苯酚红的底物液及500mm的尿素以备用。
样品溶液
用分析天平称取0.025g样品,将其加入到25ml的容量瓶中,用dmso定容。
实验设计
每一个样品做三组平行实验,dmso做空白。样品都加入后,进行孵化半小时。
尿素酶抑制剂的初选
孵化半小时后,将孔酶标板放入全自动酶标仪中,开始用计算机检测。其中设置测试循环次数为八次以减小误差。结果见表2:
表2目标产物对尿素酶的抑制结果
对抑制率在50%以上的求其ic50。
将4-甲基-2-(3-甲基苯基)-5-(4,5-二氢噻唑-2-基)-1,3-噻唑(3b)继续稀释至浓度为1000ug/ml、800ug/ml、600ug/ml、400ug/ml、200ug/ml,测定ic50。
根据样品浓度和抑制率的曲线,求其ic50为729.93ug/ml。
抑菌活性的测定
菌种选用大肠杆菌(escherichiacoli),枯草芽孢杆菌(bacillussubtilis)和金黄色葡萄球菌(staphylococcusaureus)
平板培养基用牛肉膏蛋白胨培养基,液体培养基牛肉膏蛋白胨不加琼脂培养基。
样品溶液配制[0074]
菌种分别经平板活化、液体培养基活化后涂布法接种。
抑菌活性测试采用牛津杯,样品量为200μl。
浓度为1mg/ml的链霉素为阳性对照,溶剂dmso为空白对照。
每个平板重复三次,放入细菌培养箱中于35℃培养20小时。以抑菌圈直接考察活性大小,结果见表3。
表3目标产物对的体外抑菌活性
- 还没有人留言评论。精彩留言会获得点赞!