一种miRNA功能模块的识别方法及系统与流程
本发明涉及信息生物学领域,特别是涉及一种mirna功能模块的识别方法及系统。
背景技术:
mirnas(micrornas)是一类进化上保守的非编码小分子rna,具有在翻译水平调控基因表达的功能,因此在与疾病相关的一些生物通路中起着十分重要的作用。尽管mirna在特定生物过程中的功能角色已经被发现和验证,然而,大多数mirna的调控机制与功能还不为人知。随着生物数据的日益增长,越来越多的mirna已经被人类发现其调控功能,揭示了mirna与疾病的潜在关系。由于mirna通过其调控的靶基因履行其功能,近年来出现了很多基于靶基因探索mirna功能的计算方法。
研究者们提出,有相似功能的mirna通常与相似的疾病具有一定的联系,反之亦然。因此,mirna被广泛地用于疾病-mirna的预测。尽管已有各种方法用于预测mirna与疾病之间的关联,然而,如何有效地发现mirna与疾病之间的关联仍然是生物信息学中的一大挑战。已有研究表明,功能模块是指包含在相同生物过程中的蛋白质和调控子等生物分子,它们具有相似或相近的特征,相互作用完成一组特定的功能。因此,有必要设计一种mirna功能模块的识别方法及系统来发现疾病中潜在的mirna,为医疗诊断提供有价值的线索。
技术实现要素:
本发明的目的是提供一种mirna功能模块的识别方法及系统,以识别mirna功能模块,为医疗诊断及mirna的科学研究提供技术支持。
为实现上述目的,本发明提供了如下方案:
一种mirna功能模块的识别方法,包括:
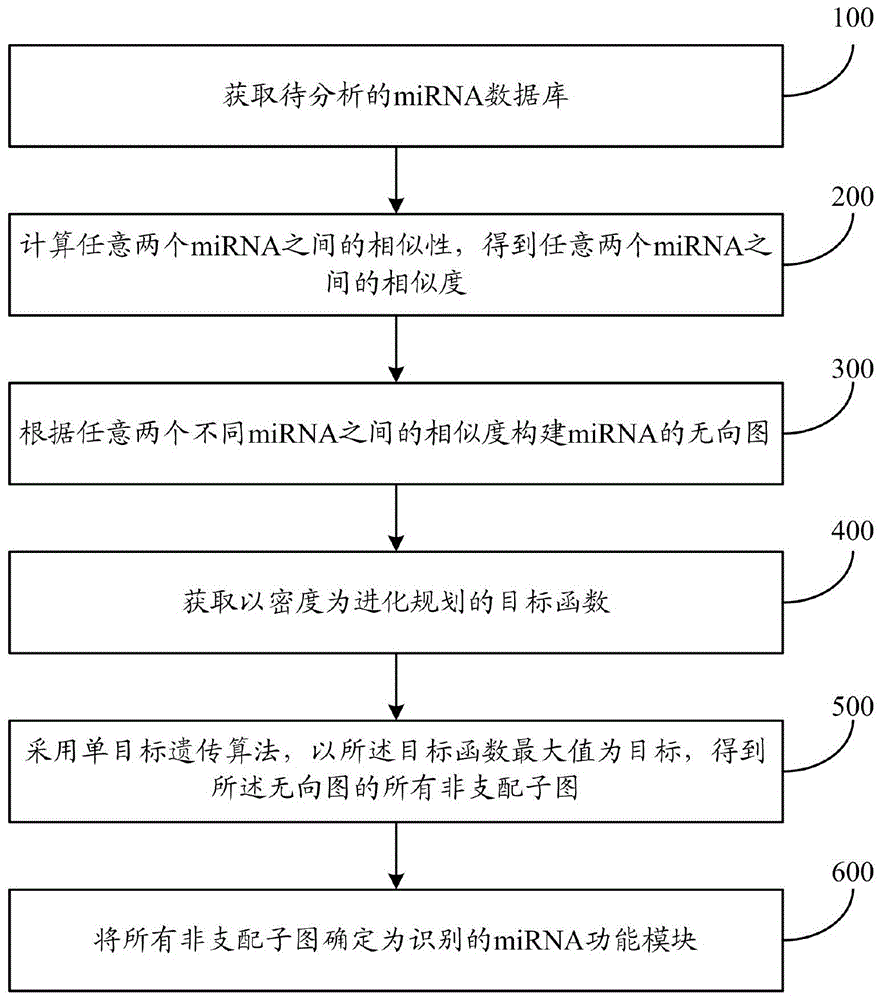
获取待分析的mirna数据库;
计算任意两个mirna之间的相似性,得到任意两个mirna之间的相似度;
根据任意两个不同mirna之间的相似度构建mirna的无向图;所述无向图中的顶点为待分析的mirna,相邻两个顶点之间的边的权重为两个顶点对应的两个不同mirna之间的相似度;
获取以密度为进化规划的目标函数;
采用单目标遗传算法,以所述目标函数最大值为目标,得到所述无向图的所有非支配子图;所述非支配子图为所述无向图的连通子图;
将所有非支配子图确定为识别的mirna功能模块。
可选的,所述计算任意两个mirna之间的相似性,得到任意两个mirna之间的相似度,具体包括:
利用函数
可选的,所述以密度为进化规划的目标函数为
可选的,所述采用单目标遗传算法,以所述目标函数最大值为目标,得到所述无向图的所有非支配子图,具体包括:
初始化子图种群,得到初始种群;所述初始种群中的个体为所述无向图的子图,且所述初始种群中的不同个体均为支配子图;
对于第i次迭代,将父代子图种群通过变异操作产生子代子图种群;当i=1时,所述父代子图种群为所述初始种群;
将子代子图种群和父代子图种群合并,去除重复的个体,生成临时种群;
获取所述临时种群中的非支配子图;
判断当前迭代是否满足迭代停止条件,得到第一判断结果;所述迭代停止条件为当前迭代次数到达最大迭代次数,或所述子代子图种群为空集;
当所述第一判断结果表示当前迭代满足迭代停止条件时,迭代停止;
当所述第一判断结果表示当前迭代不满足迭代停止条件时,根据所述目标函数计算所述临时种群中所有个体的目标函数值;
按照所有个体的目标函数值选择所述临时种群中的部分个体进行变异,得到新的子代子图种群;
将所述新的子代子图种群与所述临时种群合并,生成下一次迭代的父代子图种群,返回“将父代子图种群通过变异操作产生子代子图种群”步骤。
可选的,所述得到识别后的mirna功能模块,之后还包括:
将所述识别后的mirna功能模块与hmdd数据库比对,根据mirna功能模块内成员相似性高、功能模块间相似性弱的特征,得到mirna与疾病之间的关联。
本发明还提供一种mirna功能模块的识别系统,包括:
mirna数据库获取模块,用于获取待分析的mirna数据库;
相似性计算模块,用于计算任意两个mirna之间的相似性,得到任意两个mirna之间的相似度;
无向图构建模块,用于根据任意两个不同mirna之间的相似度构建mirna的无向图;所述无向图中的顶点为待分析的mirna,相邻两个顶点之间的边的权重为两个顶点对应的两个不同mirna之间的相似度;
目标函数获取模块,用于获取以密度为进化规划的目标函数;
非支配子图获取模块,用于采用单目标遗传算法,以所述目标函数最大值为目标,得到所述无向图的所有非支配子图;所述非支配子图为所述无向图的连通子图;
mirna功能模块确定模块,用于将所有非支配子图确定为识别的mirna功能模块。
可选的,所述相似性计算模块,利用函数
可选的,所述目标函数获取模块的目标函数为:以密度为进化规划的目标函数
可选的,所述非支配子图获取模块具体包括:
初始化单元,用于初始化子图种群,得到初始种群;所述初始种群中的个体为所述无向图的子图,且所述初始种群中的不同个体均为支配子图;
变异单元,用于对于第i次迭代,将父代子图种群通过变异操作产生子代子图种群;当i=1时,所述父代子图种群为所述初始种群;
合并单元,用于将子代子图种群和父代子图种群合并,去除重复的个体,生成临时种群;
非支配子图获取单元,用于获取所述临时种群中的非支配子图;
判断单元,用于判断当前迭代是否满足迭代停止条件,得到第一判断结果;所述迭代停止条件为当前迭代次数到达最大迭代次数,或所述子代子图种群为空集;
迭代停止单元,用于当所述第一判断结果表示当前迭代满足迭代停止条件时,迭代停止;
目标函数值计算单元,用于当所述第一判断结果表示当前迭代不满足迭代停止条件时,根据所述目标函数计算所述临时种群中所有个体的目标函数值;
选择单元,用于按照所有个体的目标函数值选择所述临时种群中的部分个体进行变异,得到新的子代子图种群;
父代子图种群生成单元,用于将所述新的子代子图种群与所述临时种群合并,生成下一次迭代的父代子图种群,返回“将父代子图种群通过变异操作产生子代子图种群”步骤。
可选的,所述系统还包括:
比对模块,用于在所述得到识别后的mirna功能模块之后,将所述识别后的mirna功能模块与hmdd数据库比对,根据mirna功能模块内成员相似性高、功能模块间相似性弱的特征,得到mirna与疾病之间的关联。
根据本发明提供的具体实施例,本发明公开了以下技术效果:
本发明从疾病-疾病的相似性来计算mirna-mirna的功能相似性构建mirna-mirna的相似性网络无向图,以密度为目标函数,通过初始化父代种群、变异、选择操作,识别mirna功能模块,然后借助于人类mirna相关的疾病数据库,利用功能模块的相似性,揭示潜在的mirna与疾病的关联,为医疗诊断提供有价值的线索。
附图说明
为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动性的前提下,还可以根据这些附图获得其他的附图。
图1为本发明mirna功能模块的识别方法的流程示意图;
图2为本发明mirna功能模块的识别系统的结构示意图;
图3是本发明单目标遗传算法的伪代码;
图4是本发明中变异操作的伪代码;
图5是本发明中选择操作的伪代码;
图6是本发明子图变异实施例1的示意图;
图7是本发明子图变异实施例2的示意图。
具体实施方式
下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
为使本发明的上述目的、特征和优点能够更加明显易懂,下面结合附图和具体实施方式对本发明作进一步详细的说明。
图1为本发明mirna功能模块的识别方法的流程示意图。如图1所示,所述识别方法包括以下步骤:
步骤100:获取待分析的mirna数据库。本发明借助人类mirna相关的疾病数据库,直接获得待分析的mirna数据库。
步骤200:计算任意两个mirna之间的相似性,得到任意两个mirna之间的相似度。利用函数
步骤300:根据任意两个不同mirna之间的相似度构建mirna的无向图。如果两个mirna之间的相似度大于0,则说明两个mirna之间是有边相联的,该分数被设置为mirna无向图中边的权重。输入一组mirna构成的网络信息,过滤其中的重复相互作用和自相互作用,根据两个不同mirna之间的相似度可以构建mirna网络的无向图g,所述无向图中的顶点为待分析的mirna,相邻两个顶点之间的边的权重为两个顶点对应的两个不同mirna之间的相似度。
步骤400:获取以密度为进化规划的目标函数。所述以密度为进化规划的目标函数为
步骤500:采用单目标遗传算法,以所述目标函数最大值为目标,得到所述无向图的所有非支配子图。所述非支配子图为所述无向图的连通子图。此步骤采用图论中的非支配子图数学表达手段对进化规划遗传算法的过程进行描述,得到非支配子图(功能模块)的结果。具体如下:
给定一无向图g,从中抽取表示连通子图的非支配子图,必须满足密度公式定义的目标,可用如下数学模型加以描述:
f(s)=(f(s))
f(s)=max.density
其中,s.t.s∈x。
f(s)∈y
上述公式中,x表示子图s的搜索空间,即mirna功能模块集合,y表示目标空间,即目标函数值的空间。上述公式中问题的解为x中最优解的集合,x描述了目标空间y中不同的权衡。本发明中,我们求解目标的最大值。为了便于比较种群中的每一对子图,利用支配概念对子图加以描述。一个解x被称为支配另一个解x’,当且仅当满足:
(i)在所有目标中,x都要好于x’;
(ii)至少在一个目标中,x严格好于x’
如果任一条件不满足,那么解x不支配解x’。为了从mirna相似性网络中挖掘所有可能的非支配子图,单目标遗传算法通过求解每个子图的目标函数值和进行排序,以便通过选择操作来选取下一代变异的子图。
具体过程如下:
初始化子图种群,得到初始种群;所述初始种群中的个体为所述无向图的子图,且所述初始种群中的不同个体均为支配子图;
对于第i次迭代,将父代子图种群通过变异操作产生子代子图种群;当i=1时,所述父代子图种群为所述初始种群;
将子代子图种群和父代子图种群合并,去除重复的个体,生成临时种群;
获取所述临时种群中的非支配子图;
判断当前迭代是否满足迭代停止条件,得到第一判断结果;所述迭代停止条件为当前迭代次数到达最大迭代次数,或所述子代子图种群为空集;
当所述第一判断结果表示当前迭代满足迭代停止条件时,迭代停止;
当所述第一判断结果表示当前迭代不满足迭代停止条件时,根据所述目标函数计算所述临时种群中所有个体的目标函数值;
按照所有个体的目标函数值选择所述临时种群中的部分个体进行变异,得到新的子代子图种群;
将所述新的子代子图种群与所述临时种群合并,生成下一次迭代的父代子图种群,返回“将父代子图种群通过变异操作产生子代子图种群”步骤。
步骤600:将迭代过程中产生的所有非支配子图确定为识别的mirna功能模块。
本发明中使用的已知人类mirna-疾病关联来自于hmdd(humanmicrornadiseasedatabase,hmdd)数据库(2014年6月发布),包括491个mirna、326种疾病,共5100对相互作用。
本发明将单目标遗传算法得到的mirna功能模块与hmdd数据库比对,利用功能模块内成员相似性较高,功能模块间相似性较弱的特征,推导出潜在的mirna与疾病之间的关联,为医疗诊断提供有价值的线索。例如,假设功能模块a包括如下mirna:hsa-mir-195、hsa-mir-26a、hsa-mir-30b以及hsa-let-7a。从hmdd数据库可知,疾病aorticvalveinsufficiency的产生与hsa-mir-195、hsa-mir-26a、hsa-mir-30b相关。利用功能模块的特性,我们可以推导出hsa-let-7a可能与aorticvalveinsufficiency的产生有关,为医疗诊断提供一定的参考依据。
图2为本发明mirna功能模块的识别系统的结构示意图。如图2所示,所述识别系统包括:
mirna数据库获取模块201,用于获取待分析的mirna数据库;
相似性计算模块202,用于计算任意两个mirna之间的相似性,得到任意两个mirna之间的相似度;
无向图构建模块203,用于根据任意两个不同mirna之间的相似度构建mirna的无向图;所述无向图中的顶点为待分析的mirna,相邻两个顶点之间的边的权重为两个顶点对应的两个不同mirna之间的相似度;
目标函数获取模块204,用于获取以密度为进化规划的目标函数;
非支配子图获取模块205,用于采用单目标遗传算法,以所述目标函数最大值为目标,得到所述无向图的所有非支配子图;所述非支配子图为所述无向图的连通子图;
mirna功能模块确定模块206,用于将所有非支配子图确定为识别的mirna功能模块。
所述相似性计算模块202,利用函数
所述目标函数获取模块204获取的目标函数为:以密度为进化规划的目标函数
所述非支配子图获取模块205具体包括:
初始化单元,用于初始化子图种群,得到初始种群;所述初始种群中的个体为所述无向图的子图,且所述初始种群中的不同个体均为支配子图;
变异单元,用于对于第i次迭代,将父代子图种群通过变异操作产生子代子图种群;当i=1时,所述父代子图种群为所述初始种群;
合并单元,用于将子代子图种群和父代子图种群合并,去除重复的个体,生成临时种群;
非支配子图获取单元,用于获取所述临时种群中的非支配子图;
判断单元,用于判断当前迭代是否满足迭代停止条件,得到第一判断结果;所述迭代停止条件为当前迭代次数到达最大迭代次数,或所述子代子图种群为空集;
迭代停止单元,用于当所述第一判断结果表示当前迭代满足迭代停止条件时,迭代停止;
目标函数值计算单元,用于当所述第一判断结果表示当前迭代不满足迭代停止条件时,根据所述目标函数计算所述临时种群中所有个体的目标函数值;
选择单元,用于按照所有个体的目标函数值选择所述临时种群中的部分个体进行变异,得到新的子代子图种群;
父代子图种群生成单元,用于将所述新的子代子图种群与所述临时种群合并,生成下一次迭代的父代子图种群,返回“将父代子图种群通过变异操作产生子代子图种群”步骤。
所述系统还包括:比对模块,用于在所述得到识别后的mirna功能模块之后,将所述识别后的mirna功能模块与hmdd数据库比对,根据mirna功能模块内成员相似性高、功能模块间相似性弱的特征,得到mirna与疾病之间的关联。
下面结合具体实施例来进一步说明本发明中单目标遗传算法的具体过程。
单目标遗传算法总体来说三个步骤:初始化;变异;选择,如图3所示,图3是本发明单目标遗传算法的伪代码。
第一步初始化父代种群
用mirna相似性网络无向图g初始化子图种群r,得到所有的连通子图,然后判断子图之间是否存在重叠的顶点。如果子图之间不存在重叠的顶点,将这样的非支配子图从种群r中剔除。
第二步变异操作
单目标遗传算法的第二步通过变异操作产生子代子图。为了产生子代子图s',对于种群r产生的每一个子图,如果两个父代子图中存在一个重叠的结点,那么通过一条边对图g中的父代子图进行扩展产生子代子图。
结合图6,图6是本发明子图变异实施例1的示意图,图中,第一层的父代子图“(p1,p2)”与子图“(p1,p3)”和“(p2,p3)”分别存在重叠顶点“p1”、“p2”,则通过边“(p1,p3)”、“(p2,p3)”产生了两个子代子图“(p2,p1,p3)”、“(p1,p2,p3)”。如果两个子图的重叠顶点个数大于1,将随机选择一条边进行变异产生子代子图。变异操作的代码如图4所示,图4是本发明中变异操作的伪代码。
结合图7,图7是本发明子图变异实施例2的示意图,图中,对于子图s1,s2,它们的重叠顶点有“p1”、“p2”,此时,对子图s1进行变异操作时可以在子图s2选择不同的边进行。因此,边“(p1,p4)”、“(p1,p5)”、“(p2,p4)”均可成为被选择的边,则位于下方对应的三个子图都为产生的子代子图。为了产生子代种群qgen,在r的所有父代个体中进行变异操作,然后在qgen中用目标函数对每一个子代子图进行评价。
第三选择操作
采用父代种群r和变异产生的子代种群qgen构建一个临时种群r∪qgen,创建一个新的父代种群。为了创建下一代种群,首先根据公式f(s)对临时种群的所有子图进行排序,然后去除重复的子图,从临时种群中以一定的概率ps选择排名靠前的子图产生下一代子图。在图6中,仅出现一个子图的原因在于其它两个是重复的子图,已被去除。在进化过程中,单目标遗传算法会产生一些非支配子图,它们与其它子图不存在重叠的顶点,如图6中的“(p4,p5)”。这些在每一代变异后产生的非支配子图,我们把它看作算法发现的mirna功能模块。为了存储这些非支配子图,声明一个外部文档变量archive,将每一代产生的非支配子图追加至该变量,并在每一代变异完成后进行更新。如图5所示,图5是本发明中选择操作的伪代码。当达到最大运行次数或不再产生非支配子图时,程序运行结束,此时archive中的支配子图集合即为产生的mirna功能模块。
以图6和图7示意的子图为例,假设种群r={(p1,p2),(p1,p3),(p2,p3),(p4,p5)},qgen=φ。
子图(p4,p5)与其它子图不存在重叠的顶点看作单目标遗传算法产生的非支配子图,不参与变异操作,直接存储到外部文档变量archive中作为产生的功能模块,archive={(p4,p5)}。此时,r={(p1,p2),(p1,p3),(p2,p3)},qgen=φ,临时种群为r∪qgen。其它三个子图有重叠的顶点,则参与变异操作。结果为ggen={(p2,p1,p3),(p1,p2,p3),(p1,p3,p2)},r∪qgen={(p1,p2),(p1,p3),(p2,p3),(p2,p1,p3),(p1,p2,p3),(p1,p3,p2)}。根据公式f(s)对r∪qgen的所有子图进行排序,然后去除重复的子图,从临时种群中以一定的概率ps选择排名靠前的子图产生下一代子图,去除重复的子图,假设得到r∪qgen={(p1,p2),(p1,p3),(p2,p3),(p2,p1,p3)}作为下一代父代种群r。以此类推,直到迭代次数达到最大运行次数或不再产生子图为止。
本发明通过调整参数pm(变异概率)和ps(选择概率),当达到迭代次数或所得子图数目不再发生改变时,输出的结果即为识别的mirna功能模块。
本说明书中各个实施例采用递进的方式描述,每个实施例重点说明的都是与其他实施例的不同之处,各个实施例之间相同相似部分互相参见即可。对于实施例公开的系统而言,由于其与实施例公开的方法相对应,所以描述的比较简单,相关之处参见方法部分说明即可。
本文中应用了具体个例对本发明的原理及实施方式进行了阐述,以上实施例的说明只是用于帮助理解本发明的方法及其核心思想;同时,对于本领域的一般技术人员,依据本发明的思想,在具体实施方式及应用范围上均会有改变之处。综上所述,本说明书内容不应理解为对本发明的限制。
- 还没有人留言评论。精彩留言会获得点赞!