铜和镍催化的脱羧硼化反应的制作方法
政府支持声明
本发明是在美国国立卫生研究院授予的gm-118176下的政府支持下完成的。政府拥有本发明的某些权利。
背景技术:
硼酸对化学科学的各个方面至关重要。尽管它们的普及和广泛应用可能可以归因于suzuki耦合器的令人难以置信的实用性,(1)但迄今为止,硼酸已经在交叉耦合以外的领域,例如材料科学,(2)化学传感器,(3)高分子科学,(2)和药物发现中找到了无数的应用。(4-5)在医学上,fda目前批准了两种烷基硼酸用于各种肿瘤适应症:ninlaro(1)和万珂(velcade)(49)。药物化学家指出,硼酸具有生物异构性,因为它们在某些情况下可以充当羧酸的替代物。(6)尽管硼酸很受欢迎,但硼酸几乎完全是通过合成获得的,这与它们试图取代的无处不在且廉价的羧酸不同。在这种情况下采用的逆合成分析本身就可以阻止将其纳入候选药物。
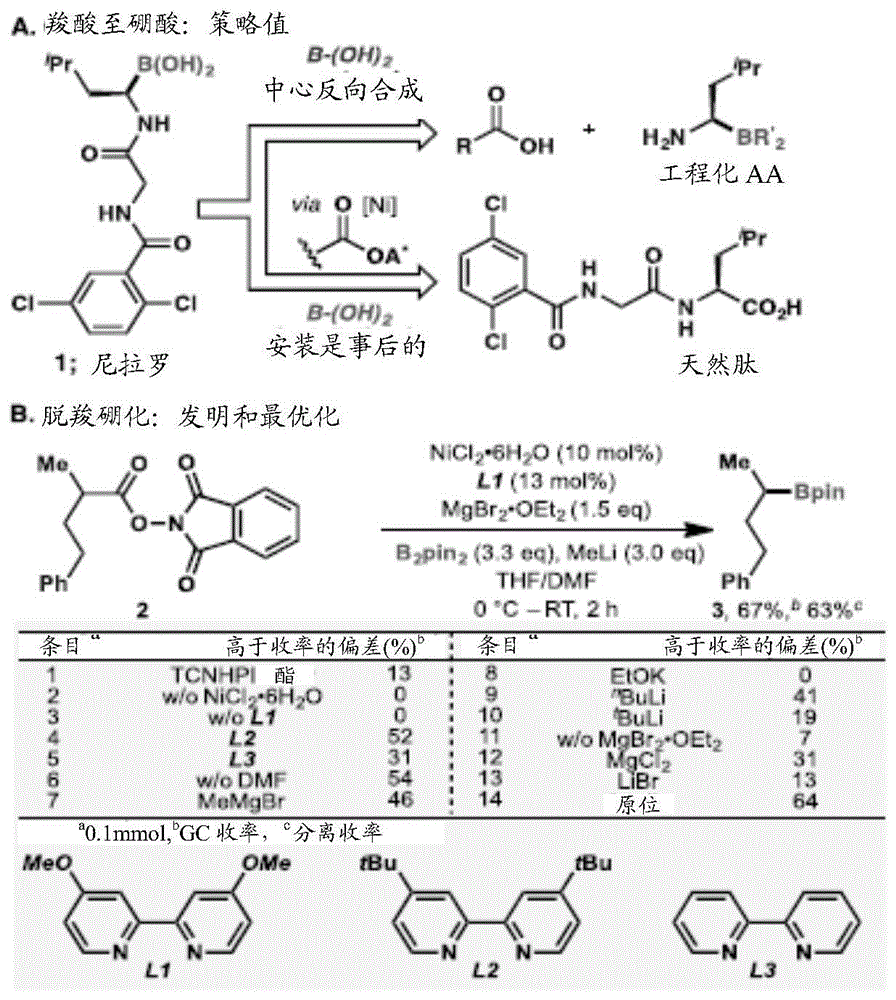
如1(图1a)所示,常规方法将所有策略注意力集中在硼原子的掺入方法上,但是这表示<5%的1的总分子量。(7)因此需要工程化氨基酸(aa)的合成,并且每个类似物必须单独制备。相反,从策略角度看,在后期将含羧酸的天然肽直接转化为相应的硼酸将容易得多,并且更加合乎逻辑。考虑到原料化学品、天然产物和药物分子中烷基羧酸的绝对数量,这种转化可以提供独特的机会,以方便地获得大量以前难以获得的硼酸,作为通用的结构单元、功能材料和有效的药物。
技术实现要素:
在多种实施方案中,本发明提供了当羧酸基团和硼酸酯基团或硼酸基团分别键合至烷基(即sp3杂化)碳原子时,将羧酸基团转化为硼酸酯基团或硼酸基团的方法。
本文所使用的术语烷基羧酸化合物rco2h是具有与烷基碳原子(即sp3杂化碳原子)键合的羧酸(-co2h)基团的化合物。分子的其他部分可包含芳基或杂芳基环、杂芳族、不饱和基团和其他官能团、以及其他烷基碳原子。
本文所用的术语烷基硼酸化合物rb(oh)2是具有与烷基碳原子(sp3杂化碳原子)键合的硼酸(-b(oh)2)基团的化合物。分子的其他部分可包含芳基或杂芳基环、杂芳族、不饱和基团和其他官能团、以及其他烷基碳原子。
烷基硼酸的频哪醇合酯(pinacolatoester)具有式
本发明在其多种实施方案中在进行所述的转化中具有以下优点,其在选择性、温和和成本有效的条件下用硼酸基团取代了羧酸基团。实施本发明的方法的一些优点包括:实用性:本发明使用的试剂廉价,而预防措施很少。因此,它可以很容易地在发现和工艺设置中采用。广泛的范围:在这种转化中使用了商业化学品和药物中最普遍存在的官能团之一的羧酸。该反应还表现出高化学选择性,因此可以容易地用于使各种各样的底物多样化。紧迫性:正越来越认识到硼酸在药物发现中的重要性,但缺乏有效且通用的硼酸合成方法。本发明填补了方法上的空白。
在多种实施方案中,本发明提供了一种将烷基羧酸化合物rco2h转化为
相应的烷基硼酸频哪醇合酯(pinacolatoester)化合物
所述方法包括:
a)形成所述烷基羧酸化合物的氧化还原活性酯(rae);然后,
b)在至少20摩尔%的mg(ii)盐和至少一摩尔当量的包含(c1-c4)烷基锂、(c1-c4)烷氧基锂或氢氧化锂的锂化合物,以及至少10摩尔%的cu或ni盐的存在下;
在与所述cu形成式(m)化合物的1,3-二羰基配体的存在下,
其中r1a和r2a各自为独立地选择的(c1-c4)烷基、三氟甲基或苯基;
或在式(l)配体存在下,所述式(l)配体包含式
使在非质子溶剂中的所述烷基羧酸化合物的氧化还原活性酯与双(频哪醇合)二硼(b2pin2)接触;
以提供所述相应的烷基硼酸频哪醇合酯化合物
更具体地,在多种实施方案中,本发明提供了一种将烷基羧酸化合物转化为相应的烷基硼酸频哪醇合酯化合物的方法,其包括:
a)形成所述烷基羧酸化合物的氧化还原活性酯(rae);然后,
b)或者:1)在氢氧化锂或(c1-c4)醇锂存在下,以及在1,3-二羰基化合物的cu(i)或cu(ii)配合物或两者存在下,该配合物具有式(m)
其中r1a和r2a各自为独立地选择的(c1-c4)烷基、三氟甲基或苯基,
或者在cu(i)或cu(ii)盐或两者和有效量的配体(l)的存在下,所述配体(l)包含式
使在质子惰性溶剂中的所述氧化还原活性酯、至少1摩尔当量的双(频哪醇合)二硼(b2pin2)和有效量的mg(ii)盐接触;
或者:2)在有效量的配体(l)的存在下,所述配体(l)包含式
使在非质子溶液中的所述氧化还原活性酯与有效量的ni(ii)盐和mg(ii)盐接触;
然后,加入包含至少1摩尔当量的有机锂化合物和至少一个摩尔当量的双(频哪醇合)二硼(b2pin2)的预混合溶液;
以提供所述相应的烷基硼酸酯化合物的频哪醇合酯。
更具体地,在实施本发明的方法中,烷基羧酸的氧化还原活性酯可以是n-羟基邻苯二甲酰亚胺(nhpi)或可以是四氯-n-羟基邻苯二甲酰亚胺(tcnhpi)。
更具体地,在实施本发明的方法中,对于cu催化的反应,cu(ii)盐可以是cu(acac)2,或者对于ni催化的反应,ni(ii)盐可以是nicl2。
更具体地,在实施本发明的方法中,mg(ii)盐可以是mgbr2或mgcl2。
更具体地,在实施本发明的方法中,对于cu催化的反应,锂化合物可以是lioh或(c1-c4)醇锂,或者对于ni催化的反应,有机锂化合物可以是甲基锂。
更具体地,在实施本发明的方法中,非质子溶剂可包含thf或二恶烷和dmf。
在多种实施方案中,本发明进一步提供了以上公开的方法,该方法还包括:步骤c),在酸性条件下裂解烷基硼酸化合物
在多种实施方案中,所述配体(l)可以是具有式l1-l5
其中
r1=ome,r2=h,l1
r1=tbu,r2=h,l2
r1=h,r2=h,l3
r1=me,r2=h,l4
r1=ome,r2=ome,l5;
或所述配体(l)可以是具有式l7-l9
其中
r3=h,r4=h,l7
r3=ph,r4=h,l8
r3=ome,r4=h,l9。
在反应的多种实施方案中,式(m)的cu配体可以是
其中r1a和r2a各自为独立地选择的(c1-c4)烷基、三氟甲基或苯基。
例如,在多种实施方案中,本发明提供了烷基硼化合物尼拉罗(ninlaro)(1)
通过从所述烷基羧酸化合物开始,执行上文所公开的所述步骤a)、b)和c)来制备。
例如,在多种实施方案中,本发明提供了制备阿托伐他汀缩酮的硼酸酯类似物的方法,该方法包括,首先,a)形成阿托伐他汀缩酮的n-羟基邻苯二甲酰亚胺(nhpi)酯以提供氧化还原活性酯,
然后,进行上述步骤b),以提供阿托伐他汀缩酮类似物的硼酸酯
本发明进一步提供作为物质组合物的具有式
本发明还提供了作为物质组合物的具有式i的
阿托伐他汀缩酮的硼酸类似物,其不含频哪醇合酯基。
例如,本发明在多个实施方案中提供了一种制备万古霉素糖苷配基的二甲基叔丁基甲硅烷基(tbs)羟基保护的硼酸类似物
转化成相应的nhpi氧化还原活化酯,然后进行上述步骤b),以提供硼酸的o-保护的硼酸频哪醇合酯,然后进行上述步骤c),裂解硼酸酯基团以提供具有式
本发明进一步提供了作为物质组合物的万古霉素糖苷配基的羟基保护的硼酸类似物
其中tbs表示二甲基叔丁基甲硅烷基o-保护基。
本发明还提供了作为物质组合物和方法的具有式
例如,本发明在多种实施方案中提供了制备硼酸mcbk319弹性蛋白酶抑制剂化合物
开始进行如上所述的步骤a),b)和c)。
本发明进一步提供作为物质组合物的具有式
例如,本发明在多种实施方案中提供了一种制备具有式
进行上述步骤b),以提供boc保护的硼酸频哪醇合酯化合物
在另外的实施方案中,boc保护的硼酸频哪醇合酯化合物
可以进一步用三氟乙酸裂解化合物的boc基,然后将所得的游离氨基与化合物
缩合,然后在hcl水溶液中用苯基硼酸裂解频哪醇合硼酸酯基团,以提供具有式
本发明进一步提供作为物质组合物的具有式
在进一步的实施方案中,上述boc保护的硼酸频哪醇合化合物可以进一步用三氟乙酸裂解该化合物的boc基团和硼酸酯,然后将所得的游离氨基与具有式
本发明进一步提供了作为物质组合物的具有式
在多种实施方案中,本发明进一步提供了制备芳霉素侧链类似物硼酸
本发明还提供了作为新的物质组合物的具有式
在多种实施方案中,本发明还提供了一种合成具有式
合成的其余部分可以如hecker中所述进行,其中在化合物hecker-12的硼酸酯交换反应中使用手性蒎烷二醇以产生手性hecker-13(请参见hecker等人的方案1)可以提供具有良好对映体纯度的中间体。这些中间体可用于制备所有的β-内酰胺酶抑制剂,例如rpx7009、化合物hecker-9a至hecker-9r(参见hecker等人的出版物的表1)。
另请参阅相关文档:“reaktionvona-lithioessigsaureesternmitbernsteinsaureanhydriden”vonfranz-petermontfortsundsilvioofner,angew.chem.(1979)91,no.8,p.656;“highlyenantioselectivehydrogenationofβ-ketoestersundermildconditions”markj.burk,*t.gregoryp.harper,andchristophers.kalberg,(1995)j.am.chem.soc.,117,4423-4424;以及“asymmetrichydrogenationofα-ketocarboxylicesters.apractical,purelychemicalaccesstoα-hydroxyestersinhighenantiomericpurity”r.noyori,*t.ohkuma,andm.kitamura(1987),j.am.chem.soc.,vol.109,no.19,5856。
尽管本发明已经进行了足够详细的描述和例示以使本领域的技术人员对其进行制造和使用,但是在不脱离权利要求的精神和范围的情况下,各种替代、修改和改进方案对于本领域技术人员将是显而易见的。
使用本领域众所周知的化学基团的标准缩写,例如;例如,me=甲基,et=乙基,i-pr=异丙基,bu=丁基,t-bu=叔丁基,ph=苯基,bn=苄基,ac=乙酰基,bz=苯甲酰基等。
附图说明
图1a和1b。脱羧硼化反应的发展。(图1a)从羧酸与硼酸:策略价值。(图1b)脱羧硼化:发明和最优化。
图2a和2b。镍催化的氧化还原活性酯的脱羧硼化反应的范围。图2a标准反应条件:氧化还原活性nhpi酯(1.0当量),nicl2·6h2o(10mol%),l1(13mol%),mgbr2·oet2(1.5当量),b2pin2(3.3当量),meli(3.0当量),thf/dmf(2.5:1),0℃–rt,2h;初级和次级硼酸盐的示例;图2b是叔硼酸盐和天然产物的示例。
图3a、3b和3c。脱羧硼化反应的应用。(图3a)脱羧硼化使立普妥(lipitor)的后期多样化成为可能。(图3b)硼万古霉素类似物的合成和生物学评估。(图3c)人弹性蛋白酶抑制剂的合成和生物学评估。
图4a和4b。发现新型人类嗜中性粒细胞弹性蛋白酶(hne)抑制剂,图4a被测试的化合物;图4bhne抑制剂50-58的抑制活性与浓度的函数关系。
具体实施方式
在本报告中,提出了一种镍催化脱羧硼化的简单方法,该方法温和、可扩展且通用于各种伯、仲、叔肽,甚至天然存在的底物。直接从相应的羧酸中提供了本来(otherwise)需要漫长的从头合成的各种硼酸盐。这种方法能够将天然肽直接转化为α-氨基硼酸的功能已经导致发现了三种有效的小分子弹性蛋白酶抑制剂。
在我们实验室中的最新研究表明,衍生自烷基羧酸的氧化还原活性酯(rae,例如n-羟基邻苯二甲酰亚胺酯2)在镍或铁催化的交叉偶联反应中用作烷基卤化物的方便替代物。这些多用途的中间体(最常用于酰胺键形成反应中)已实现了各种形式的形成c-c键的可行的手段,包括脱羧negishi(22-23)、suzuki(24)和kumada(25)偶联以及giese反应(26)。尽管raes尚未用于碳-杂原子的交叉偶联反应中,但我们的早期发现,加上fu(10)在镍催化的烷基卤化物的miyaura硼化反应方面的开创性工作(11-14),促使我们研究了利用它们形成c-b键的可能性,从而将烷基羧酸直接转化为硼酸衍生物。
这种看似简单的转换的实现需要大量的实验。图1b提供了关于2-甲基-4-苯基丁酸的最佳反应参数,以及最优化过程的简图。简单的nhpi酯(2)被证明是用于与双(频哪醇合)双硼(b2pin2)
掌握了最佳条件后,随后探索了该方法的范围。发现广泛衍生自伯、仲和叔羧酸的rae都是可行的底物(图2)。这些包括无环、环状、笼状、桥头、氟代烷基和苄基酸,它们被平滑地转化为相应的bpin硼酸酯。通过以克为单位制备29可以明显看出反应的可扩展性。另外,当使用仅2.5摩尔%的镍催化剂(3.3摩尔%的配体)时,以相当的收率获得了12种产品(3、4、7、11、12、13、16、19、25、29、35、38),从而进一步证明了该方法在工艺设置中的适应性。
当甲基锂与b2pin2预混合以形成盐配合物时,强烈的亲核/碱性有机金属物质被从底物中螯合:大量的官能团,例如醚(30、31、35、37、41),酯(5、8、21、22、39、41),氨基甲酸酯/酰胺(8、15、28、36、37、1),酮(34、38、39、40),烯烃(39、40、41)和羟基(40、41)在温和的反应条件下没有损失。实际上,即使是对碱高度敏感的fmoc基团也可以耐受(参见8)。与烷基溴化物(7)和氯化物(33)的相容性表明该反应与基于卤化物的miyaura硼化反应的正交性(orthogonality)。以相似的收率获得了由烯诺酮衍生的硼酸酯39和40,表明游离羟基对反应的影响最小。如前所述,rae的离散隔离是不必要的。当原位产生rae时,叔和仲硼酸酯可以直接由羧酸制备。这种一锅法也适用于伯(primary)底物,但收率较低。
尽管本文介绍的某些产品(例如4、17、19、20、23)可以通过miyaura硼化反应从类似的卤化物合成,但起始有机卤化物通常不可商购,并且需要额外的步骤来制备(通常是从相应的酒精)。相反,使用现成的羧酸很大程度上避免了这个问题。图2中的大部分产品直接来源于可商购的酸。例如,可以方便地从基于立方烷的羧酸制备21,而已报道的类似卤化物的合成在相同的酸上引发了苛刻的hundsdiecker反应(br2和hgo)(29)。此外,该硼化方案的范围可以扩展至氨基酸衍生物以提供α-氨基硼酸酯,例如15。通过基于卤化物的miyaura硼化来合成15是完全不可行的,因为相应的α-氨基卤化物原料将是不稳定的。在这方面,脱羧硼化策略可以探索以前难以捉摸的化学空间。
烷基羧酸的普及型通过在450多种批准的药物分子中的存在得以证明(30)。为此,该反应令人印象深刻的化学选择性提供了独特的机会,以对以活性功能进行致密修饰的生物分子进行后期改性。超过10个含羧酸盐的药物分子/天然产物已成功转化为频哪醇硼酸酯(28-41),否则其只能通过多步官能团互变或从头合成来获得。
硼酸酯可以方便地水解成相应的硼酸(例如4a、3a、33a、1)(图2)。这使得能将生物活性羧酸转化为其二羟硼基-生物电子等排体(borono-bioisoteres),以鉴定具有优异效力或药代动力学性质的化合物。替代地,硼酸酯可以多样化为多种结构基序(31-33)。作为说明性实例,在用适当的氧化剂处理后,可以方便地将立普妥(lipitor)衍生的bpin酯(36)精加工成相应的醇(36a)或氨基甲酸酯(36b)(34)(图3a)。在aggarwal报告的条件下,通过与芳基锂物质反应直接获得36c和36d(35)。脱羧硼化也可以将交联中的亲电子试剂rae转化为在suzuki反应中用作亲核试剂的bpin酯(例如36至36e和36至36f)(36)。在36e的情况下,这种“极性反转(umpolung)”方法尤其具有策略意义,因为缺乏稳定性,因此2-吡啶基硼酸或有机锌物质通常不是可行的suzuki底物。
此外,在天然肽的c-末端进行选择性脱羧硼化,可以快速获得令人垂涎的α-氨基硼酸,这是优先的药物化学基序(18、37)。例如,ninlaro(1)从简单的肽中分三步获得(图2)。这为基于肽的疗法的研究开辟了独特的领域:在也许最显著的示例中,万古霉素通过42的脱羧硼化作用转变为硼酸类似物(44)(图3b)(38)。在四个甲基化苯氧基、两个tbs保护的羟基、两个芳基氯化物、六个仲酰胺、一个伯酰胺、一个仲胺和七个可差向异构的立体中心的存在下,该过程顺利进行。尽管44与母体酸43相比显示出较小的活性,但是这种显著的化学选择性仍然证明了该反应的潜在效用。
自由基过程的不可预测的立体选择性通常会阻碍其在药物前导物或天然产物的后期修饰中的广泛采用。在该基于自由基的脱羧硼化反应中,作为单一非对映异构体获得了配合物α-氨基硼酸44。该结果促使我们研究关于几种二肽的脱羧硼化的立体选择性(图3d)。我们发现,增加n-末端残基的空间体积会导致更好的非对映选择性:尽管对于45,两种非对映异构体几乎相等地被提供,但对于46和47却观察到更高的选择性。同时,从boc-l-val-l-val和boc-l-val-d-val以相同的非对映异构体比例获得46。较低的反应温度也可用于增强立体选择性。在-15℃时,48以大于5比1的非对映异构体(d.r.)提供,从而可以以较短的顺序进行velcade(49)的立体选择性合成。
通过将硼酸盐的丰富药用潜力与烷基羧酸的普遍存在相结合,脱羧硼化反应可以在药物开发中开拓新的前景。例如,将脱羧硼化反应应用于易于获得的二肽可以方便地制备50–52的制剂,这些制剂形成为单个非对映异构体,被发现是人类嗜中性粒细胞弹性蛋白酶(hne)的有效抑制剂(图4a和b)。值得注意的是,发现50的羧酸前体(50b)没有任何抑制活性,而50和51与其三氟酮同类物(50a和51a)相比则显示出显著增强的效力,后者已在肺部疾病(例如囊性纤维化)ii期临床试验中进行了研究(39-45)。hne是一种高度活跃的丝氨酸蛋白酶,其通过破坏人体细胞基质以及外源蛋白质的机械重要结构而在免疫应答、组织重塑和炎症发作/消退中起关键作用(46)。尽管已经在多种炎症性肺病(例如,囊性纤维化、肺气肿和支气管扩张)中对五代hne抑制剂进行了临床评估,但在人类中却没有一种具有压倒性的功效以对这些疾病产生显著作用(46)。
为此,针对纯化的hne,52表现出ic50=15pm(ki=3.7pm),而51表现出ic50=30pm(ki=34pm)。与其他临床前和临床验证的hne抑制剂并列确定ic50值(53–57),其他临床前和临床验证的hne抑制剂包括bay85-8501(54,据报道ki=80pm的领先临床候选药物)(47)、55(pol6014,基于i期肽的囊性纤维化的临床候选药物(48)以及56和57(chiesipharmaceuticals报告)(49)。此外,51和52在囊性纤维化(cf)和慢性阻塞性肺疾病(copd)患者的痰标本中保留了其抑制活性中的许多,从而在与传统生化分析方法相比更具病理生理相关性的环境的背景中突显了它们的潜能。相反,虽然来自阿斯利康(astrazeneca)的二聚体化合物58(ic50=11pm,ki=2.7pm)(50)和bay85-8501(54)表现出较低的ic50值,但在源自患者的痰液中,其效力却大大降低。copd痰中lipe值的比较显示,52的优越效力并非由增加的亲脂性驱动(10.2对57的9.45)(51)。
此外,发现随着孵育时间的增加(5-60分钟之间),52的ic50值保持不变,而在相同条件下,58(非共价抑制剂)的ic50值则显示其效力增加了55倍。这些数据保留了预期的特性:化合物52的性能类似于基于部分机理的抑制剂(或共价可逆抑制剂),这可能是由于α-氨基硼酸的潜在缓慢释放速率所致。这和在不同于许多可逆的弹性蛋白酶抑制剂(即58)的其他氨基硼酸化合物中观察到的更紧密的结合和更长的停留时间有关(52)。临床上,该机制已通过velcade(49)成功证明,其抑制26s蛋白酶体的催化位点-硼酸酯和亲核氧之间的共价可逆键导致缓慢的解离速率(53-54)。由于大多数临床弹性蛋白酶抑制剂(例如54,bay85-8501,迄今为止报道的最有效的分子之一)是非反应性、可逆的过渡态抑制剂,因此52的高效力和氨基硼酸的内在机理可以帮助解决这些限制。通过这种“混合”酶抑制方法(基于fischer的锁(lock)和钥匙(key)模型/ehrlich的药效团(pharmacophore)模型),硼酸(如52)将快速有效的结合和缓慢的解离速率组合,可以有效地恢复在临床设置中蛋白酶与抗蛋白酶的平衡。因此,可以迅速将其调整为针对肺的特异性临床应用。
为了进一步评估51和52的治疗潜力,探查了体外adme特性,以确定是否会发现酮的硼酸酯替代物有任何有害作用(图4c)。这些氨基硼酸显示出与三氟甲基酮类似物(51a)相当的动力学溶解度。发现在2小时内cd-1小鼠血浆中51和52的大部分(分别为90%和79%)是完整的。51和52显示出与三氟酮51a相似的代谢稳定性。51和51a在caco-2细胞中也显示出相似的渗透性水平(请参见补充信息的第176页)。这些数据表明,新型硼酸盐仅能提高效力,而不会改变其酮同类物的类药物性质。
方法摘要:
在程序上,氧化还原活性酯向硼酸酯的转化是通过三个步骤实现的:即催化剂混合物的制备,[b2pin2me]li配合物的制备以及镍催化的脱羧硼化反应。本文提供了一个简短的实验规程和图形指南。补充信息中可以发现有关商业来源和化学物质纯度或不同底物类别的实验细节变化的全面信息。
nicl2·6h2o/配体储备溶液或悬浮液的制备:
将装有nicl2·6h2o(1.0当量)和配体(l1或l2,1.3当量)的烧瓶抽空,并用氩气回填三次。加入thf(nicl2·6h2o的浓度为0.025m)或dmf(nicl2·6h2o的浓度为0.050m)。将所得混合物在室温下搅拌过夜(或直至未观察到颗粒状的nicl2·6h2o),得到绿色溶液或悬浮液。[注:所有在氩气下保存的溶液或悬浮液可以使用几天,而反应收率不会明显下降。]
[b2pin2me]li配合物的制备:
在氩气下于0℃向b2pin2(1.1当量)的thf溶液(b2pin2的浓度为1.1m)中添加meli(1.6m,在et2o中,1.0当量)。将反应混合物温热至室温,并搅拌1h以得到乳白色悬浮液。
镍催化的脱羧硼化:
将装有氧化还原活性酯(1.0当量)和mgbr2·oet2(1.5当量)的烧瓶抽空,并用氩气回填三次。经由注射器添加催化剂溶液或悬浮液(含有10mol%的nicl2·6h2o和13mol%的配体)。当使用dmf中的催化剂悬浮液/溶液时,向反应容器中添加另一部分thf(所需dmf悬浮液/溶液体积的两倍),然后添加催化剂混合物[此过程可能会大量放热。(用冰/水浴)冷却可能是必要的]。剧烈搅拌所得混合物,直到在反应容器的底部没有观察到可见的固体[发现这通过超声处理加速了]。将该混合物冷却至0℃,然后一次性加入[b2pin2me]li在thf(3当量)中的悬浮液。在0℃下搅拌1小时后,将反应温热至室温并再搅拌1小时。当薄层色谱法(tlc)分析表明反应完成时,将反应用hcl水溶液(0.1m)或饱和nh4cl水溶液淬灭,并用乙醚(et2o)或乙酸乙酯(etoac)萃取。替代地,如图5所示的情况,以较大规模将反应混合物直接倒入et2o上,然后将所得悬浮液通过硅胶和硅藻土垫过滤。通过柱色谱法纯化,得到所需的硼酸酯。
通过专门使用n-羟基邻苯二甲酰亚胺氧化还原活性酯,已经实现了一种简单的方法来相互转化有机化学中最重要的两个官能团。通过包括药物分子在内的众多复杂的底物说明了实用性和化学选择性。体现在以良好的立体选择性将天然肽直接转化为二羟硼基-电子等排体(borono-isoteres)的能力的广泛范围与以卤化物为基础的miyaura-suzuki方案不匹配。硼酸烷基酯可以在合成的任何阶段引入,从而改变了其制备的策略范式。通过将硼酸盐的丰富医学潜力与烷基羧酸的普遍存在相结合,该方法可能会在药物开发中开辟新的前景。这已经从mcbk320的发现中得到证实,mcbk320是一种高效的弹性蛋白酶抑制剂,其在癌症治疗中具有潜力。
所引用的文件
1.a.suzuki,angew.chem.int.ed.50,6722(2011).
2.w.l.a.brooks,b.s.sumerlin,chem.rev.116,1375(2016).
3.s.d.bull,etal.acc.chem.res.46,312(2013).
4.p.c.trippier,c.mcguiganmed.chem.commun.1,183(2010).
5.a.draganov,d.wang,b.wang,top.med.chem.17,1(2016).
6.c.ballatore,d.m.huryn,a.b.smith,chemmedchem8,385(2013).
7.r.smoum,a.rubinstein,v.m.dembitsky,m.srebnik,chem.rev.112,4156(2012).
8.h.c.brown,hydroboration(benjamin/cummings,1980).
9.c.m.vogels,s.a.westcott,curr.org.chem.9,687(2005).
10.a.s.dudnik,g.c.fu,j.am.chem.soc.134,10693(2012).
11.t.c.atack,r.m.lecker,s.p.cook,j.am.chem.soc.136,9521(2014).
12.r.b.bedfordetal.,organometallics33,5940(2014).
13.c.-t.yangetal.,angew.chem.int.ed.51,528(2012).
14.h.ito,k.kubota,org.lett.14,890(2012).
15.h.c.brown,t.e.cole,organometallics2,1316(1983).
16.k.-s.lee,a.r.zhugralin,a.h.hoveyda,j.am.chem.soc.131,7253(2009).
17.j.a.schniffner,k.müther,m.oestreich,angew.chem.int.ed.49,1194(2010).
18.p.andrés,g.ballano,m.isabelcalaza,c.cativiela,chem.soc.rev.45,2291(2016).
19.m.a.beenen,c.an,j.a.ellman,j.am.chem.soc.130,6910(2008).
20.i.a.mkhalid,j.h.barnard,t.b.marder,j.m.murphy,j.f.hartwig,chem.rev.110,890(2010).e.j.olhava,m.d.danca,us7442830b1(2008).
21.t.qinetal.,science352,801(2016).
22.j.cornellaetal.,j.am.chem.soc.138,2174(2016).
23.j.wangetal.,angew.chem.int.ed.55,9676(2016).
24.f.toriyamaetal.,j.am.chem.soc.138,11132(2016).
25.t.qinetal.,angew.chem.int.ed.55,266(2016).
26.t.hatakeyamaetal.,j.am.chem.soc.132,10674(2010).
27.r.b.bedfordetal.,chem.eur.j.20,7935(2014).
28.r.a.hussainyetal.,j.med.chem.54,3480(2011).
29.p.lassalasetal.,acsmed.chem.lett.59,3183(2016).
30.j.schmidt,j.choi,a.liu,m.slusarczyk,g.c.fu,science354,1265(2016).
31.y.xi,j.hartwig,j.am.chem.soc.138,6703(2016).
32.g.a.molander,n.ellis,acc.chem.res.40,275(2007).
33.s.n.mlyanrski,a.s.karns,j.p.morken,j.am.chem.soc.134,16449(2012).
34.a.bonet,m.odachowski,d.leonori,s.essafi.v.k.aggarwal,nat.chem.6,584(2014).
35.s.laulhé,j.m.blackburn,j.l.roizen,org.lett.18,4440(2016).v.m.dembitsky,m.srebnik,tetrahedron59,579(2003).j.j.mcatee,s.l.castle,q.jin,d.l.boger,bioorg.med.chem.lett.12,1319(2002).
36.p.r.bernsteinetal.,j.med.chem.37,1259(1994).
37.c.a.vealeetal.,j.med.chem.40,3173(1997).
38.p.r.bernsteinetal.,j.med.chem.38,212(1995).
39.j.p.burkhartetal.,j.med.chem.37,223(1995).
40.p.d.edwardsetal.,j.med.chem.40,1876(1997).
41.k.hemmi,i.shima,k.imai,h.tanaka,ep0494071a2(1992).
42.t.kinoshita,i.nakanishi,a.sato,t.tada,bioorg.med.chem.lett.13,21(2003).
43.f.vonnussbaum,v.m.-j.li,bioorg.med.chem.lett.25,4370(2015).
44.f.vonnussbaum,etal.,chemmedchem10,1163(2015).
45.f.otto,etal.,wo2015096873(2015).
46.t.j.blench,etal.,wo2013037809a1(2013).
47.l.
48.m.d.schultz,bioorg.med.chem.lett.23,5992(2013).
49.a.zervosen,etal.,j.am.chem.soc.133,10839(2011).
50.m.groll,c.r.berkers,h.l.ploegh,h.ovaa,structure14,451(2006).
51.m.d.schultz,bioorg.med.chem.lett.23,5992(2013).
52.a.zervosen,etal.,j.am.chem.soc.133,10839(2011).
53.m.groll,c.r.berkers,h.l.ploegh,h.ovaa,structure14,451(2006).
54.54.a.f.kisselev,w.a.vanderlinden,h.s.overkleeft,chem.biol.19,99(2012).
55.55.s.p.thomas,m.d.greenhalgh,chem.commun.49,11230(2013).
56.y.wen,j.xie,c.deng,c.li,j.org.chem.80,4142(2015).
57.s.roesner,etal.,chem.commun.50,4053(2014).
58.j.yi,etal.,adv.synth.catal.354,1685(2012).
59.j.hu,etal.,j.org.chem.81,14(2016).
60.a.chen,l.ren,c.m.crudden,j.org.chem.64,9704(1999).
61.t.furukawa,etal.,bioorg.med.chem.20,2002(2012).
62.d.l.boger,etal.,j.am.chem.soc.120,8920(1998).
63.clsidocumentm07-a9ofmethodsfordilutionantimicrobialsusceptibilitytestsforbacteriathatgrowaerobically(clinicalandlaboratorystandardsinstitute:wayne,pa,9thed.,2012).
64.s.kokinaki,l.leondiadia,n.ferderigos,org.lett.7,1723(2005).
65.t.miyazawa,s.hiramatsu,y.tsuboi,t.yamada,s.kuwata,bull.chem.soc.jpn.58,1976(1985).
66.j.liu,k.r.west,c.r.bondy,k.m.sanders,org.biomol.chem.5,778(2007).
67.a.w.buesking,v.bacauanu,i.cai,j.a.ellman,j.org.chem.79,3671(2014).
68.r.a.copeland,evaluationofenzymeinhibitorsindrugdiscoveryaguideformedicinalchemistsandpharmacologists(wiley,newyork,2005).
实施例
通过本发明的方法制备的人嗜中性粒细胞弹性蛋白酶的硼酸抑制剂
公开的化合物(人类嗜中性粒细胞弹性蛋白酶的ic50)
人嗜中性粒细胞弹性蛋白酶的硼酸抑制剂的合成途径
发现所有三种化合物(mcbk319、mcbk320和mcbk323)在硼酸基序中具有高的三聚化倾向。这些三聚体的生物活性可能需要进一步的研究。这些新型的弹性蛋白酶抑制剂具有潜在的应用前景,可用于治疗癌症、循环纤维化和支气管扩张。
这些化合物与作为弹性酶抑制剂的筛选出的先导化合物相比,显示出更高的效价(与类似的三氟喹啉先导化合物相比,提高了100-1000倍)。硼酸基序的独特的理化性质可以引起有利的药代动力学特性。本文公开的制备方法是简明的并且易于扩大规模。
一般信息
通过将预先脱气的溶剂传送通过活化的氧化铝柱获得四氢呋喃(thf)、n,n-二甲基甲酰胺(dmf)、乙腈(ch3cn)二氯甲烷(ch2cl2)。除非另有说明,否则以最高商业质量购买其他溶剂和试剂,无需进一步纯化即可使用。除非另有说明,否则收率是指色谱和光谱(1h-nmr)均质材料。通过gc/ms、lc/ms和薄层色谱法(tlc)监控反应。tlc使用0.25mme.merck硅胶板(60f-254),使用短波紫外光作为可视化剂,高锰酸钾(kmno4)或铈酸钼酸铵(cam)和热作为显影剂进行。nmr光谱在brukerdrx-600、drx-500或dpx-400仪器上记录并使用残留的未氘化溶剂进行校准(1h:对于cdcl3,δ7.26,对于meoh-d4,δ3.31,对于thf-d8,δ3.58,1.73,对于dmso-d6,δ2.50,对于丙酮-d6,δ2.05;13c:对于cdcl3,δ77.16,对于meoh-d4,δ49.0,对于thf-d8,δ67.6,25.5,对于dmso-d6,δ39.50,对于丙酮-d6,δ29.84)。以下缩写用于解释多重性:s=单重态,d=双重态,t=三重态,q=四重态,m=多重态,br=宽峰。使用e.merck硅胶(60,粒径0.043-0.063mm)进行柱色谱,并且在0.25mme.merck硅胶板(60f-254)上进行制备型tlc。通过电喷雾电离反射电子飞行时间实验在agilentlc/msdtof质谱仪上记录高分辨率质谱(hrms)。使用配备了尺寸为200×50mm的phenomenexgemini10μmc18色谱柱的agilentsd-1prepstar系统进行制备型高效液相色谱(hplc)。熔点记录在fisher-johns12-144熔点仪上,并且未经校正。收集所有x射线衍射数据并通过ucsd小分子x射线设备进行分析。通过将硅胶和去离子水混合,然后剧烈振摇直至观察到蓬松的粉末,来制备失活的硅胶(35wt%h2o)。
b2pin2是双(频哪醇合)二硼。
nhpi是n-羟基邻苯二甲酰亚胺。tcnhpi是四氯-n-羟基邻苯二甲酰亚胺。使用例如本领域众所周知的化学基团的标准缩写;例如,me=甲基,et=乙基,i-pr=异丙基,bu=丁基,t-bu=叔丁基,ph=苯基,bn=苄基,ac=乙酰基,bz=苯甲酰基,tbs=二甲基-t-丁基甲硅烷基,boc=叔丁氧羰基等。
使用配备有尺寸为200×50mm的phenomenexgemini10μmc18色谱柱的agilentsd-1prepstar系统进行制备型hplc。熔点记录在fisher-johns12-144熔点仪上,并且未经校正。收集所有x射线衍射数据并通过ucsd小分子x射线设备进行分析。通过将硅胶和去离子水混合,然后剧烈振摇直至观察到蓬松的粉末,来制备失活的硅胶(35wt%h2o)。
氧化还原活性酯(rae)合成的通用程序(通用程序a)
在圆底烧瓶中装入羧酸(1.0当量)、n-羟基邻苯二甲酰亚胺(nhpi,1.0当量)或四氯-n-羟基邻苯二甲酰亚胺(tcnhpi,1.0当量)和dmap(0.1当量)。加入ch2cl2(0.2m),然后加入n,n’-二异丙基碳二亚胺(dic,1.1当量),两者均在室温下进行。将混合物在室温下搅拌直至所有酸被消耗掉(通过tlc指示)。快速过滤得到的混合物,并用更多的ch2cl2冲洗固体残余物。真空浓缩滤液,并通过快速柱色谱纯化,得到相应的氧化还原活性酯,除非另有说明,否则其无需进一步纯化即可使用。
优化细节
基于0.1mmol规模筛选所有反应。优化从s1开始。tcnhpi酯用于初始筛选,因为较早的条件表明其性能优于nhpi酯(nhpi酯最终在优化条件下使用。
表1
*利用十二烷作为内标,通过gc-fid确定的收率。
表2
*利用十二烷作为内标,通过gc-fid确定的收率。
表3
*利用十二烷作为内标,通过gc-fid确定的收率。
表4
*利用十二烷作为内标,通过gc-fid确定的收率。
表5
*利用十二烷作为内标,通过gc-fid确定的收率。
表6
*利用十二烷作为内标,通过gc-fid确定的收率。
表7
*利用十二烷作为内标,通过gc-fid确定的收率。
然而,在上述用于s1的最优化条件下,与s2的nhpi酯相比,s2a的脱羧硼化以较低的收率进行。
表8
*利用十二烷作为内标,通过gc-fid确定的收率。
为了确定更一般的条件组,对nhpi酯s2进行了进一步的优化。
表9
*利用十二烷作为内标,通过gc-fid确定的收率。
表10
*利用十二烷作为内标,通过gc-fid确定的收率。
对于s2(1°rae)的脱羧硼化的这一优化条件组更为通用,也适用于2(2°rae)。
进一步的筛选表明,使用thf作为唯一溶剂,叔羧酸(3°rae)的收率最高。
表11
*利用十二烷作为内标,通过gc-fid确定的收率。
氧化还原活性酯的镍催化硼化的一般步骤
第i部分。nicl2·6h2o/配体储备溶液或悬浮液的制备
(1)悬浮液a:在thf(0.025m)中的nicl2·6h2o/di-meobipy(l1)
将装有nicl2·6h2o(23.8mg,0.1mmol)和4,4'-二甲氧基-2,2'-联吡啶(l1,28.1mg,0.13mmol)的旋盖培养管抽空,并用氩气回填三次。加入thf(4.0ml),并将所得混合物在室温搅拌过夜(或直至观察不到颗粒状的nicl2·6h2o),得到浅绿色悬浮液。
(2)悬浮液b:在dmf(0.05m)中的nicl2·6h2o/di-meobipy(l1)。
将装有nicl2·6h2o(23.8mg,0.1mmol)和4,4'-二甲氧基-2,2'-联吡啶(l1,28.1mg,0.13mmol)的旋盖培养管抽空,并用氩气回填三次。加入dmf(2.0ml),并将所得混合物在室温搅拌过夜,得到浅绿色悬浮液。
(3)悬浮液c:在thf(0.025m)中的nicl2·6h2o/di-tbubipy(l2)。
将装有nicl2·6h2o(23.8mg,0.1mmol)和4,4'-二叔丁基-2,2'-联吡啶(l2,34.8mg,0.13mmol)的旋盖培养管抽空并回填氩气三次。加入thf(4.0ml),并将所得混合物在室温搅拌过夜(或直至观察不到颗粒状的nicl2·6h2o),得到浅绿色悬浮液。
(4)溶液d:在dmf(0.05m)中的nicl2·6h2o/di-tbubipy(l2)。
将装有nicl2·6h2o(23.8mg,0.1mmol)和4,4'-二叔丁基-2,2'-联吡啶(l2,34.8mg,0.13mmol)的旋盖培养管抽空并回填氩气三次。加入dmf(2.0ml),并将所得混合物在室温搅拌2h,得到绿色溶液。
注意:所有在氩气下保存的溶液或悬浮液都可以使用几天,而反应收率不会明显下降。
第ii部分。[b2pin2me]li悬浮液的制备
在0℃,在氩气下,向b2pin2(168mg,0.66mmol)的thf(0.6ml)溶液中加入meli(0.38ml,1.6m的et2o溶液,0.6mmol)。将反应混合物温热至室温并搅拌1小时以得到悬浮液(有时我们也经历过获得该复合物为澄清溶液)。
注意:所得混合物可在搅拌下保存数小时,而不会明显变质。
第iii部分。镍催化交叉偶联反应
通用程序b
将装有氧化还原活性酯(0.2mmol,1.0当量)和mgbr2·oet2(77mg,0.3mmol,1.5当量)的旋盖培养管抽空,并用氩气回填3次。加入thf(0.8ml),搅拌混合物直到(约10min)未观察到的颗粒状mgbr2·oet2,通过注射器加入悬浮液b(0.4ml,nicl2·6h2o(10mol%)/di-meobipy(13mol%)的dmf溶液),或溶液d(0.4ml,nicl2·6h2o(10mol%)/di-tbubipy(13mol%)的dmf溶液)。剧烈搅拌所得混合物,直到在反应容器的底部(约10分钟)未观察到可见的固体。将该混合物冷却至0℃,然后一次性加入[b2pin2me]li在thf中的悬浮液(3eq,1.1ml)(注意:不要滴加!)。在0℃下搅拌1小时后,将反应温热至室温并再搅拌1小时,然后用0.1nhcl(10ml)淬灭。所得混合物用et2o或etoac(3ml×2)萃取。将合并的有机层真空浓缩,并将粗产物通过快速柱色谱法纯化。对于酸不稳定的底物,可替代地用饱和nh4cl水溶液(10ml)淬灭反应。
通用程序c
将装有氧化还原活性酯(0.2mmol,1.0当量)和mgbr2·oet2(77mg,0.3mmol,1.5当量)的旋盖培养管抽空,并用氩气回填3次。经由注射器添加悬浮液a(0.8ml,在thf中的nicl2·6h2o(10mol%)/di-meobipy(13mol%))或c(0.8ml,在thf中的nicl2·6h2o(10mol%)/di-tbubipy(13mol%))。将混合物在室温下剧烈搅拌直至(约15分钟)观察不到颗粒状的mgbr2·oet2。将此悬浮液冷却至0℃,然后一次性添加[b2pin2me]li悬浮液(注意:请勿逐滴添加!)。在0℃下搅拌1小时后,将反应物温热至室温并再搅拌1小时。将反应混合物用et2o(10ml)稀释,通过硅胶和硅藻土的短垫过滤(顶层:硅藻土,底层:硅胶,v/v硅藻土:硅胶=1:1),用et2o洗涤(50毫升)。将滤液浓缩,并将粗产物通过柱色谱法纯化。
对于极性底物,如肽,用0.1nhcl(10ml)或饱和nh4cl水溶液(10ml)淬灭反应。然后用etoac(3ml×2)萃取。合并的有机层经na2so4干燥,真空浓缩并通过快速柱色谱法纯化。
氧化还原活性酯的克级镍催化硼化的通用程序(布洛芬的硼化)。
根据通用程序c略微修改了克规模程序。将装有b2pin2(2.57g,10.1mmol,3.3当量)的火焰干燥的圆底烧瓶抽空,并用氩气回填三次。添加thf(9.2ml),并且当逐滴添加meli(5.8ml,在et2o中的1.6m,9.3mmol,3.0当量)时,将澄清溶液冷却至0℃。然后将反应混合物温热至室温并搅拌1小时。
将布洛芬s18(1.08g,3.07mmol)和mgbr2·oet2(粉末,792mg,3.07mmol,1.0当量)的nhpi氧化还原活性酯顺序添加到另一个火焰干燥的圆底烧瓶中。将该烧瓶抽空并用氩气回填3次,并冷却至0℃。加入thf(12ml),将混合物超声处理直至未观察到颗粒状的mgbr2·oet2。加入nicl2·6h2o(73mg,0.31mmol)和di-meobipy(l2,86mg,0.40mmol)在thf(12ml)中的悬浮液,再次超声处理所得混合物,直至在烧瓶底部上没有可见的固体。接着将混合物冷却至0℃,然后一次性加入[b2pin2me]li在thf中的悬浮液。在0℃下搅拌1小时后,将反应混合物温热至室温并再搅拌1小时。
然后将反应混合物倒入et2o(100ml)中,并将烧瓶用另外的et2o(100ml)冲洗。将所得混合物通过硅胶和硅藻土塞过滤(顶层:硅藻土,底层:硅胶,v/v硅藻土:硅胶=1:1),将固体残余物用et2o(350ml)洗涤,将滤液真空浓缩。通过快速柱色谱法纯化(硅胶,己烷至1∶30et2o∶己烷),得到产物(709mg,80%),其为无色油状物。
烷基羧酸通过原位生成的rae进行镍催化脱羧硼化的通用程序(通用程序d)
在带有搅拌棒的旋盖培养管中装入烷基羧酸(0.2mmol)、n-羟基邻苯二甲酰亚胺或四氯-n-羟基邻苯二甲酰亚胺(0.2mmol,1.0当量)和n,n'-二环己基碳二亚胺(0.2mmol,1.0当量)。然后将试管抽空并用氩气回填三次。加入ch2cl2(2.0ml),并将所得混合物在室温搅拌2h,然后真空除去挥发物。加入mgbr2·oet2(77mg,0.3mmol,1.5当量)。将该管抽真空并用氩气回填三次。加入悬浮液a(0.8ml,在thf中的nicl2·6h2o(10mol%)/l1(13mol%)溶液)或悬浮液c(0.8ml,nicl2·6h2o(10mol%)/l2(13mol%)。将混合物在室温下剧烈搅拌15分钟(或直到未观察到颗粒状mgbr2·oet2),随后冷却至0℃,然后一次性添加[b2pin2me]li在thf(1.1ml)中的悬浮液(注:不要逐滴加入!)。搅拌1小时后,将反应物温热至室温,再搅拌1小时。然后将反应混合物用0.1nhcl(10ml)淬灭,并用et2o(5ml×2)萃取。合并的有机层经na2so4干燥,真空浓缩并通过柱色谱法纯化,得到所需产物。
烷基羧酸通过原位生成的rae进行镍催化的脱羧硼化反应的实施例按照通用程序d在六种烷基羧酸上证明了这种原位程序。
通用程序d对伯羧酸的效果较差(通常约20%的收率)。
用2.5mol%的镍对氧化还原活性酯进行ni催化的硼化反应的通用程序。
第i部分。nicl2·6h2o/配体储备溶液或悬浮液的制备。
(1)悬浮液e:在thf(6.25mm)中的nicl2·6h2o/di-meobipy(l1)。
将装有nicl2·6h2o(23.8mg,0.1mmol)和4,4'-二甲氧基-2,2'-联吡啶(l1,28.1mg,0.13mmol)的旋盖培养管抽空,并用氩气回填三次。加入thf(16.0ml),并将所得混合物在室温搅拌过夜(或直至观察不到颗粒状的nicl2·6h2o),得到浅绿色悬浮液。
(2)溶液f:在dmf(12.5mm)中的nicl2·6h2o/di-meobipy(l1)。
将装有nicl2·6h2o(23.8mg,0.1mmol)和4,4'-二甲氧基-2,2'-联吡啶(l1,28.1mg,0.13mmol)的旋盖培养管抽空,并用氩气回填三次。加入dmf(8.0ml),并将所得混合物在室温搅拌过夜,得到浅绿色溶液。
(3)悬浮液g:在thf(6.25mm)中的nicl2·6h2o/di-tbubipy(l2)。
将装有nicl2·6h2o(23.8mg,0.1mmol)和4,4'-二叔丁基-2,2'-联吡啶(l2,34.8mg,0.13mmol)的旋盖培养管抽空并回填氩气三次。加入thf(16.0ml),并将所得混合物在室温搅拌过夜(或直至观察不到颗粒状的nicl2·6h2o),得到浅绿色悬浮液。
注意:所有在氩气下保存的溶液或悬浮液均可使用两周,而反应收率没有明显下降。
第ii部分。镍催化交叉偶联反应。
通用程序b/c之后,利用悬浮液e/溶液f/悬浮液g,以2.5摩尔%镍负载对氧化还原活性酯进行硼化。
利用2.5mol%镍对氧化还原活性酯进行ni催化的硼化反应的实施例。
a使用thf作为溶剂。b使用l2作为配体以及thf作为溶剂。
c使用tcnhpi酯
标准反应条件:氧化还原活性nhpi酯(1.0当量),nicl2·6h2o(2.5mol%),l1(3.3mol%),mgbr2·oet2(1.5当量),b2pin2(3.3当量),meli(3.0当量),thf/dmf(2.5:1),0℃–rt,2小时。
氧化还原活性酯的铜催化的硼化反应。
在本发明的另外的实施方案中,本文公开并要求保护烷基羧酸的铜催化的硼酸酯化以产生烷基硼酸。本发明需要通过氧化还原活性酯的铜催化的羧基化将羧酸酯官能团转化成相应的硼衍生物。在各种实施方案中,这种转化使得能够对药物或其类似物进行后期修饰;其使得能方便地合成含硼的生物活性分子,包括fda批准的药物;其使得能制备广泛用于药物成分合成的含硼结构单元。与其他方法相比,它使得程序能简单并且试剂便宜,从而很容易在工艺化学中采用。
针对硼酸酯化催化活性,筛选cu(ii)盐,该cu(ii)盐包含与各种配体络合的铜,这些配体包括式l(例如4,4'-二-t-bu-2,2'-联吡啶基,l2,参见上文)的联吡啶基型配体和与铜离子(cu(i)或cu(ii))形成式m配合物(例如丙酮基丙酮酸酯,m1)的1,3-二羰基型配体。
m1r1a=r2a=me,cu(acac)2
m2r1a=r2a=tbu
m3r1a=r2a=ipr
m4r1a=r2a=ph
m5r1a,r2a=tbu,me
m6r1a=r2a=cf3
m7r1a,r2a=tbu,cf3
下表12显示了在进行本发明的硼化反应中使用具有各种碱和配体的铜盐(cu(i)或cu(ii))的初步筛选结果。可以看出,即使与tbubipy(配体l2)结合使用膦配体(如pph3和pcy3)导致产品收率仍为零或收率低,但是带有和不带有mgbr2的tbubipy的cu(i)盐的效果更好。
表13显示了使用不同负载的mgbr2和各种cu盐得到的结果的研究。可以看出,mgbr2似乎抑制了该反应。以tbubipy为配体,无mgbr2,以cucl、cucl2、cucl2.6h2o,cui和cu(oac)2作为铜源,可获得最佳结果;cu(i)和cu(ii)态在该系统中均具有催化活性。
表14显示了溶剂对在tbubipy配体l2存在下使用叔丁醇锂、cu(oac)2(即cu(ii)盐)的铜催化的硼化反应的影响。尽管dmf与其他溶剂(例如ch2cl2、etoac或甲苯)的混合物也很有效,但最有效的溶剂混合物似乎是dmf加醚(例如二噁烷、thf或甘醇二甲醚(glyme),二甘醇二甲醚或二乙醚)。有趣的是,纯dmf提供较低的收率。
表15显示了使用lioh和liotbu,甚至在存在mgbr2的情况下,以cu(oac)2作为铜源和tbubipyl2作为配体所获得的收率显著提高。
表16显示了在lioh/mgbr2-et2o存在下,使用优选的二噁烷/dmf溶剂体系的变体,以及cu(oac)2/tbubipy催化配合物,对溶剂收率的影响。作为二噁烷的替代品,其他醚和酯似乎是最有效的。作为dmf的替代品,其他偶极非质子传递溶剂也很有效,吡啶和丙酮也是如此。
表17显示了在二氧六环-dmf溶剂系统中,在lioh和mgbr2-et2o存在下,使用cu(oac)2作为铜源对催化剂和铜源收率的影响。有效的配体与以前一样多,联吡啶配体l2比膦类配体更有效,但有趣的是,注意到,当tbubipyl2是铜配合物的主要配体时,添加1,3-二羰基配体丙酮基丙酮酸酯(acac)提高了收率。
表18和19显示了在lioh和mgbr2.et2o存在下,在二噁烷-dmf溶剂体系中使用如上定义的各种1,3-二羰基型配体m1-m7对收率的影响。配体m1,丙酮基丙酮酸铜本身以及其他1,3-二羰基配体m2-m7是广泛地有效的,而当以cu(acac)2为主要催化剂时,其他cu盐的存在并不会在获得的收率方面带来很大的变化。
表20示出了在lioh存在下并且在不存在任何联吡啶基配体l2,具有各种mg来源和lioh负载的情况下,在二噁烷-dmf溶剂体系中铜催化的硼化反应的收率。发现用mgcl2可以代替更昂贵的mgbr2,而没有收率损失,而超过约15当量的lioh负载量不会产生明显的收率增加。
表21显示了在二噁烷/dmf中存在lioh和mgcl2的情况下使用cu(acac)2m1进行的反应的最终优化,并进行了生产成本估算比较。
表12:铜催化的氧化还原活性酯的硼化;配体筛选
表13:铜催化的氧化还原活性酯的硼化;mrbr2负载和铜盐的影响
表14:铜催化的氧化还原活性酯的硼化
表15:铜催化的氧化还原活性酯的硼化;lioh对liotbu,第二配体效应
表16:铜催化的氧化还原活性酯的硼酸酯化;溶剂效果
表17:铜催化的氧化还原活性酯的硼化;催化剂和铜源
表18:铜催化的氧化还原活性酯的硼化;1,3-二羰基配体
tbubipy=化合物l2;acac=乙酰丙酮
表19:铜催化的氧化还原活性酯的硼化
表20:铜催化的氧化还原活性酯的硼化;镁源和lioh负载
表21:cu催化与ni催化的硼化的反应条件的优化和成本估算的比较
铜催化的硼化程序:优化
程序:向配有搅拌棒的15ml培养管中加入氧化还原活性酯(0.2mmol)、b2pin2(76mg,1.5eq(当量))、lioh·h2o(126mg,15eq)、cu(acac)2(10.4mg,20mol%)和mgcl2(28.5mg,1.5eq)。将该管抽空并用氩气回填3次。加入脱气的二噁烷/dmf(来自acros超干瓶,4/1,1.4ml),将所得混合物在室温以1000rpm搅拌30分钟,然后用etoac(7ml)稀释,并用饱和nh4cl(7ml)洗涤。有机相经无水na2so4干燥、蒸发并通过硅胶色谱法纯化,得到所需产物。
克规模的程序:向装配有搅拌棒的50ml烧瓶中加入氧化还原活性酯(1.2g,2.5mmol)、b2pin2(953mg,1.5eq),lioh·h2o(1.58g,15eq),cu(acac)2(130mg,20mol%)和mgcl2(356mg,1.5eq)。将烧瓶抽空并用氩气回填3次。将烧瓶中的固体搅拌2分钟,然后加入脱气的二噁烷/dmf(来自acros超干瓶,4/1,17.5ml),并将得到的混合物在室温以1000rpm搅拌30分钟,然后用et2o稀释(50ml),并依次用饱和nh4cl(30ml)和盐水(30ml)洗涤。收集有机相,用无水na2so4干燥,蒸发并通过硅胶色谱法纯化,得到硼酸化产物(625mg,60%)。
对于上面所示的己二酸底物,以0.2mmol的规模获得69%的gc收率,而以3.5mmol的规模获得55%的分离收率。下图显示了在该反应系统中获得的其他底物和收率。
阿霉素侧链类似物硼酸
实验:将起始羧酸(50mg,0.044mmol)和n-羟基苯甲酰亚胺(6.0mg,0.047mmol,1.1eq)放入装有搅拌棒的烤箱干燥培养管中。将其抽空并用氩气回填3次。通过注射器向其中加入dcm(0.5ml),产生悬浮液。在搅拌下,通过注射器加入n,n’-二异丙基碳二亚胺(9.5l,7.7mg,0.047mmol,1.1eq.)。通过tlc监测起始消耗(约1小时)后,在氮气流下吹出溶剂,并在高真空下放置2小时。之后,将cu(acac)2(11.5mg,0.044mmol,1.0eq.)、b2pin2(83.8mg,0.33mmol,7.5eq.)、lioh·h2o(55.4mg,1.32mmol,30eq.)和mgcl2(31.4mg,0.33mmol,7.5eq.)快速加入管中并重新密封。将其抽空并用氩气回填3次。将二噁烷/dmf的6∶1混合物(0.5ml)加入反应管。然后将所得混合物在室温下剧烈搅拌45分钟。然后将其用2ml饱和nh4cl水溶液淬灭。将其用etoac萃取3次。将合并的有机层用盐水冲洗并用mgso4干燥。减压除去溶剂。粗物质通过快速闪蒸色谱柱(sio2–3%dcm的meoh溶液)纯化,得到31.2mg半纯品,为灰白色固体(约50%收率)。将该物质溶于0.4ml的二噁烷中。向其中加入3ml的3mhcl(水溶液)。将得到的混合物在室温搅拌24小时。将反应物在旋转蒸发仪上浓缩。将得到的残余物溶于3ml的1:1mecn/h2o中,并通过制备型hplc(c18;h2o至mecn的梯度,各自包含0.1%的甲酸)纯化,得到2.1mg。
铜催化的硼化实施例
a:cu(acac)2(20mol%),b2pin2(15eq),二噁烷/dmf4/1;b:cu(acac)2(30mol%),b2pin2(30eq),二噁烷/dmf4/1;
c:cu(acac)2(30mol%),b2pin2(30eq),二噁烷/dmf2/1;d:cu(acac)2(30mol%),b2pin2(3.0eq),二噁烷/dmf1/2
氧化还原活性酯的实验程序和表征数据
化合物2
1,3-二氧代异二氢吲哚-2-基-2-甲基-4-苯基丁酸酯(2)
在8.75mmol规模上,按照通用程序a加入2-甲基-4-苯基丁酸。通过快速柱色谱法纯化(硅胶,1∶9的etoac∶己烷),得到2(2.31g,82%)。物理状态:无色油状物;
rf=0.60(硅胶,3:7的etoac:己烷);
1hnmr(600mhz,cdcl3):δ7.92-7.88(m,2h),7.81-7.78(m,2h),7.32-7.29(m,2h),7.27-7.25(m,2h),7.22-7.20(m,1h),2.90-2.74(m,3h),2.20-2.14(m,1h),1.96-1.90(m,1h),1.40(d,j=7.2hz,3h)ppm;
13cnmr(151mhz,cdcl3):δ172.7,162.2,141.3,134.9,129.2,128.7,128.6,126.2,124.1,36.7,35.7,33.1,17.2ppm;
hrms(esi-tof,m/z):计算值c19h18no4[m+h]+324.1230;实测值324.1230.
化合物s1
4,5,6,7-四氯-1,3-二氧代异二氢吲哚-2-基2-甲基-4-苯基丁酸(s1)
在13mmol规模下,按照通用程序a加入2-甲基-4-苯基丁酸。通过快速柱色谱法纯化(硅胶,1:10的etoac:己烷),得到黄色产物。然后将该化合物从ch2cl2/meoh中重结晶,得到s1(4.12g,69%)。
物理状态:白色固体;
m.p.=80–81℃;
rf=0.63(硅胶,1∶4的etoac∶己烷);
1hnmr(600mhz,cdcl3):δ7.32–7.28(m,2h),7.26–7.19(m,3h),2.89–2.72(m,3h),2.20–2.13(m,1h),1.95–1.88(m,1h),1.39(d,j=8.4hz,3h)ppm;
13cnmr(151mhz,cdcl3):δ172.3,157.8,141.1,141.0,130.6,128.6,126.3,124.9,36.6,35.5,33.1,17.3ppm;
hrms(esi-tof,m/z):计算值c19h14cl4no4[m+h]+459.9671;实测值459.9659.
化合物s3
1,3-二氧代异二氢吲哚-2-基3-(2-溴苯基)丙酸酯(s3)
在5.0mmol规模上,按照通用程序a加入3-(2-溴苯基)丙酸。通过快速柱色谱法纯化(硅胶,1:9的etoac:己烷),得到s3(1.63g,87%)。
物理状态:白色固体;
m.p.=158–160℃;
rf=0.36(硅胶,1:4的etoac∶己烷);
1hnmr(600mhz,cdcl3):δ7.90–7.87(m,2h),7.80–7.77(m,2h),7.56(dd,j=1.2hz,7.8hz,1h),7.34(dd,j=7.8hz,1.8hz,1h),7.28(dt,j=7.8hz,1.2hz,1h),7.12(dt,j=7.8hz,1.8hz,1h),3.21(t,j=7.2hz,2h),3.02(t,j=7.2hz,2h)ppm;
13cnmr(151mhz,cdcl3):δ168.8,162.0,138.5,134.9,133.1,130.1,129.0,128.7,127.9,124.4,124.1,31.2,31.0ppm;
hrms(esi-tof,m/z):计算值c17h13brno4[m+h]+374.0022;实测值374.0022.
化合物s4
1,3-二氧代异二氢吲哚-2-基6-溴己酸酯(s4)
在5.0mmol规模上,按照通用程序a加入6-溴己酸。通过快速柱色谱法纯化(硅胶,1:10的etoac:己烷),得到s4(1.52g,89%)。
物理状态:白色固体;
m.p.=60–62℃;
rf=0.45(硅胶,1:4的etoac∶己烷);
1hnmr(600mhz,cdcl3):δ7.89–7.86(m,2h),7.79–7.77(m,2h),3.42(t,j=7.2hz,2h),2.68(t,j=7.2hz,2h),1.94–1.89(m,2h),1.84–1.79(m,2h),1.63–1.57(m,2h)ppm;
13cnmr(151mhz,cdcl3):δ169.4,162.0,134.9,129.0,124.1,33.3,32.3,30.9,27.5,24.0ppm;
hrms(esi-tof,m/z):计算值c14h15brno4[m+h]+340.0179;实测值340.0178.
化合物s5
1-(叔丁基)5-(1,3-二氧代异二氢吲哚-2-基)(((9h-芴-9-基)甲氧基)羰基)-l谷氨酸酯(s5)
以3.0mmol的规模,按照通用程序a,用fmoc-glu-otbu进行。通过快速柱色谱法纯化(硅胶,1:3的etoac:己烷),得到s5(1.53g,89%)。
物理状态:白色泡沫;
rf=0.49(硅胶,1:4的etoac∶己烷);
1hnmr(600mhz,cdcl3):δ7.90–7.86(m,2h),7.80–7.76(m,4h),7.67–7.61(m,2h),7.42–7.38(m,2h),7.31(dt,j=7.2hz,1.2hz,2h),5.52(brd,j=7.8hz,1h),4.50(dd,j=10.8hz,7.2hz,1h),4.39–4.36(m,2h),4.23(t,j=7.2hz,1h),2.82–2.77(m,1h),2.73–2.67(m,1h),2.40–2.34(m,1h),2.15–2.09(m,1h),1.50(s,9h)ppm;
13cnmr(151mhz,cdcl3):δ170.6,169.1,162.0,156.2,143.9,141.5,134.9,129.0,127.9,127.2,125.4,125.2,124.2,120.1,83.1,67.2,53.7,47.4,28.1,28.0,27.6ppm;
hrms(esi-tof,m/z):计算值c32h30n2nao8[m+na]+593.1894;实测值593.1895;
[α]d20=+5.4(c1.0,chcl3).
化合物s6
4,5,6,7-四氯-1,3-二氧代异二氢吲哚-2-基2-(4-溴苯基)乙酸酯(s6)
在5.0mmol的规模上,按照通用程序a加入2-(4-溴苯基)乙酸。反应完成后,反应混合物通过短硅胶垫填充,并用etoac/己烷(1∶8)洗涤。浓缩滤液,并在用ch2cl2/meoh(1.52g,61%)重结晶后获得s6。
物理状态:淡黄色固体;
m.p.=212–213℃;
rf=0.57(硅胶,1:4的etoac∶己烷);
1hnmr(600mhz,dmso-d6):δ7.61–7.59(m,2h),7.37–7.35(m,2h),4.25(s,2h)ppm;
13cnmr(151mhz,dmso-d6):δ167.7,157.5,139.3,131.7,131.6,131.6,129.0,125.2,120.9,35.8ppm;
hrms(esi-tof,m/z):计算值c16h7brcl4no4[m+h]+495.8307;实测值495.8323。
化合物s7
4,5,6,7-四氯-1,3-二氧代异二氢吲哚-2-基2-甲基-3-苯基丙酸酯(s7)
在5.0mmol的规模上,按照通用程序a加入2-甲基-3-苯基丙酸。通过快速柱色谱纯化(硅胶,1:10的etoac:己烷),得到黄色产物,将其从ch2cl2/meoh中重结晶,得到s7(1.45g,65%)。
物理状态:淡黄色固体;
m.p.=127–128℃
rf=0.63(硅胶,1:4的etoac∶己烷);
1hnmr(600mhz,cdcl3):δ7.35–7.32(m,2h),7.28–7.23(m,3h),3.25(dd,j=13.8hz,6.6hz,1h),3.14–3.08(m,1h),2.82(dd,j=13.8hz,7.8hz,1h),1.34(d,j=7.2hz,3h)ppm;
13cnmr(151mhz,cdcl3):δ171.9,157.7,141.2,137.8,130.6,129.2,128.8,127.0,124.9,39.3,39.0,16.6ppm;
hrms(esi-tof,m/z):计算值c18h12cl4no4[m+h]+445.9515;实测值445.9516.
化合物s8
1,3-二氧代异二氢吲哚-2-基2-苯基丙酸酯(s8)
在5.0mmol的规模上,按照通用程序a加入2-苯基丙酸。通过快速柱色谱法纯化(硅胶,1:10的etoac:己烷),得到s8(1.19g,81%)。
物理状态:无色油状物;
rf=0.21(硅胶,1:4的etoac∶己烷);
1hnmr(600mhz,cdcl3):δ7.87–7.85(m,2h),7.79–7.76(m,2h),7.43–7.39(m,4h),7.34–7.31(m,1h),4.13(q,j=7.2hz,1h),1.68(d,j=7.2hz,3h)pm;
13cnmr(151mhz,cdcl3):δ170.9,162.0,138.5,134.9,129.1,129.1,127.9,127.7,124.1,43.1,19.1ppm;
hrms(esi-tof,m/z):计算值c17h14no4[m+h]+296.0917;实测值296.0920.
化合物s9
1,3-二氧代异二氢吲哚-2-基2,2-二苯基乙酸酯(s9)
在1.5mmol的规模上,按照通用程序a加入二苯乙酸。通过快速柱色谱法纯化(硅胶,1∶4的etoac∶己烷),得到s9(0.46g,86%)。
物理状态:白色固体;
m.p.=135–137℃;
rf=0.33(硅胶,1:4的etoac∶己烷)
1hnmr(600mhz,cdcl3):δ7.89–7.86(m,2h),7.80-7.77(m,2h),7.42–7.37(m,8h),7.34–7.31(m,2h),5.42(s,1h)ppm;
13cnmr(151mhz,cdcl3):δ169.2,162.0,136.9,134.9,129.1,129.0,128.9,128.0,124.1,54.2ppm;
hrms(esi-tof,m/z):计算值c22h16no4[m+h]+358.1074;实测值358.1078.
化合物s10
1,3-二氧代异二氢吲哚-2-基-双环[2.2.1]庚烷-2-羧酸酯(s10)
在3.0mmol的规模上,按照通用程序a加入双环[2.2.1]庚烷-2-羧酸(内和外异构体的混合物)。通过快速柱色谱法(硅胶,1:19至1:9的etoac:己烷)纯化,得到s10(0.75g,88%),为外/内异构体的混合物。
物理状态:白色固体;
rf=0.41(硅胶,1:4的etoac∶己烷);
1hnmr(600mhz,cdcl3):δ7.90–7.86(m,2h),7.80–7.77(m,2h),3.15–3.11(m,0.82h),2.81(brs,0.82h),2.77(brd,j=4.2hz,0.18h),2.70(dd,j=9.6hz,6.0hz,0.18h),2.38(brt,j=4.2hz,0.18h),2.35-2.33(br,m,0.82h),2.00-1.96(m,0.18h),1.86–1.81(m,0.82h),1.74–1.70(m,0.82h),1.63–1.67(m,3.28h),1.51–1.44(m,1.64h),1.38–1.25(m,1.26h)ppm;
13cnmr(151mhz,cdcl3):δ171.5,162.3,134.8,129.2,124.0,43.4,41.0,40.5,37.0,32.7,29.0,24.9ppm(主要异构体);172.3,162.3,134.8,129.2,124.0,43.7,41.7,36.7,36.2,34.6,29.5,28.6ppm(次要异构体).
hrms(esi-tof,m/z):计算值c16h16no4[m+h]+286.1074;实测值286.1071.
化合物s11
4,5,6,7-四氯-1,3-二氧代异二氢吲哚-2-基反式-2-苯基环丙烷-1-羧酸酯(s11)
在3.0mmol的规模上,按照通用程序a加入反式-2-苯基环丙烷-1-羧酸。起始材料完全消耗(tlc)后,将反应混合物通过硅藻土过滤,用ch2cl2(100ml)洗涤,并在减压下浓缩。粗产物通过结晶(ch2cl2/meoh)纯化,得到s11(949mg,71%)。
物理状态:淡黄色针状
m.p.=203–205℃;
rf=0.48((硅胶,1:9的etoac∶己烷);
1hnmr(600mhz,cdcl3):δ7.34–7.31(m,2h),7.28–7.25(m,1h),7.18–7.16(m,2h),2.80–2.77(m,1h),2.22–2.19(m,1h),1.84(dt,j=10.2hz,5.4hz,1h),1.69–1.66(m,1h)ppm;
13cnmr(151mhz,cdcl3):δ169.4,157.7,141.2,138.3,130.6,128.8,127.4,126.5,124.8,28.8,21.0,18.6ppm;
hrms(esi-tof,m/z):计算值c18h10cl4no4[m+h]+443.9358;实测值443.9356.
化合物s12
1,3-二氧代异二氢吲哚-2-基2,2-二甲基-3-苯基丙酸酯(s12)
在5.0mmol规模上,按照通用程序a加入2,2-二甲基-3-苯基丙酸。通过快速柱色谱法纯化(硅胶,1:10的etoac:己烷),得到s12(1.36g,84%)。
物理状态:白色固体;
m.p.=70–72℃;
rf=0.45(硅胶,1:4的etoac∶己烷);
1hnmr(600mhz,cdcl3):δ7.92–7.88(m,2h),7.81–7.78(m,2h),7.36–7.31(m,4h),7.29–7.26(m,1h),3.10(s,2h),1.40(s,6h)ppm;
13cnmr(151mhz,cdcl3):δ173.7,162.2,136.5,134.8,130.6,129.1,128.3,127.0,124.0,45.8,43.3,25.0ppm;
hrms(esi-tof,m/z):计算值c19h18no4[m+h]+324.1230;实测值324.1232.
化合物s13
1,3-二氧代异二氢吲哚-2-基1-苯基环己烷-1-甲酸酯(s13)
在5.0mmol规模上,按照通用程序a加入2-苯基丙酸。通过快速柱色谱法纯化(硅胶,1:9的etoac:己烷),得到s13(1.64g,81%)。
物理状态:白色固体;
m.p.=108–109℃;
rf=0.39(硅胶,1:4的etoac∶己烷);
1hnmr(600mhz,cdcl3):δ7.87–7.84(m,2h),7.78–7.75(m,2h),7.54–7.52(m,2h),7.44–7.41(m,2h),7.34–7.31(m,1h),2.64(brd,j=13.2hz,2h),1.89–1.73(m,7h),1.37–1.30(m,1h)ppm;
13cnmr(151mhz,cdcl3):δ171.8,162.2,142.3,134.8,129.2,128.9,127.7,126.1,124.0,51.3,35.5,25.6,23.6ppm;
hrms(esi-tof,m/z):计算值c21h20no4[m+h]+350.1387;实测值350.1387.
化合物s14
1,3-二氧代异二氢吲哚-2-基2-甲基-2-苯基丙酸酯(s14)
在5.0mmol的规模上,按照通用程序a加入2-甲基-2-苯基丙酸。通过快速柱色谱法纯化(硅胶,1:8的etoac:己烷),得到s14(1.32g,85%)。
物理状态:白色固体;
m.p.=73–74℃;
rf=0.36(硅胶,1:4的etoac∶己烷);
1hnmr(600mhz,cdcl3):δ7.88–7.85(m,2h),7.79–7.75(m,2h),7.51–7.49(m,2h),7.44–7.41(m,2h),7.34–7.31(m,1h),1.79(s,6h)ppm;
13cnmr(151mhz,cdcl3):δ173.4,162.1,142.7,134.8,129.1,128.8,127.5,125.9,124.046.5,27.0ppm;
hrms(esi-tof,m/z):计算值c18h16no4[m+h]+310.1074;实测值310.1082.
化合物s15
1,3-二氧代异二氢吲哚-2-基2-(1-(((叔丁氧羰基)氨基)甲基)环己基)乙酸酯(s15)
在0.44mmol的规模上,按照通用程序a加入boc保护的加巴喷丁(gabapentin)。通过快速柱色谱法纯化(硅胶,1:5的etoac:己烷),得到s15(165mg,85%)。
物理状态:白色固体;
m.p.=76–79℃;
rf=0.32(硅胶,1:5的etoac∶己烷);
1hnmr(600mhz,cdcl3):δ7.90–7.87(m,2h),7.82–7.77(m,2h),4.95(brt,j=7.2hz,1h),3.38(d,j=6.6hz,2h),2.63(s,2h),1.65–1.43(m,10h),1.44(s,9h)ppm;
13cnmr(151mhz,cdcl3):δ168.3,162.1,156.6,135.0,129.0,124.2,79.3,46.9,39.1,37.8,33.9,28.5,26.0,21.6ppm;
hrms(esi-tof,m/z):计算值c22h29n2o6[m+h]+417.2020;实测值417.2022.
化合物s16
1,3-二氧代异二氢吲哚-2-基2-(4-异丁基苯基)丙酸酯(s16)
在5.0mmol的规模上,按照通用程序a加入布洛芬。通过快速柱色谱法纯化(硅胶,1:9的etoac:己烷),得到s16(1.48g,84%)。
物理状态:无色油状;
rf=0.42(硅胶,1:4的etoac∶己烷);
1hnmr(600mhz,cdcl3):δ7.87–7.85(m,2h),7.79–7.76(m,2h),7.31(d,j=8.4hz,2h),7.17(d,j=8.4hz,2h),4.10(q,j=7.2hz,1h),2.48(d,j=7.2hz,2h),1.91–1.84(m,1h),1.67(d,j=7.2hz,3h),0.91(d,j=6.6hz,6h)ppm;
13cnmr(151mhz,cdcl3):δ171.1,162.0,141.4,135.7,134.8,129.8,129.1,127.4,124.0,45.2,42.7,30.3,22.5,19.2ppm;
hrms(esi-tof,m/z):计算值c21h22no4[m+h]+352.1543;实测值352.1544.
化合物s17
1,3-二氧代异二氢吲哚-2-基5-(2,5-二甲基苯氧基)-2,2-二甲基戊酸酯(s17)
在1.0mmol的规模上,按照通用程序a加入吉非贝齐(gemfibrozil)。通过快速柱色谱法纯化(硅胶,1:25的etoac:己烷),得到s17(0.33g,84%)。
物理状态:白色固体;
m.p.=65–67℃;
rf=0.50(硅胶,1:4的etoac∶己烷)
1hnmr(600mhz,cdcl3):δ7.90–7.87(m,2h),7.80–7.77(m,2h),7.01(d,j=7.8hz,1h),6.67(d,j=7.8hz,1h),6.66(s,1h),4.02(t,j=6.0hz,2h),2.32(s,3h),2.20(s,3h),1.95–2.00(m,4h),1.46(s,6h)ppm;
13cnmr(151mhz,cdcl3):δ173.9,162.2,157.1,136.6,134.8,130.4,129.2,124.0,123.8,120.8,112.1,67.9,42.1,37.5,31.7,25.3,25.1,21.5,15.9ppm;
hrms(esi-tof,m/z):计算值c23h26no5[m+h]+396.1805;实测值396.1803.
化合物s18
1,3-二氧代异二氢吲哚-2-基-2-(6-甲氧基萘-2-基)丙酸酯(s18)
在5.0mmol的规模上,按照常规方法a用萘普生(naproxen)。通过快速柱色谱法纯化(硅胶,1:7的etoac:己烷),得到s18(1.65g,88%)。
物理状态:白色固体;
m.p.=110–111℃;
rf=0.53(硅胶,2:3etoac:己烷);
1hnmr(600mhz,cdcl3):δ7.86(brs,2h),7.80–7.75(m,5h),7.49(dd,j=8.4hz,1.8hz,1h),7.17(dd,j=8.4hz,2.4hz,1h),7.14(d,j=2.4hz,1h),4.26(q,j=7.2hz,1h),3.92(s,3h),1.75(d,j=7.2hz,3h)ppm;
13cnmr(151mhz,cdcl3):δ171.1,162.0,158.0,134.9,134.1,133.6,129.6,129.1,127.7,126.5,126.0,124.1,119.3,105.8,55.5,43.1,19.2ppm;
hrms(esi-tof,m/z):计算值c22h18no5[m+h]+376.1179;实测值376.1183.
化合物s19
双(1,3-二氧代异二氢吲哚-2-基)壬二酸酯(s19)
在5.0mmol的规模上,按照通用程序a加入壬二酸(5.0mmol,1.0当量(equiv)),nhpi(10mmol,2.0当量),dic(11mmol,2.2当量)和dmap(1mmol,0.2当量)。通过快速柱色谱法纯化(硅胶,1:10的etoac:ch2cl2),得到s19(1.52g,64%)。
物理状态:白色固体;
m.p.=103–105℃;
rf=0.55(硅胶,1:1的etoac∶己烷);
1hnmr(600mhz,cdcl3):δ7.90–7.87(m,4h),7.81–7.78(m,4h),2.69(t,j=4.8hz,4h),1.85–1.80(m,4h),1.53–1.48(m,4h),1.45–1.42(m,2h)ppm;
13cnmr(151mhz,cdcl3):δ169.1,161.5,134.3,128.5,123.5,30.5,28.1,28.0,24.1ppm;
hrms(esi-tof,m/z):计算值c25h23n2o8[m+h]+479.1449;实测值479.1451.
化合物s20
1,3-二氧代异二氢吲哚-2-基4-(4-(双(2-氯乙基)氨基)苯基)丁酸酯(s20)
在1.0mmol的规模上,按照通用程序a加入苯丁酸氮芥。通过快速柱色谱法纯化(硅胶,1∶4的etoac∶己烷),得到s20(431mg,96%)。
物理状态:黄色油状;
rf=0.23(硅胶,1:4的etoac∶己烷);
1hnmr(600mhz,cdcl3):δ7.91–7.87(m,2h),7.81–7.77(m,2h),7.12(d,j=8.5hz,2h),6.65(d,j=9.0hz,2h),3.66(abt,j=6.7hz,4h),3.63(bat,j=6.7hz,4h),2.67(dt,j=7.5hz,16hz,4h),2.09–2.02(m,2h)ppm;
13cnmr(151mhz,cdcl3):δ169.6,162.1,144.7,134.9,130.0,129.1,124.1,112.4,53.8,40.7,33.6,30.3,26.6ppm;
hrms(esi-tof,m/z):计算值c22h23cl2n2o4[m+h]+449.1029;实测值449.1009.
化合物s21
1,3-二氧代异二氢吲哚-2-基2-(3-苯甲酰基苯基)丙酸酯(s21)
在5.0mmol的规模上,按照通用程序a加入酮洛芬(ketoprofen)。通过快速柱色谱法纯化(硅胶,1:3的etoac:己烷),得到s21(1.91g,96%)。
物理状态:白色固体;
m.p.=118–120℃;
rf=0.45(硅胶,1:2的etoac∶己烷);
1hnmr(600mhz,cdcl3):δ7.88–7.84(m,5h),7.80–7.76(m,3h),7.67(dt,j=8.4hz,1.2hz,1h),7.61–7.58(m,1h),7.55–7.48(m,3h),4.20(q,j=7.2hz,1h),1.71(d,j=7.2hz,3h)ppm;
13cnmr(151mhz,cdcl3):δ196.4,170.6,161.9,138.7,138.4,137.5,134.9,132.7,131.7,130.3,129.8,129.5,129.1,129.1,128.5,124.1,43.0,19.0ppm;
hrms(esi-tof,m/z):计算值c24h18no5[m+h]+400.1179;实测值400.1181.
化合物s22
1,3-二氧代异二氢吲哚-2-基2-(3-苯氧基苯基)丙酸酯(s22)
在5.0mmol的规模上,按照通用程序a加入芬诺洛芬(fenoprofen)。通过快速柱色谱法纯化(硅胶,1:8的etoac:己烷),得到s22(1.83g,94%)。
物理状态:无色油状;
rf=0.50(硅胶,1:4的etoac∶己烷)
1hnmr(600mhz,cdcl3):δ7.89–7.85(m,2h),7.79–7.76(m,2h),7.37–7.33(m,3h),7.16(dt,j=7.8hz,1.2hz,1h),7.13–7.10(m,1h),7.09(t,j=2.4hz,1h),7.07-7.04(m,2h),6.95(ddd,j=7.8hz,2.4hz,0.6hz,1h),4.09(q,j=7.2hz,1h).1.67(d,j=7.2hz,3h)ppm;
13cnmr(151mhz,cdcl3):δ170.6,161.9,157.8,157.1,140.3,134.9,130.3,129.9,129.1,124.1,123.5,122.5,119.2,118.4,118.2,42.9,19.0ppm;
hrms(esi-tof,m/z):计算值c23h18no5[m+h]+388.1179;实测值388.1178.
化合物s23
1,3-二氧代异二氢吲哚-2-基2-((4r,6r)-6-(2-(2-(4-氟苯基)-5-异丙基-3-苯基-4-(苯基氨基甲酰基)-1h-吡咯-1-基)乙基)-2,2-二甲基-1,3-二噁烷-4-基)乙酸酯(s23)
在0.5mmol的规模上,按照通用程序a加入阿托伐他汀的缩酮酯。通过快速柱色谱法纯化(硅胶,1∶2的etoac∶己烷),得到s23(0.35g,95%)。
物理状态:黄色泡沫;
rf=0.35(硅胶,1:2的etoac∶己烷);
1hnmr(600mhz,cdcl3):δ7.90–7.87(m,2h),7.81–7.77(m,2h),7.21–7.15(m,9h),7.08(d,j=8.4hz,2h),7.02–6.97(m,3h),6.88(brs,1h),4.33–4.28(m,1h),4.14–4.07(m,1h),3.89–3.84(m,1h),3.75–3.71(m,1h),3.61–3.56(m,1h),2.85(dd,j=15.6hz,6.6hz,1h),2.69(dd,j=15.0hz,6.6hz,1h),1.76–1.70(m,2h),1.55–1.53(m,7h),1.40(s,3h),1.35(s,3h),1.18(dd,j=12.0hz,5.4hz,1h)ppm;
13cnmr(151mhz,cdcl3):δ166.9,164.9,162.4(d,j=247.8hz),161.9,141.6,138.5,134.9,134.8,133.3(d,j=8.0hz),130.6,129.0,128.9,128.8,128.4,128.3(d,j=3.6hz),126.7,124.1,123.6,121.9,119.7,115.5(d,j=21.3hz),99.2,66.4,65.6,40.9,38.4,38.1,35.8,29.9,26.2,21.9,21.7,19.7ppm;
19fnmr(376mhz,cdcl3):δ-113.91ppm;
hrms(esi-tof,m/z):计算值c44h43fn3o7[m+h]+744.3080;实测值744.3061.
[α]d20=+25.1(c1.0,chcl3).
化合物s24
1,3-二氧代异二氢吲哚-2-基(2s,4as,6as,6br,8ar,10s,12as,12br,14br)-10-乙酰氧基-2,4a,6a,6b,9,9,12a-七甲基-13-氧代-1,2,3,4,4a,5,6,6a,6b,7,8,8a,9,10,11,12,12a,12b,13,14b-二十碳氢苉-2-羧酸酯(s24)
在1.0mmol的规模上,按照通用程序a加入甘草次酸乙酰酯(acetylenoxolone)。通过快速柱色谱法(硅胶,1:5的etoac:己烷)纯化,得到s24(0.49g,75%)。
物理状态:白色固体;
m.p.=264℃;
rf=0.57(硅胶,2:3的etoac∶己烷);
1hnmr(600mhz,cdcl3):δ7.89–7.86(m,2h),7.80–7.77(m,2h),5.76(s,1h),4.51(dd,j=11.8,4.6hz,1h),2.79(dt,j=13.7,3.7hz,1h),2.45(ddd,j=13.7,4.3,1.7hz,1h),2.35(s,1h),2.15–2.11(m,1h),2.11–2.00(m,2h),2.04(s,3h),1.86(td,j=13.7,4.7hz,1h),1.79(t,j=13.7hz,1h),1.74–1.55(m,4h),1.51–1.40(m,4h),1.43(s,3h),1.37(s,3h),1.20(ddd,j=13.8,4.6,2.4hz,1h),1.15(s,3h),1.14(s,3h),1.10–1.01(m,3h),0.90(s,3h),0.87(s,6h),0.80(dd,j=11.9,1.8hz,1h)ppm;
13cnmr(151mhz,cdcl3):δ200.0,172.7,171.1,168.5,162.2,134.8,129.2,129.0,124.0,80.8,61.9,55.2,47.9,45.5,44.0,43.3,41.3,38.9,38.2,37.4,37.1,32.9,32.0,31.6,28.5,28.2,28.1,26.6,26.6,23.7,23.4,21.5,18.8,17.5,16.8,16.6ppm;
hrms(esi-tof,m/z):计算值c40h52no7[m+h]+658.3738;实测值658.3736;
[α]d20=+191.0(c1.0,chcl3).
化合物s25
1,3-二氧代异二氢吲哚-2-基(e)-6-(4-羟基-6-甲氧基-7-甲基-3-氧代-1,3-二氢异苯并呋喃-5-基)-4-甲基己-4-烯酸酯(s25)
在1.0mmol的规模上,按照通用程序a加入霉酚酸(mycophenolicacid)。通过快速柱色谱法纯化(硅胶,1∶4的etoac∶己烷),得到s25(0.36g,78%)。
物理状态:白色固体;
m.p.=126–128℃;
rf=0.40(硅胶,1:3的etoac∶己烷);
1hnmr(600mhz,cdcl3):δ7.88–7.85(m,2h),7.79–7.77(m,2h),7.68(s,1h),5.34(t,j=7.2hz,1h),5.19(s,2h),3.77(s,3h),3.42(d,j=6.6hz,2h),2.76(t,j=7.8hz,2h),2.45(t,j=7.8hz,2h),2.15(s,3h),1.85(s,3h)ppm;
13cnmr(151mhz,cdcl3):δ173.0,169.3,163.8,162.0,153.8,144.2,134.9,133.1,129.0,124.1,123.9,122.0,116.8,106.5,70.2,61.2,34.1,29.9,22.8,16.2,11.7ppm;
hrms(esi-tof,m/z):计算值c25h24no8[m+h]+466.1496;实测值466.1499。
已经报道了以下rae的制备和光谱数据。i22-26
硼化产物的实验步骤和表征数据
化合物3
4,4,5,5-四甲基-2-(4-苯基丁-2-基)-1,3,2-二氧杂硼杂环戊烷(4,4,5,5-tetramethyl-2-(4-phenylbutan-2-yl)-1,3,2-dioxaborolane)(3)
在0.2mmol的规模上,按照通用程序b加入nhpi酯(2)和悬浮液b(dmf中的nicl2·6h2o/di-meobipy)。通过快速柱色谱法纯化(硅胶,己烷至1:35的et2o:己烷),得到3(32.7mg,63%)。
物理状态:无色油状;
rf=0.49(硅胶,1:12的etoac∶己烷);
1hnmr(600mhz,cdcl3):δ7.28–7.25(m,2h),7.20–7.14(m,3h),2.66–2.58(m,2h),1.82–1.76(m,1h),1.62–1.57(m,1h),1.25(s,12h),1.10–1.05(m,1h),1.02(d,j=7.2hz,3h)ppm;
13cnmr(151mhz,cdcl3):δ143.2,128.6,128.3,125.6,83.0,35.5,35.4,25.0,24.9,15.7ppm;
光谱数据与文献报道相符2
化合物4
4,4,5,5-四甲基-2-(4-苯基丁基)-1,3,2-二氧杂硼杂环戊烷(4)
在0.2mmol的规模上,按照通用程序b加入nhpi酯(s2)和溶液b(dmf中的nicl2·6h2o/di-meobipy)。通过快速柱色谱法纯化(硅胶,己烷至1∶30的et2o∶己烷),得到4(34.0mg,65%)。
物理状态:无色油状;
rf=0.50(硅胶,1:12etoac:己烷);
1hnmr(600mhz,cdcl3):δ7.28–7.25(m,2h),7.18–7.15(m,3h),2.61(t,j=7.8hz,2h),1.66–1.61(m,2h),1.50–1.45(m,2h),1.24(s,12h),0.82(t,j=7.8hz,2h)ppm;
13cnmr(151mhz,cdcl3):δ143.1,128.5,128.3,125.6,83.0,35.9,34.3,25.0,23.9ppm;
光谱数据与文献报道相符。3
化合物5
5-(4,4,5,5-四甲基-1,3,2-二氧杂硼杂环戊烷-2-基)戊酸甲酯(5)
在0.2mmol的规模上,按照通用程序b加入nhpi酯(s26)和溶液b(在dmf中的nicl2·6h2o/di-meobipy)。通过快速柱色谱法纯化(硅胶,己烷至1:100的ch2cl2:己烷),得到5(25.2mg,52%)。
物理状态:无色油状;
rf=0.55(硅胶,1:6的etoac∶己烷);
1hnmr(600mhz,cdcl3):δ3.64(s,3h),2.29(t,j=7.2hz,2h),1.64–1.59(m,2h),1.45–1.40(m,2h),1.23(s,12h),0.78(t,j=7.8hz,2h)ppm;
13cnmr(151mhz,cdcl3):δ174.4,83.1,51.6,34.1,27.7,25.0,23.8ppm;
hrms(esi-tof,m/z):计算值c12h24bo4[m+h]+243.1762;实测值243.1765.
化合物6
2-(2-溴苯乙基)-4,4,5,5-四甲基-1,3,2-二氧杂硼杂环戊烷(6)
在0.2mmol的规模上,按照通用程序b加入nhpi酯(s3)和溶液b(在dmf中的nicl2·6h2o/di-meobipy)。通过快速柱色谱法(硅胶,己烷至1:30的et2o:己烷)纯化,得到6(34.3mg,55%)。
物理状态:无色油状;
rf=0.55(硅胶,1:12的etoac∶己烷);
1hnmr(600mhz,cdcl3):δ7.50(d,j=7.8hz,1h),7.27(d,j=7.8hz,1h),7.21(t,j=7.8hz,1h),7.02(t,j=7.8hz,1h),2.84(t,j=7.8hz,2h),1.24(s,12h),1.15
(t,j=7.8hz,2h)ppm;
13cnmr(151mhz,cdcl3):δ143.7,132.8,129.9,127.4,127.4,124.5,83.32,30.6,25.0ppm;
hrms(esi-tof,m/z):计算值c14h21bbro2[m+h]+313.0798;实测值313.0799.
化合物7
2-(5-溴戊基)-4,4,5,5-四甲基-1,3,2-二氧杂硼杂环戊烷(7)
在0.2mmol的规模上,按照通用程序b加入nhpi酯(s4)和溶液b(在dmf中的nicl2·6h2o/di-meobipy)。通过快速柱色谱法纯化(硅胶,己烷至1:30的et2o:己烷),得到7(36.0mg,65%)。
物理状态:无色油状;
rf=0.55(硅胶,1:12的etoac∶己烷)
1hnmr(600mhz,cdcl3):δ3.40(t,j=7.2hz,2h),1.88–1.83(m,2h),1.45–1.42(m,4h),1.24(s,12h),0.80–0.77(m,2h)ppm;
13cnmr(151mhz,cdcl3):δ83.1,34.2,32.8,31.0,25.0,23.4ppm;
hrms(esi-tof,m/z):计算值c11h23bbro2[m+h]+277.0969;实测值277.0968.
化合物8
2-((((9h-芴-9-基)甲氧基)羟基)氨基)4-(4,4,5,5-四甲基-1,3,2-二氧杂硼杂环戊烷-2-基)丁酸叔丁酯(8)
在0.2mmol的规模上,按照通用程序b加入nhpi酯(s5)和溶液b(在dmf中的nicl2·6h2o/di-meobipy)。通过快速柱色谱法纯化(硅胶,1:12的etoac:己烷至1:6的etoac:己烷至1:4的etoac:己烷),得到8(37.6mg,37%)。
物理状态:无色油状;
rf=0.40(硅胶,1:4的etoac∶己烷)
1hnmr(600mhz,cdcl3):δ7.76(d,j=7.2hz,2h),7.61(d,j=7.2hz,2h),7.41–7.38(m,2h),7.33–7.30(m,2h),5.53(d,j=8.4hz,1h),4.34–4.24(m,2h),4.23–4.19(m,2h),1.97–1.91(m,1h),1.84–1.78(m,1h),1.47(s,9h),1.23(s,12h),0.89–0.78(m,2h)ppm;
13cnmr(151mhz,cdcl3):δ171.8,156.2,144.2,144.1,141.4,127.8,127.2,125.3,120.1,83.5,81.9,67.0,56.1,47.4,28.2,27.0,25.0,24.9ppm;
hrms(esi-tof,m/z):计算值c29h38bnnao6[m+na]+530.2684;实测值530.2685;
[α]d20=+2.3(c0.35,chcl3).
化合物9
2-(4-溴苄基)-4,4,5,5-四甲基-1,3,2-二氧杂硼杂环戊烷(9)
在0.2mmol的规模上,按照通用程序c加入tcnhpi酯(s6)和悬浮液c(在thf中的nicl2·6h2o/di-tbubipy)。通过快速柱色谱法纯化(硅胶,1∶40至1∶20的et2o∶己烷),得到9(30.5mg,51%)。
物理状态:无色油状;
rf=0.30(硅胶,1:19的etoac∶己烷);
1hnmr(600mhz,cdcl3):δ7.35–7.33(m,2h),7.06–7.04(m,2h),2.23(s,2h),1.23(s,12h)ppm;
13cnmr(151mhz,cdcl3):δ137.8,131.4,130.9,118.7,83.7,24.9ppm;
光谱数据与文献报道相符。4
化合物10
4,4,5,5-四甲基-2-(1-苯基丙-2-基)-1,3,2-二氧杂硼杂环戊烷(10)
在0.2mmol的规模上,按照通用程序c加入tcnhpi酯(s7)和悬浮液c(在thf中的nicl2·6h2o/di-tbubipy)。通过快速柱色谱法纯化(硅胶,己烷至1∶35的et2o∶己烷),得到10(33.1mg,67%)。
物理状态:无色油状;
rf=0.53(硅胶,1:12的etoac∶己烷);
1hnmr(600mhz,cdcl3):δ7.26–7.13(m,5h),2.81(dd,j=13.8hz,7.8hz,1h),2.54(dd,j=13.8hz,7.8hz,1h),1.41–1.34(m,1h),1.19(s,6h),1.18(s,6h),0.97(d,j=7.8hz,3h)ppm;
13cnmr(151mhz,cdcl3):δ142.5,129.0,128.1,125.7,83.1,39.1,24.9,15.3ppm;
光谱数据与文献报道相符。5
化合物11
4,4,5,5-四甲基-2-(1-苯乙基)-1,3,2-二氧杂硼杂环戊烷(11)
在0.2mmol的规模上,按照通用程序c加入nhpi酯(s8)、mgbr2·oet2(52mg,0.2mmol,1当量)和悬浮液a(在thf中的nicl2·6h2o/di-meobipy)。通过快速柱色谱法纯化(硅胶,己烷至1∶30的et2o∶己烷),得到11(33.8mg,73%)。
物理状态:无色油状;
rf=0.33(硅胶,1:19的etoac∶己烷);
1hnmr(600mhz,cdcl3):δ7.27–7.21(m,4h),7.15–7.12(m,1h),2.44(q,j=7.8hz,1h),1.33(d,j=7.8hz,3h),1.21(s,6h),1.20(s,6h)ppm;
13cnmr(151mhz,cdcl3):δ145.1,128.4,127.9,125.2,83.4,24.8,24.7,17.2ppm;
光谱数据与文献报道相符。2
化合物12
2-二苯甲基-4,4,5,5-四甲基-1,3,2-二氧杂硼杂环戊烷(12)
在0.2mmol的规模上,按照通用程序c加入nhpi酯(s9)和悬浮液a(在thf中的nicl2·6h2o/di-meobipy)。通过快速柱色谱法纯化(硅胶,己烷至1∶30的et2o∶己烷),得到12(41.0mg,69%)。
物理状态:无色油状;
rf=0.41(硅胶,1:9的etoac∶己烷);
1hnmr(600mhz,cdcl3):δ7.30–7.25(m,8h),7.19–7.15(m,2h),3.88(s,1h),1.24(s,12h)ppm;
13cnmr(151mhz,cdcl3):δ142.2,129.2,128.5,125.7,83.9,24.7ppm;
光谱数据与文献报道相符。6
化合物13
2-(4,4-二氟环己基)-4,4,5,5-四甲基-1,3,2-二氧杂硼杂环戊烷(13)
在0.2mmol的规模上,按照通用程序c加入tcnhpi酯(s27)和悬浮液c(在thf中的nicl2·6h2o/di-tbubipy)。通过快速柱色谱纯化(硅胶,己烷至1:45的et2o:己烷),得到13(23.0mg,47%)。
物理状态:无色油状;
rf=0.45(硅胶,1:9的etoac∶己烷);
1hnmr(600mhz,cdcl3):δ2.02–1.95(m,2h),1.82-1.78(m,2h),1.75–1.58(m,4h),1.23(s,12h),1.00–0.96(m,1h)ppm;
13cnmr(151mhz,cdcl3):δ123.9(t,j=239.9hz),83.4,34.5(t,j=23.3hz),24.9,24.4(t,j=4.6hz)ppm;
hrms(esi-tof,m/z):无法获得此化合物的高分辨率质谱数据。
化合物14
2-(庚-3-基)-4,4,5,5-四甲基-1,3,2-二氧杂硼杂环戊烷(14)
在0.2mmol的规模上,按照通用程序c加入tcnhpi酯(s26)和悬浮液c(在thf中的nicl2·6h2o/di-tbubipy)。通过快速柱色谱法(硅胶,己烷至1:40的et2o:己烷)纯化,得到14(25.6mg,57%)。
物理状态:无色油状;
rf=0.42(硅胶,1:19的etoac∶己烷);
1hnmr(600mhz,cdcl3):δ1.45–1.22(m,20h),0.90–0.86(m,7h)ppm;
13cnmr(151mhz,cdcl3):δ82.9,31.7,30.8,25.0,24.4,23.1,14.3,13.9ppm;
光谱数据与文献报道相符。7
化合物15
2-(4,4,5,5-四甲基-1,3,2-二氧杂硼杂环戊烷-2-基)吡咯烷-1-羧酸叔丁酯(15)
在0.2mmol的规模上,按照通用程序c加入nhpi酯(s29)和悬浮液c(在thf中的nicl2·6h2o/di-tbubipy)。通过快速柱色谱法(第一快速柱色谱法:使硅胶失活,己烷至1:9的etoac:己烷)进行纯化;第二快速柱色谱法(减活的硅胶,ch2cl2),得到15(39.2mg,66%)。
物理状态:无色油状;
rf=0.45(硅胶,1:4的etoac∶己烷);
1hnmr(600mhz,cdcl3):δ3.42–2.99(m,3h),2.09–1.65(m,4h),1.43(s,9h),1.26–1.22(m,12h)ppm;
13cnmr(151mhz,cdcl3):δ155.1,83.6,79.1,46.1,28.7,27.9,27.325.2,25.0,24.6ppm;
hrms(esi-tof,m/z):计算值c15h29bno4[m+h]+298.2184;实测值298.2179;
[α]d20=0(c0.3,chcl3).
化合物16
2-(双环[2.2.1]庚-2-基)-4,4,5,5-四甲基-1,3,2-二氧杂硼杂环戊烷(16)
在0.2mmol的规模上,按照通用程序b加入nhpi酯(s10)和溶液b(在dmf中的nicl2·6h2o/di-meobipy)。通过快速柱色谱法纯化(硅胶,己烷至1:40的et2o:己烷至1:20的et2o:己烷),得到16(24.4mg,55%),为外型/内型异构体的混合物。
物理状态:无色油状;
rf=0.38(硅胶,1:19的etoac∶己烷);
1hnmr(600mhz,cdcl3):δ2.28–2.27(m,1h),2.22–2.21(m,1h),1.56–1.44(m,3h),1.37–1.33(m,1h),1.26–1.21(m,14h),1.20–1.14(m,2h),0.89–0.86(m,1h)ppm;
13cnmr(151mhz,cdcl3):δ82.9,38.9,38.3,36.8,32.4,32.3,29.4,24.9ppm(外型(exo));83.0,41.1,39.1,37.2,32.3,30.0,28.0,25.1,25.0ppm(内型(endo));
光谱数据与文献报道相符。8
化合物17
2-金刚烷-2-基-4,4,5,5-四甲基-1,3,2-二氧杂硼杂环戊烷(17)
在0.2mmol的规模上,按照通用程序c加入nhpi酯(s28)和悬浮液a(在thf中的nicl2·6h2o/di-meobipy)。通过快速柱色谱法纯化(硅胶,己烷至1:30的et2o:己烷),得到17(30.9mg,59%)。
物理状态:无色油状;
rf=0.55(硅胶,1:9的etoac∶己烷);
1hnmr(600mhz,cdcl3):δ2.06–2.04(m,2h),1.90–1.67(m,12h),1.37-1.35(m,1h),1.25(s,12h)ppm;
13cnmr(151mhz,cdcl3):δ82.9,39.5,37.9,36.4,29.5,28.4,28.3,25.0ppm;
光谱数据与文献报道相符。2
化合物18
反式-4,4,5,5-四甲基-2-(2-苯基环丙基)-1,3,2-二氧杂硼杂环戊烷(18)
在0.2mmol的规模上,按照通用程序b加入tcnhpi酯(s11)和溶液d(在dmf中的nicl2·6h2o/di-tbubipy)。通过快速柱色谱法纯化(硅胶,己烷至1∶30的et2o∶己烷),得到18(11.3mg,23%,dr>20∶1)
物理状态:无色油状;
rf=0.48(硅胶,1:9的etoac∶己烷);
1hnmr(600mhz,cdcl3):δ7.25–7.22(m,2h),7.15–7.11(m,1h),7.09–7.06(m,2h),2.10(dt,j=7.8hz,5.4hz,1h),1.25(s,6h),1.24(s,6h),1.17–1.14(m,1h),1.02–0.99(m,1h),0.32–0.29(m,1h)ppm;
13cnmr(151mhz,cdcl3):δ143.5,128.4,125.8,125.7,83.3,24.9,24.8,22.0,15.2ppm;
hrms(esi-tof,m/z):计算值c15h22bo2[m+h]+245.1707;实测值245.1714.
化合物19
4,4,5,5-四甲基-2-(2-甲基-1-苯基丙烷-2-基)-1,3,2-二氧杂硼杂环戊烷(19)
在0.2mmol的规模上,按照通用程序c加入nhpi酯(s12)和悬浮液a(在thf中的nicl2·6h2o/di-meobipy)。通过快速柱色谱法纯化(硅胶,己烷至1∶30的et2o∶己烷),得到19(35.3mg,68%)。
物理状态:无色固体;
m.p.=36–37℃;
rf=0.50(硅胶,1:12的etoac∶己烷);
1hnmr(600mhz,cdcl3):δ7.24–7.19(m,4h),7.17–7.14(m,1h),2.61(s,2h),1.21(s,12h),0.94(s,6h)ppm;
13cnmr(151mhz,cdcl3):δ140.6,130.3,127.8,125.8,83.24,46.5,24.9ppm.
光谱数据与文献报道相符。2
化合物20
2-金刚烷-1-基-4,4,5,5-四甲基-1,3,2-二氧杂硼杂环戊烷(20)
在0.2mmol的规模上,按照通用程序c加入nhpi酯(s31)和悬浮液c(在thf中的nicl2·6h2o/di-tbubipy)。通过快速柱色谱法纯化(硅胶,1∶60的et2o∶己烷至1∶40的et2o∶己烷),得到20(29.2mg,56%)。
物理状态:白色无定形固体;
rf=0.60(硅胶,1:9的etoac∶己烷);
1hnmr(600mhz,cdcl3):δ1.84(brs,3h),1.75(brt,j=3.6hz,12h),1.20(s,12h)ppm;
13cnmr(151mhz,cdcl3):δ82.7,38.1,37.6,27.7,24.8ppm;
光谱数据与文献报道相符。2
化合物21
4-(4,4,5,5-四甲基-1,3,2-二氧杂硼杂环戊烷-2-基)立方烷-1-羧酸甲酯(21)
在0.2mmol的规模上,按照通用程序c加入nhpi酯(s32)和悬浮液a(在thf中的nicl2·6h2o/di-meobipy)。通过快速柱色谱法纯化(硅胶,己烷至1∶15∶15的et2o∶ch2cl2:己烷),得到21(26.2mg,46%)。
物理状态:白色固体;
m.p.=152–155℃;
rf=0.45(硅胶,1:6的etoac∶己烷);
1hnmr(600mhz,cdcl3):δ4.30–4.28(m,3h),4.03–4.01(m,3h),3.70(s,3h),1.26(s,12h);
13cnmr(151mhz,cdcl3):δ172.8,83.4,55.4,51.6,47.0,45.2,24.9ppm;
hrms(esi-tof,m/z):计算值c16h22bo4[m+h]+289.1606;实测值289.1607.
化合物22
4-(4,4,5,5-四甲基-1,3,2-二氧杂硼杂环戊烷-2-基)双环[2.2.2]辛烷-1-羧酸甲酯(22)
在0.2mmol的规模上,按照通用程序c加入nhpi酯(s34)和悬浮液c(在thf中的nicl2·6h2o/di-tbubipy)。通过快速柱色谱法纯化(硅胶,己烷至1∶9的et2o∶己烷),得到22(31.1mg,53%)。
物理状态:无色固体;
在100℃升华;
rf=0.39(硅胶,1:5的etoac∶己烷);
1hnmr(600mhz,cdcl3):δ3.62(s,3h),1.72–1.65(m,6h),1.62–1.54(m,6h),1.19(s,12h)ppm;
13cnmr(151mhz,cdcl3):δ179.0,83.0,51.7,38.6,27.9,26.7,24.8ppm;
hrms(esi-tof,m/z):计算值c16h28bo4[m+h]+295.2075;实测值295.2077.
化合物23
4,4,5,5-四甲基-2-(1-甲基环己基)-1,3,2-二氧杂硼杂环戊烷(23)
在0.2mmol的规模上,按照通用程序c加入nhpi酯(s33)和悬浮液a(在thf中的nicl2·6h2o/di-meobipy)。通过快速柱色谱法纯化(硅胶,己烷至1∶30的et2o∶己烷),得到23(27.8mg,62%)。
物理状态:无色油状;
rf=0.50(硅胶,1:12的etoac∶己烷);
1hnmr(600mhz,cdcl3):δ1.84–1.80(m,2h),1.64–1.57(m,3h),1.29–1.21(m,14h),1.16–1.08(m,1h),0.92–0.87(m,5h)ppm;
13cnmr(151mhz,cdcl3):δ82.9,37.2,26.6,26.0,25.7,24.8ppm;
光谱数据与文献报道相符。2
化合物24
4,4,5,5-四甲基-2-(1-苯基环己基)-1,3,2-二氧杂硼杂环戊烷(24)
在0.2mmol的规模上,按照通用程序c加入nhpi酯(s13)和悬浮液c(在thf中的nicl2·6h2o/di-tbubipy)。通过快速柱色谱法纯化(硅胶,己烷至1∶30的et2o∶己烷),得到24(28.5mg,50%)。
物理状态:白色固体;
m.p.=87–88℃;
rf=0.60(硅胶,1:9的etoac∶己烷);
1hnmr(600mhz,cdcl3):δ7.36–7.34(m,2h),7.29–7.26(m,2h),7.13–7.11(m,1h),2.36–2.32(m,2h),1.82–1.78(m,2h),1.70–1.66(m,1h),1.49–1.38(m,4h),1.21–1.14(m,1h),1.17(s,12h)ppm;
13cnmr(151mhz,cdcl3):δ147.6,128.2,126.3,125.1,83.4,35.0,26.4,25.9,25.7ppm;
hrms(esi-tof,m/z):计算值c18h28bo2[m+h]+287.2177;实测值287.2184.
化合物25
叔丁基二甲基(2-甲基-2-(4,4,5,5-四甲基-1,3,2-二氧杂硼杂环戊烷-2-基)丙氧基)硅烷(25)
在0.2mmol的规模上,按照通用程序c加入nhpi酯(s30)和悬浮液a(在thf中的nicl2·6h2o/di-meobipy)。通过快速柱色谱法纯化(硅胶,己烷至1∶30的et2o∶己烷),得到25(41.2mg,66%)。
物理状态:无色油状;
rf=0.40(硅胶,1:12的etoac∶己烷);
1hnmr(600mhz,cdcl3):δ3.39(s,2h),1.22(s,12h),0.90(s,6h),0.88(s,9h),0.01(s,6h)ppm;
13cnmr(151mhz,cdcl3):δ83.0,72.0,26.1,24.9,21.4,18.5,-5.34ppm;
hrms(esi-tof,m/z):计算值c16h36bo3si[m+h]+315.2521;实测值315.2523.
化合物26
4,4,5,5-四甲基-2-(2-苯基丙-2-基)-1,3,2-二氧杂硼杂环戊烷(26)
在0.2mmol的规模上,按照通用程序c加入nhpi酯(s14)和悬浮液c(在thf中的nicl2·6h2o/di-tbubipy)。通过快速柱色谱法纯化(硅胶,己烷至1∶30的et2o∶己烷),得到26(23.3mg,47%)。
物理状态:无色油状;
rf=0.51(硅胶,1:9的etoac∶己烷);
1hnmr(600mhz,cdcl3):δ7.33–7.27(m,4h),7.15–7.12(m,1h),1.35(s,6h),1.20(s,12h)ppm;
13cnmr(151mhz,cdcl3):δ148.8,128.2,126.4,125.1,83.4,25.7,24.7ppm;
光谱数据与文献报道相符。9
化合物27
2-(1-(4-氯苯基)环丙基)-4,4,5,5-四甲基-1,3,2-二氧杂硼杂环戊烷(27)
在0.2mmol的规模上,按照通用程序c加入nhpi酯(s34)和悬浮液a(thf中的nicl2·6h2o/di-meobipy)。通过快速柱色谱法纯化(硅胶,己烷至1:30的et2o:己烷),得到27(32.9mg,59%)。
物理状态:白色固体;
m.p.=83–85℃;
rf=0.37(硅胶,1:19的etoac∶己烷);
1hnmr(600mhz,cdcl3):δ7.19(s,4h),1.21(s,12h),1.11(dd,j=6.0hz,3.6hz,2h),0.87(dd,j=6.0hz,3.6hz,2h)ppm;
13cnmr(151mhz,cdcl3):δ143.5,131.0,130.5,128.2,83.6,24.7,13.6ppm;
hrms(esi-tof,m/z):计算值c15h21bclo2[m+h]+279.1318;实测值279.1319.
化合物28
((1-((4,4,5,5-四甲基-1,3,2-二氧杂硼杂环戊烷-2-基)甲基)环己基)甲)基氨基甲酸叔丁酯(28)
在0.1mmol的规模上,按照通用程序b加入nhpi酯(s15)和溶液b(在dmf中的nicl2·6h2o/di-meobipy)。通过快速柱色谱法(硅胶,1:20的etoac:己烷)纯化,得到28(22.5mg,64%)。
物理状态:白色固体;
m.p.=92–96℃;
rf=0.28(硅胶,1:20的etoac∶己烷);
1hnmr(600mhz,cdcl3):δ5.32(brs,1h),3.08(d,j=6.0hz,2h),1.52–1.41(m,4h),1.43(s,9h),1.38–1.31(m,6h),1.25(s,12h),0.81(s,2h)ppm;
13cnmr(151mhz,cdcl3):δ156.5,83.4,78.7,50.0,36.7,36.3,28.6,26.4,25.0,21.9ppm;
hrms(esi-tof,m/z):计算值c19h37bno4[m+h]+354.2810;实测值354.2809.
化合物29
2-(1-(4-异丁基苯基)乙基)-4,4,5,5-四甲基-1,3,2-二氧杂硼杂环戊烷(29)
在0.2mmol的规模上,按照通用程序c加入nhpi酯(s16),mgbr2·oet2(52mg,0.2mmol,1当量)和悬浮液a(在thf中的nicl2·6h2o/di-meobipy)。通过快速柱色谱法纯化(硅胶,己烷至1∶30的et2o∶己烷),得到29(43.0mg,75%)。
物理状态:无色油状;
rf=0.59(硅胶,1:9的etoac∶己烷);
1hnmr(600mhz,cdcl3):δ7.12–7.10(m,2h),7.04–7.02(m,2h),2.42(d,j=7.2hz,2h),2.40(q,j=7.2hz,1h),1.79-1.88(m,1h),1.31(d,j=7.2hz,3h),1.21(s,6h),1.20(s,6h),0.89(d,j=6.6hz,6h)ppm;
13cnmr(151mhz,cdcl3):δ142.1,138.4,129.2,127.6,83.4,45.2,30.4,24.8,24.7,22.6,17.2ppm;
光谱数据与文献报道相符。10
化合物30
2-(5-(2,5-二甲基苯氧基)-2-甲基戊-2-基)-4,4,5,5-四甲基-1,3,2-二氧杂硼杂环戊烷(30)
在0.2mmol的规模上,按照通用程序c加入nhpi酯(s17)和悬浮液a(thf中的nicl2·6h2o/di-meobipy)。通过快速柱色谱法(硅胶,己烷至1∶30的et2o∶己烷)纯化,得到30(36.3mg,55%)。
物理状态:无色固体;
m.p.=59–61℃;
rf=0.60(硅胶,1:12的etoac∶己烷);
1hnmr(600mhz,cdcl3):δ6.99(d,j=7.8hz,1h),6.64(d,j=7.2hz,1h),6.62(s,1h),3.92(t,j=6.6hz,2h),2.30(s,3h),2.18(s,3h),1.78–1.73(m,2h),1.41–1.44(m,2h),1.23(s,12h),0.96(s,6h)ppm;
13cnmr(151mhz,cdcl3):δ157.3,136.5,130.4,123.8,120.6,112.2,83.1,68.8,37.4,26.6,25.0,24.9,21.6,16.0ppm;
hrms(esi-tof,m/z):计算值c20h34bo3[m+h]+333.2595;实测值333.2598.
化合物31
2-(1-(6-甲氧基萘-2-基)乙基)-4,4,5,5-四甲基-1,3,2-二氧杂硼杂环戊烷
在0.2mmol的规模上,按照通用程序c加入nhpi酯(s18)、mgbr2·oet2(52mg,0.2mmol,1当量)和悬浮液a(在thf中的nicl2·6h2o/di-meobipy)。通过快速柱色谱法(硅胶,己烷至1:25的et2o:己烷)纯化,得到31(50.0mg,80%)。
物理状态:白色固体;
m.p.=82–84℃;
rf=0.62(硅胶,1:4的etoac∶己烷);
1hnmr(600mhz,cdcl3):δ7.64–7.67(m,2h),7.57(s,1h),7.35(dd,j=8.4,1.8hz,1h),7.09–7.11(m,2h),3.90(s,3h),2.57(q,j=7.2hz,1h),1.41(d,j=7.2hz,3h),1.21(s,6h),1.20(s,6h)ppm;
13cnmr(151mhz,cdcl3):δ157.1,140.3,132.8,129.5,129.1,127.8,126.7,125.3,118.5,105.8,83.5,55.4,24.8,24.8,17.1ppm;
hrms(esi-tof,m/z):计算值c19h26bo3[m+h]+313.1969;实测值313.1970.
化合物32
1,7-双(4,4,5,5-四甲基-1,3,2-二氧杂硼杂环戊烷-2-基)庚烷(32)
在0.2mmol的规模上,按照通用程序b加入nhpi酯(s19)和溶液b(nicl2·6h2o(20mol%)/di-meobipy(26%mol%)的dmf(0.8ml)溶液)。通过快速柱色谱法(硅胶,己烷至1∶20的et2o∶己烷)纯化,得到32(26.5mg,38%)。
物理状态:无色油状;
rf=0.45(硅胶,1:8的etoac∶己烷);
1hnmr(600mhz,cdcl3):δ1.41–1.36(m,4h),1.29–1.24(m,6h),1.24(s,24h),0.75(t,j=7.8hz,4h)ppm;
13cnmr(151mhz,cdcl3):δ83.0,32.5,29.4,25.0,24.2ppm;
hrms(esi-tof,m/z):计算值c19h39b2o4[m+h]+353.3029;实测值353.3030.
化合物33
n,n-双(2-氯乙基)-4-(3-(4,4,5,5-四甲基-1,3,2-二氧杂硼杂环戊烷-2-基)丙基)苯胺33)
在0.2mmol的规模上,按照通用程序c加入nhpi酯(s20)和悬浮液a(在thf中的nicl2·6h2o/di-meobipy)。通过快速柱色谱法(硅胶,己烷至1:19的etoac:己烷)纯化,得到33(20.7mg,26%)。
物理状态:黄色油状;
rf=0.36(硅胶,1:9的etoac∶己烷);
1hnmr(600mhz,cdcl3):δ7.09-7.04(m,2h),6.63-6.59(m,2h),3.69(t,j=7.1hz,4h),3.61(t,j=7.1hz,4h),2.54-2.48(t,j=7.8hz,2h),1.68(p,j=7.8hz,2h),1.24(s,12h),0.81(t,j=7.8hz,2h)ppm;
13cnmr(151mhz,cdcl3):δ144.2,132.2,129.9,112.2,83.1,53.8,40.7,37.6,26.5,25.0ppm;
hrms(esi-tof,m/z):计算值c19h31bcl2no2[m+h]+386.1819;实测值386.1815.
化合物34
苯基(3-(1-(4,4,5,5-四甲基-1,3,2-二氧杂硼杂环戊烷-2-基)乙基)苯基)甲酮(34)
在0.2mmol的规模上,按照通用程序c加入nhpi酯(s21)、mgbr2·oet2(52mg,0.2mmol,1当量)和悬浮液a(在thf中的nicl2·6h2o/di-meobipy)。通过快速柱色谱法(硅胶,己烷至1∶15的etoac∶己烷)纯化,得到34(51.9mg,77%)。
物理状态:无色油状;
rf=0.45(硅胶,1:6的etoac∶己烷);
1hnmr(600mhz,cdcl3):δ7.83–7.81(m,2h),7.66(t,j=1.8hz,1h),7.59–7.56(m,2h),7.49–7.44(m,3h),7.38(t,j=7.8hz,1h),2.51(q,j=7.8hz,1h),1.35(d,j=7.8hz,3h),1.21(s,6h),1.21(s,6h)ppm;
13cnmr(151mhz,cdcl3):δ197.2,145.4,138.0,137.7,132.4,132.2,130.3,129.7,128.4,128.3,127.2,83.6,24.8,24.8,17.1ppm;
hrms(esi-tof,m/z):计算值c21h26bo3[m+h]+337.1969;实测值337.1971.
化合物35
4,4,5,5-四甲基-2-(1-(3-苯氧基苯基)乙基)-1,3,2-二氧杂硼杂环戊烷(35)
在0.2mmol的规模上,按照通用程序c加入nhpi酯(s22)、mgbr2·oet2(52mg,0.2mmol,1当量)和悬浮液a(在thf中的nicl2·6h2o/di-meobipy)。通过快速柱色谱法(硅胶,己烷至1∶30的et2o∶己烷)纯化,得到35(52.6mg,81%)。
物理状态:无色油状;
rf=0.50(硅胶,1:12的etoac∶己烷);
1hnmr(600mhz,cdcl3):δ7.32(t,j=7.8hz,2h),7.22(t,j=7.8hz,1h),7.07(t,j=7.8hz,1h),7.01(d,j=7.2hz,2h),6.97(d,j=7.2hz,1h),6.91(t,j=1.8hz,1h),6.79(dd,j=7.8hz,2.4hz,1h),2.42(q,j=7.8hz,1h),1.31(d,j=7.8hz,3h),1.20(s,6h),1.19(s,6h)ppm;
13cnmr(151mhz,cdcl3):δ157.7,157.2,147.3,129.7,129.6,123.0,123.0,118.8,118.7,115.9,83.5,24.8,24.7,17.0ppm;
hrms(esi-tof,m/z):计算值c20h26bo3[m+h]+325.1969;实测值325.1970.
化合物36
1-(2-((4r,6s)-2,2-二甲基-6-((4,4,5,5-四甲基-1,3,2-二氧杂硼杂环戊烷-2-基)甲基)-1,3-二噁烷-4-基)乙基)-5-(4-氟苯基)-2-异丙基-n,4-二苯基-1h-吡咯-3-羧酰胺(36)
在0.2mmol的规模上,按照通用程序b加入nhpi酯(s23)和溶液b(在dmf中的nicl2·6h2o/di-meobipy)。通过快速柱色谱(硅胶,己烷至1:9的etoac:己烷)纯化,得到36(77.4mg,57%)。
物理状态:白色泡沫;
rf=0.52(硅胶,1:4的etoac∶己烷);
1hnmr(600mhz,cdcl3):δ7.21–7.14(m,9h),7.07(brd,j=8.4hz,2h),7.00–6.97(m,3h),6.85(brs,1h),4.08–4.03(m,1h),4.00–3.96(m,1h),3.85–3.80(m,1h),3.69–3.65(m,1h),3.60–3.55(m,1h),1.68–1.64(m,2h),1.55(d,j=1.8hz,3h),1.53(d,j=1.8hz,3h),1.34(dt,j=13.2hz,1.2hz,1h),1.34(s,3h),1.30(s,3h),1.23(s,12h),1.08–1.03(m,2h),0.98–0.94(m,1h)ppm;
13cnmr(151mhz,cdcl3):δ165.0,162.4(d,j=247.6hz),141.7,138.6,134.8,134.5,133.3(d,j=8.2hz),130.7,128.9,128.8,128.5,128.4(d,j=3.8hz),126.7,123.8,123.6,121.8,119.7,115.4(d,j=21.3hz),98.6,83.3,66.7,66.7,41.0,38.4,38.3,30.3,26.2,24.9,24.9,21.9,21.7,20.0ppm;
hrms(esi-tof,m/z):计算值c41h51bfn2o5[m+h]+681.3870;实测值681.3870;
[α]d20=+4.0(c0.68,chcl3).
化合物37
(4-氯苯基)(5-甲氧基-2-甲基-3-((4,4,5,5-四甲基-1,3,2-二氧杂硼杂环戊烷-2-基)甲基)-1h-吲哚-1-基)甲酮(37)
在0.1mmol的规模上,按照通用程序c加入nhpi酯(s37)和悬浮液a(在thf中的nicl2·6h2o/di-meobipy)。通过快速柱色谱法(硅胶,己烷至1:17的etoac:己烷)纯化,得到37(22.1mg,50%)。
物理状态:黄色油状;
rf=0.5(硅胶,1:4的etoac∶己烷);
1hnmr(600mhz,cdcl3):δ7.64(dt,j=9.0hz,1.8hz,2h),7.45(m,dt,j=8.4hz,1.8hz,2h),6.96–6.93(m,2h),6.64(dd,j=9.0hz,2.6hz,1h),3.84(s,3h),2.29(s,3h),2.18(s,2h),1.23(s,12h)ppm;
13cnmr(151mhz,cdcl3):δ168.3,156.0,138.8,134.7,133.2,132.0,131.2,131.1,129.1,116.7,115.0,111.3,101.7,83.7,55.8,29.9,25.0,13.9ppm;
hrms(esi-tof,m/z):计算值c24h28bclno4[m+h]+440.1794;实测值440.1794.
化合物38
(5s,8r,9s,10s,13r,14s,17r)-10,13-二甲基-17-((r)-4-(4,4,5,5-四甲基-1,3,2-二氧杂硼杂环戊烷-2-基)丁-2-基)十二氢-3h-环戊[a]菲-3,7,12(2h,4h)-三酮(38)
在0.2mmol的规模上,按照通用程序b加入nhpi酯(s38)和溶液b(在dmf中的nicl2·6h2o/di-meobipy)。通过快速柱色谱(硅胶,己烷至1:5的etoac:己烷)纯化,得到38(63.0mg,65%)。
物理状态:白色固体;
rf=0.40(硅胶,1:3的etoac∶己烷);
m.p.=230-232℃;
1hnmr(600mhz,cdcl3):δ2.92–2.82(m,3h),2.35–2.19(m,6h),2.14–2.09(m,2h),2.05–1.94(m,4h),1.80–1.85(m,1h),1.56–1.63(m,2h),1.39(s,3h),1.35–1.12(m,16h),1.06(s,3h),0.87–0.81(m,4h),0.68–0.62(m,1h)ppm;
13cnmr(151mhz,cdcl3):δ212.1,209.2,208.9,83.0,57.1,51.9,49.2,47.0,45.8,45.7,45.1,42.9,38.8,38.2,36.6,36.1,35.4,29.4,27.8,25.4,25.0,24.9,22.1,18.6,12.0ppm;
hrms(esi-tof,m/z):计算值c29h45bo5[m+h]+485.3433;实测值485.3435.
[α]d20=+16.9(c0.62,chcl3).
化合物39
(3s,4ar,6ar,6bs,8ar,11s,12ar,14ar,14bs)-4,4,6a,6b,8a,11,14b-七甲基-14-氧代-11-(4,4,5,5-四甲基-1,3,2-二氧杂硼杂环戊烷-2-基)-1,2,3,4,4a,5,6,6a,6b,7,8,8a,9,10,11,12,12a,14,14a,14b-二十碳氢苉-3-基乙酸酯(39)
在0.2mmol的规模上,按照通用程序c加入nhpi酯(s24)和悬浮液a(在thf中的nicl2·6h2o/di-meobipy)。通过快速柱色谱法(硅胶,1:12:3的etoac:己烷:ch2cl2)纯化,得到39(82.0mg,69%,d.r.=11.8∶1)。
物理状态:无色膜;
rf=0.34(硅胶,1:5的etoac∶己烷);
1hnmr(600mhz,cdcl3):主要异构体δ5.57(s,1h),4.51(dd,j=11.8,4.7hz,1h),2.79(dt,j=13.7,3.6hz,1h),2.35(s,1h),2.20(ddd,j=13.3,4.4,1.7hz,1h),2.12(td,j=13.7,4.6hz,1h),2.04(s,3h),1.96(t,j=13.6hz,1h),1.80(td,j=13.7,4.6hz,1h),1.75–1.38(m,7h),1.37(s,3h),1.27–1.13(m,5h),1.20(d,j=1.8hz,12h),1.15(s,3h),1.12(s,3h),1.02(td,j=13.5,3.6hz,1h),0.99(s,3h),0.94(ddt,j=13.7,4.5,2.2hz,1h),0.87(s,6h),0.84(s,3h),0.81–0.76(m,1h)ppm;
13cnmr(151mhz,cdcl3):主要异构体δ200.1,171.1,171.1,128.3,83.0,80.8,61.8,55.2,45.5,45.3,43.6,38.9,38.5,38.2,37.1,34.2,32.9,32.7,29.1,28.2,27.8,26.7,26.6,24.8,24.7,23.7,23.4,21.5,18.9,17.7,17.6,16.8,16.6ppm;
hrms(esi-tof,m/z):计算值c37h60bo5[m+h]+595.4528;实测值595.4520;
[α]d20=+65.8(c1.0,chcl3).
化合物40
(2s,4ar,6as,6br,8ar,10s,12as,12br,14br)-10-羟基-2,4a,6a,6b,9,9,12a-七甲基-2-(4,4,5,5-四甲基-1,3,2-二氧杂硼杂环戊烷-2-基)-1,3,4,4a,5,6,6a,6b,7,8,8a,9,10,11,12,12a,12b,14b-十八氢苉-13(2h)-酮(40)
在0.2mmol的规模上,按照通用程序c加入nhpi酯(s39)和悬浮液a(在thf中的nicl2·6h2o/di-meobipy)。通过快速柱色谱法(硅胶,第一快速柱色谱法:1:5.7至1:4的etoac:己烷;第二快速柱色谱法,1:6:3至2:6:3的etoac:己烷:ch2cl2)纯化,得到40(72.1mg,65%,d.r.=11.3:1)。
物理状态:无色膜;
rf=0.46(硅胶,3:7的etoac∶己烷);
1hnmr(600mhz,cdcl3):主要异构体δ5.59(s,1h),3.27–3.18(m,1h),2.81(dt,j=13.5,3.6hz,1h),2.36(s,1h),2.22(ddd,j=13.5,4.5,1.7hz,1h),2.14(td,j=13.7,4.6hz,1h),1.99(t,j=13.6hz,1h),1.83(td,j=13.7,4.7hz,1h),1.74–1.58(m,4h),1.55(td,j=13.8,4.0hz,1h),1.51–1.35(m,2h),1.41(s,3h),1.33–1.15(m,7h),1.22(s,6h),1.22(s,6h),1.15(s,3h),1.15(s,3h),1.02(s,3h),1.01(s,3h),1.00–0.94(m,1h),0.86(s,3h),0.82(s,3h),0.71(dd,j=11.8,1.9hz,1h)ppm;
13cnmr(151mhz,cdcl3):主要异构体δ200.3,171.1,128.3,83.0,79.0,61.9,55.2,45.5,45.3,43.6,39.3,39.3,38.5,37.2,34.2,33.0,32.7,29.1,28.3,27.8,27.5,26.7,26.6,24.8,24.7,23.5,18.9,17.7,16.5,15.7ppm;
hrms(esi-tof,m/z):计算值c35h58bo4[m+h]+553.4422;实测值553.4423;
[α]d20=+73.4(c1.0,chcl3).
化合物40a
(3s,4ar,6ar,6bs,8ar,11s,12ar,14ar,14bs)-4,4,6a,6b,8a,11,14b-七甲基-14-氧代-11-(4,4,5,5-四甲基-1,3,2-二氧杂硼杂环戊烷-2-基)-1,2,3,4,4a,5,6,6a,6b,7,8,8a,9,10,11,12,12a,14,14a,14b-二十碳氢苉-3-基3,5-二硝基苯甲酸酯(40a)
将40(30mg,0.054mmol,1.0当量)、3,5-二硝基苯甲酰氯(50mg,0.22mmol,4.1当量)和dmap(1.3mg,0.011mmol,0.2当量)装入培养管。加入ch2cl2(0.3ml)和et3n(30μl,0.22mmol,4.1mmol),并将得到的混合物在室温搅拌1h。将该混合物直接装载到硅胶柱上,通过快速柱色谱法(1:11的etoac:己烷)纯化,得到40a(39.0mg,96%,d.r.=11.3∶1)。纯产物从己烷/ch2cl2中结晶。
物理状态:淡黄色固体(主要异构体是白色固体);
m.p.295℃在下分解;
rf=0.45(硅胶,1:5.7的etoac∶己烷);
1hnmr(600mhz,cdcl3):主要异构体δ9.22(t,j=2.2hz,1h),9.13(d,j=2.2hz,2h),5.60(s,1h),4.88(dd,j=11.9,4.7hz,1h),2.92(dt,j=13.7,3.6hz,1h),2.40(s,1h),2.26–2.19(m,1h),2.13(td,j=13.7,4.5hz,1h),1.98(t,j=13.6hz,1h),1.95–1.87(m,1h),1.87–1.75(m,2h),1.74–1.59(m,3h),1.56–1.48(m,2h),1.45(dt,j=12.8,3.1hz,1h),1.40(s,3h),1.30–1.09(m,5h),1.23(s,3h),1.21(s,6h),1.21(s,6h),1.16(s,3h),1.08(s,3h),1.00(s,3h),0.97(s,3h),0.97-0.94(m,1h),0.89(dd,j=11.8,1.9hz,1h),0.86(s,3h)ppm;
13cnmr(151mhz,cdcl3):主要异构体δ199.9,171.4,162.3,148.8,134.8,129.5,128.3,122.4,84.3,83.1,61.7,55.3,45.6,45.3,43.6,38.9,38.6,38.5,37.1,34.2,32.8,32.7,29.2,28.5,27.8,26.7,26.6,24.8,24.7,23.8,23.4,18.9,17.7,17.6,17.2,16.6ppm;
hrms(esi-tof,m/z):计算值c42h60bn2o9[m+h]+747.4386;实测值747.4385;
[α]d20=+60.5(c1.0,chcl3).
化合物41
(e)-7-羟基-5-甲氧基-4-甲基-6-(3-甲基-5-(4,4,5,5-四甲基-1,3,2-二氧杂硼杂环戊烷-2-基)戊-2-烯-1-基)异苯并呋喃-1(3h)-酮(41)
在0.2mmol的规模上,按照通用程序b加入nhpi酯(s25)和溶液b(在dmf中的nicl2·6h2o/di-meobipy)。通过快速柱色谱法纯化(硅胶,己烷至1:6:6的etoac:己烷:ch2cl2),得到41(37.0mg,46%)。
物理状态:白色固体;
m.p.=122–124℃;
rf=0.40(硅胶,1:2的etoac∶己烷);
1hnmr(600mhz,cdcl3):δ7.64(s,1h),5.21-5.18(m,3h),3.75(s,3h),3.37(d,j=6.6hz,2h),2.13(s,3h),2.08(t,j=7.8hz,2h),1.77(s,3h),1.17(s,12h),0.86(t,j=7.8hz,2h)ppm;
13cnmr(151mhz,cdcl3):δ173.1,163.9,153.8,143.9,137.8,122.8,120.6,116.8,106.4,83.0,70.1,61.1,33.6,24.9,22.7,16.3,11.7ppm;
hrms(esi-tof,m/z):计算值c22h32bo6[m+h]+403.2286;实测值403.2289.
硼酸的实验步骤和表征数据
化合物4a
(4-苯基丁基)硼酸(4a)
在氩气下,将频哪醇硼酸酯4(70mg,0.27mmol)溶于ch2cl2(5ml)中,并将其在干冰/丙酮浴中冷却至-78℃。逐滴加入bcl3(0.81ml,在ch2cl2中为1.0m,3.0当量),然后将混合物在-78℃搅拌1h。然后将混合物温热至室温,并在真空中除去挥发物。加入无水甲醇(5ml),并在真空中除去甲醇时将所得混合物搅拌10分钟。加入另外一部分的甲醇(5ml);将混合物搅拌10分钟,然后真空浓缩。此过程再重复三次。然后将所得粗产物用制备型薄层色谱法纯化,得到白色固体状的4a(41.8mg,87%)。
1hnmr(600mhz,dmso-d6/d2o100/1):δ7.28–7.22(m,2h),7.15(ddt,j=13.9hz,6.9hz,1.5hz,3h),2.56–2.51(m,2h),1.51(tt,j=7.8,6.7hz,2h),1.38–1.28(m,2h),0.60(t,j=7.9hz,2h).
13cnmr(151mhz,dmso-d6/d2o100/1):δ142.60,128.32,128.27,125.58,35.26,34.21,23.98,15.27(br);
hrms(esi-tof)计算值c10h16bo2[m+h]+179.1238;实测值179.1236.
化合物3a
(4-苯基丁-2-基)硼酸(3a)
在氩气下,将频哪醇硼酸酯3(30mg,0.12mmol)溶于ch2cl2(2ml)中,并将溶液在干冰/丙酮浴中冷却至-78℃。逐滴加入bcl3(0.36ml,在ch2cl2中为1.0m,3.0当量),然后将混合物在-78℃搅拌1h。然后将混合物温热至室温,并在真空中除去挥发物。加入无水甲醇(5ml),将所得混合物搅拌10分钟,然后真空除去甲醇。加入另外一部分的甲醇(5ml);将混合物搅拌10分钟,然后真空浓缩。此过程再重复三次。然后将所得粗产物用制备型薄层色谱法纯化,得到白色固体状的4a(15.4mg,75%)。
1hnmr(600mhz,dmso-d6/d2o100/1):δ7.24(t,j=7.6hz,2h),7.18–7.10(m,3h),2.53–2.48(m,2h),1.74–1.63(m,1h),1.42(ddt,j=13.0hz,9.9hz,6.5hz,1h),0.90(d,j=7.2hz,3h),0.89–0.81(m,1h)ppm;
13cnmr(151mhz,dmso-d6/d2o100/1):δ143.03,128.34,125.61,35.65,35.05,20.33(br),16.35ppm;
hrms(esi-tof)计算值c10h16bo2[m+h]+179.1238;实测值179.1232.
化合物33a
(3-(4-(双(2-氯乙基)氨基)苯基)丙基)硼酸(33a)
在-78℃下,向频哪醇硼酸酯34(76.2mg,0.2mmol)的ch2cl2(1ml)溶液中滴加bcl3(0.79ml,1.0m,在ch2cl2中)。将反应混合物在-78℃下搅拌30分钟,然后在室温下再搅拌30分钟。反应物用甲醇(2ml)淬灭,并真空浓缩。向残余物中加入meoh(2ml),随后将其真空除去。此过程再重复三次。通过制备型反相hplc(在30分钟内20-80%ch3cn/h2o,ch3cn和h2o均含有0.1%tfa)纯化所得残余物,得到33a(27mg,50%),为无色油状物。
1hnmr(600mhz,dmso-d6/d2o10/1):δ7.00(d,j=12.6hz,2h),6.65(d,j=12.6hz,2h),3.71–3.65(m,8h),2.39(t,j=7.8hz,2h),1.58–1.53(m,2h),0.59(t,j=8.4hz,2h)ppm;
13cnmr(151mhz,dmso-d6/d2o10/1):δ144.2,130.9,129.3,111.8,52.3,41.2,37.2,26.6ppm;
hrms(esi-tof)计算值c13h21bcl2no2[m+h]+304.1037;实测值304.1030.
方案s1。尼拉罗(ninlaro)(艾沙佐米(ixazomib))的合成
尼拉罗(ixazomib)的合成
化合物s41
(2,5-二氯苯甲酰基)甘氨酰亮氨酸(s41)
boc的脱保护:在室温下向boc-gly-leu-ome11(3.1g,10.26mmol)的ch2cl2(30ml)溶液中加入tfa(15ml),将反应混合物搅拌1h,然后浓缩真空。残留物直接用于下一步。
形成酰胺键:在–15℃下向2.5-二氯苯甲酸(2.94g,15.4mmol)的thf(70ml)溶液中加入4-甲基吗啉(4.0ml,35.9mmol),将反应混合物在该温度下搅拌10分钟。向所得的白色悬浮液中滴加氯甲酸异丁酯(2.0ml,15.4mmol),并将混合物在–15℃下搅拌另外30分钟。在相同温度下缓慢加入在thf(35ml)中的粗tfa盐(来自脱保护步骤)。将反应混合物温热至室温并搅拌6小时。将所得混合物用etoac(100ml)稀释,用水(100ml)、饱和nahco3水溶液(100ml)和盐水(100ml)洗涤。合并的有机层经无水na2so4干燥,过滤并浓缩。通过快速柱色谱法(二氧化硅,3:2己烷/etoac)纯化,得到所需的酯,其不是非常纯净,但无需进一步纯化即可用于下一步。
甲酯的水解:向前述酯的thf(50ml)溶液中加入lioh水溶液(1m,50ml)。将反应混合物在室温搅拌2h,然后用etoac(60ml)洗涤。水层用1nhcl(65ml)酸化,并用etoac(100ml)萃取。有机层用盐水(100ml)洗涤,其中水层用etoac(100ml)反萃取。将合并的有机相真空浓缩。当所需产物s41沉淀出时,向残余物中加入ch2cl2(30ml),并通过过滤收集(2.31g,63%,历经3个步骤)。
m.p.=137-138℃;
1hnmr(600mhz,meoh-d4):δ7.63(dd,j=1.8hz,0.6hz,1h),7.45–7.48(m,2h),4.50(dd,j=9.6hz,5.4hz,1h),4.08(dd,j=37.8hz,16.8hz,2h),1.79–1.72(m,1h),1.70–1.62(m,2h),0.98(d,j=6.6hz,3h),0.95(d,j=6.6hz,3h)ppm;
13cnmr(151mhz,meoh-d4):δ175.8,170.9,168.7,138.4,134.0,132.6,132.3,130.6,130.2,52.1,43.6,41.9,26.0,23.4,21.9ppm;
hrms(esi-tof)计算值c15h19cl2n2o4[m+h]+361.0716;实测值361.0706;
[α]d20=-14.0(c1.0,meoh).
化合物s42
1,3-二氧异二氢吲哚-2-基(2,5-二氯苯甲酰基)甘氨酰亮氨酸酯(s42)
在2.0mmol的规模上,按照通用程序a加入s41。通过快速柱色谱法(去活的硅胶,3:7的etoac:己烷)纯化,得到s42(940mg,79%)。
物理状态:白色固体;
m.p.=164℃;
rf=0.55(硅胶,3:2的etoac∶己烷);
1hnmr(600mhz,thf-d8):δ8.05(brs,1h),7.99–7.97(m,1h),7.91–7.89(m,2h),7.87–7.85(m,2h),7.58(dd,j=2.4hz,0.5hz,1h),7.42–7.38(m,2h),5.10–5.06(m,1h),4.14(dd,j=16.8hz,6.0hz,1h),3.99(dd,j=16.8hz,6.0hz,1h),1.92-1.83(m,2h),1.80–1.75(m,1h),1.02(d,j=6.0hz,3h),1.00(d,j=6.0hz,3h)ppm;
13cnmr(151mhz,thf-d8):δ170.5,169.4,166.0,162.4,139.2,135.9,133.5,132.3,131.5,130.7,130.5,130.2,124.7,49.7,43.6,42.3,25.8,23.4,22.1ppm;
hrms(esi-tof)计算值c23h22cl2n3o6[m+h]+506.0880;实测值506.0875;
[α]d20=-1.0(c1.0,thf).
化合物1
在0.2mmol的规模上,按照常规步骤c,使用具有s42的悬浮液c(在thf中的nicl2·6h2o/di-tbubipy)。快速柱色谱法(硅胶,己烷至2:3的etoac:己烷至4:1的etoac:己烷),得到频哪醇氨基硼酸酯s43,其无需进一步纯化即可用于下一步。
在氩气下将频哪醇氨基硼酸酯s43溶于ch2cl2(5ml)中,并在干冰/丙酮浴中将溶液冷却至-78℃。逐滴加入bcl3(0.6ml,在ch2cl2中为1.0m,3.0当量),之后将混合物在-78℃搅拌1h。然后将混合物温热至室温,并在真空中除去挥发物。加入无水甲醇(5ml),并在真空中除去甲醇时将混合物搅拌10分钟。加入另外一部分的甲醇(5ml);将混合物搅拌10分钟,然后将其真空浓缩。将该过程重复三次。然后将所得残余物通过制备型反相hplc(35分钟内10-60%ch3cn/h2o,ch3cn和h2o均含有0.1%tfa)纯化,得到ninlaro(1,23.0mg,32%,历经2个步骤)。
1hnmr(600mhz,meoh-d4):δ7.60(t,j=1.5hz,1h),7.49–7.47(m,2h),4.24(s,2h),2.79(t,j=7.6hz,1h),1.68(ddt,j=14.7hz,13.0hz,6.4hz,1h),1.38(tdd,j=13.8hz,10.4hz,5.9hz,2h),0.94(dd,j=6.6hz,1.5hz,6h);
13cnmr(151mhz,meoh-d4):δ175.6,168.8,138.0,134.0,132.7,132.5,130.7,130.2,44.7(br,α至硼),40.9,40.2,27.1,23.7,22.4.
hrms(esi-tof,m/z):计算值c14h18bcl2n2o3[m-h2o+h]+343.0782;实测值343.0779;
[α]d20=-0.6(c1.0,meoh).
脱羧硼化使立普妥的后期多样化成为可能
化合物36a
5-(4-氟苯基)-1-(2-((4r,6r)-6-(羟甲基)-2,2-二甲基-1,3-二噁烷-4-基)乙基)-2-异丙基-n,4-二苯基-1h-吡咯-3-羧酰胺(36a)
在室温下向空气开放的36(50mg,0.073mmol)的thf/h2o(1∶1,0.73ml)溶液中加入nabo3·4h2o(56mg,0.37mmol)。将混合物剧烈搅拌3小时,然后加入h2o(10ml)。将所得混合物用etoac(10ml×3)萃取。合并的有机萃取物经na2so4干燥,过滤并浓缩。通过快速柱色谱法(硅胶,2:3的etoac:己烷)纯化,得到36a(40mg,86%)。
m.p.=166–170℃;
rf=0.27(硅胶,2:3的etoac∶己烷);
1hnmr(600mhz,cdcl3):δ7.21–7.15(m,9h),7.07(d,j=8.0hz,2h),7.02–6.96(m,3h),6.86(s,1h),4.13–4.05(m,1h),3.92–3.81(m,2h),3.73–3.66(m,1h),3.61–3.52(m,2h),3.45(dd,j=11.4hz,6.1hz,1h),1.74–1.61(m,2h),1.54(s,3h),1.53(s,3h),1.37(s,3h),1.34(s,3h),1.74–1.61(m,2h);
13cnmr(151mhz,cdcl3):δ164.9,162.4(d,j=247.6hz),141.7,138.5,134.8,133.3(d,j=8.0hz),130.6,128.9,128.8,128.5,128.4(d,j=3.5hz),126.7,123.6,121.9,119.7,115.5(d,j=21.4hz),98.9,69.4,66.2,66.0,41.0,38.3,31.9,30.0,26.2,21.9,21.7,20.0;
19fnmr(376mhz,丙酮-d6):δ-114.00ppm;
hrms(esi-tof,m/z):计算值c35h40fn2o4[m+h]+571.2966;实测值571.2963;
[α]d20=-4.6(c1.0,chcl3).
化合物36b
(((4r,6r)-6-(2-(2-(4-氟苯基)-5-异丙基-3-苯基-4-(苯基氨基甲酰基)-1h-吡咯-1-基)乙基)-2,2-二甲基-1,3-二噁烷-4-基)甲基)氨基甲酸叔丁酯(36b)
胺化是按照文献程序12,稍作修改进行的。用thf(1ml)稀释o-甲基羟胺的溶液(63μl,在thf中为2.8m,6.0当量)。在–78℃下添加正丁基锂(72μl,在己烷中为2.45m,6.0当量),并将所得混合物在该温度下搅拌1小时。在–78℃下滴加频哪醇硼酸酯36(20mg,0.03mmol)的thf(1ml)溶液。温热至室温后,将反应混合物加热至65℃并搅拌36h。冷却至室温后,加入boc2o(66mg,10.0当量)。将所得混合物在室温搅拌1小时,然后真空除去挥发物。通过制备型薄层色谱法(硅胶,15:1的dcm:et2o)纯化所得残余物,得到36b(10.7mg,54%),为无色油状物。
物理状态:无色油状;
rf=0.4(硅胶,1:3的己烷:etoac);
1hnmr(600mhz,cdcl3):δ7.24–7.09(m,9h),7.07(d,j=8.0hz,2h),7.04–6.94(m,3h),6.86(s,1h),4.84(s,1h),4.07(ddd,j=15.3hz,10.7hz,5.1hz,1h),3.82(ddt,j=15.1hz,10.3hz,6.5hz,2h),3.66(tt,j=8.2hz,3.6hz,1h),3.57(p,j=7.2hz,1h),3.24(d,j=8.7hz,1h),2.98(ddd,j=13.8hz,6.8hz,5.1hz,1h),1.70-1.63(m,2h),1.53(d,j=7.1hz,6h),1.44(s,9h),1.34(s,3h),1.31(s,3h),1.27–1.20(m,1h),1.07(q,j=12.0hz,1h);
13cnmr(151mhz,cdcl3):δ164.9,162.4(d,j=247.9hz),156.2,141.6,138.5,134.8,133.3(d,j=8.1hz),130.6,128.9,128.8,128.5,128.4(d,j=3.5hz),126.7,123.6,121.9,119.7,115.5(d,j=21.3hz),98.8,79.6,68.2,66.3,45.4,41.0,38.3,33.4,30.5,30.0,28.5,26.2,21.9,21.7,20.0;
19fnmr(376mhz,cdcl3):δ-113.93ppm;
hrms(esi-tof,m/z):计算值c40h49fn3o5[m+h]+670.3651;实测值670.3646;
[α]d20=-2.0(c0.74,chcl3).
化合物36c
1-(2-((4r,6r)-2,2-二甲基-6-(噻吩-2-基甲基)-1,3-二噁烷-4-基)乙基)-5-(4-氟苯基)-2-异丙基-n,4-二苯基-1h-吡咯-3-羧酰胺(36c)
在–78℃下,向噻吩(23μl,0.28mmol)的thf(1.0ml)溶液中添加正丁基锂(0.1ml,2.5m的己烷溶液,0.25mmol)。将所得混合物温热至室温并在将一些所得黄色溶液(0.33ml)转移至反应管中时,搅拌1h。在–78℃下滴加36(12.4mg,0.018mmol)的thf(0.3ml)溶液。当添加nbs(14.4mg,0.081mmol)的thf(0.3ml)溶液时,将所得混合物在相同温度下搅拌1.5小时。在相同温度下搅拌1小时后,将反应用物用na2s2o3水溶液(1ml)淬灭,然后升温至室温。将所得混合物用etoac(1ml×3)萃取。合并的有机层经无水na2so4干燥并真空浓缩。通过快速柱色谱法(硅胶,1:9的etoac:己烷)和ptlc(硅胶,1:6的etoac:己烷)纯化,得到36c(6.5mg,56%)。
物理状态:白色泡沫;
rf=0.61(硅胶,2:3的etoac∶己烷);
1hnmr(600mhz,丙酮-d6):δ8.29(brs,1h),7.45(d,j=7.8hz,2h),7.30–7.27(m,2h),7.24(dd,j=5.4hz,1.2hz,1h),7.20(t,j=7.8hz,2h),7.13-7.09(m,6h),7.08–7.05(m,1h),6.99–6.96(m,1h),6.92(dd,j=5.4hz,3.6hz,1h),6.85–6.84(m,1h),4.11–4.06(m,1h),4.05–4.00(m,1h),3.91–3.86(m,1h),3.85–3.81(m,1h),3.43–3.39(m,1h),2.93–2.90(m,1h),2.87–2.83(m,1h),1.75–1.63(m,2h),1.47(d,j=1.2hz,3h),1.45(d,j=1.2hz,3h),1.36(dt,j=12.6hz,3.0hz,1h),1.36(s,3h),1.28(s,3h)1.05–0.99(m,1h)ppm;
13cnmr(151mhz,丙酮-d6):δ166.4,163.1(d,j=245.6hz),140.9,140.6,139.4,136.1,134.5(d,j=8.2hz),130.8,129.9(d,j=3.3hz),129.3,128.9,128.6,127.3,126.7,126.7,124.9,123.8,122.4,120.2,118.0,116.0(d,j=21.6hz),99.2,70.2,67.3,41.3,39.1,37.3,36.5,30.5,26.922.4,22.3,20.1ppm;
19fnmr(376mhz,丙酮-d6):δ-114.91ppm;
hrms(esi-tof)计算值c39h42fn2o3s[m+h]+637.2895;实测值637.2892;
[α]d20=+19.2(c0.5,chcl3).
化合物36d
1-(2-((4r,6r)-6-(苯并呋喃-2-基甲基)-2,2-二甲基-1,3-二噁烷-4-基)乙基)-5-(4-氟苯基)-2-异丙基-n,4-二苯基-1h-吡咯-3-羧酰胺(36d)
在–78℃下,向2,3-苯并呋喃(30μl,0.27mmol)的thf(1.0ml)溶液中添加正丁基锂(0.1ml,在己烷中为2.5m,0.25mmol)。将所得溶液加热至室温。将所得混合物温热至室温并在将一些所得黄色溶液(0.33ml)转移至反应管中时,搅拌1h。在–78℃下滴加36(12.0mg,0.018mmol)的thf(0.3ml)溶液。当添加nbs(14.4mg,0.081mmol)的thf(0.3ml)溶液时,将所得混合物在相同温度下搅拌1h。在相同温度下搅拌1小时后,将反应用饱和na2s2o3水溶液(1ml)淬灭,然后升温至室温。将所得混合物用etoac(1ml×3)萃取。合并的有机层经无水na2so4干燥并真空浓缩。通过快速柱色谱法(硅胶,1:9的etoac:己烷)和制备型薄层色谱法(硅胶,1:9的etoac:己烷)纯化,得到36d(6.1mg,52%)。
物理状态:无色油状;
rf=0.64(硅胶,2:3的etoac∶己烷);
1hnmr(600mhz,丙酮-d6):δ8.29(brs,1h),7.54–7.52(m,1h),7.44(d,j=7.8hz,2h),7.45–7.42(m,1h),7.31–7.27(m,2h),7.24–7.17(m,4h),7.13–7.05(m,7h),6.99–6.96(m,1h),6.58(dd,j=1.2hz,0.6hz,1h),4.29–4.24(m,1h),4.11–4.06(m,1h),3.91–3.85(m,2h),3.44–3.37(m,1h),2.93(dd,j=15.6hz,6.6hz,1h),2.79(dd,j=15.6hz,6.6hz,1h),1.76–1.65(m,2h),1.46(s,3h),1.45(s,3h),1.46–1.51(m,1h),1.39(d,j=0.6hz,3h),1.27(d,j=0.6hz,3h),1.14–1.08(m,1h)ppm;
13cnmr(151mhz,丙酮-d6):δ166.4,163.1(d,j=245.7hz),156.5,155.5,140.6,139.4,136.1,134.5(d,j=8.3hz),130.8,129.9(d,j=3.3hz),129.9,129.3,128.9,128.6,126.7,124.2,123.8,123.4,122.4,121.3,120.2,120.1,118.0,116.0(d,j=21.6hz),111.4,104.6,99.3,68.1,67.3,41.3,39.1,37.0,36.2,30.4,26.9,22.4,22.3,20.1ppm;
19fnmr(376mhz,丙酮-d6):δ-114.95ppm;
hrms(esi-tof)计算值c43h44fn2o4[m+h]+671.3280;实测值671.3274;
[α]d20=+28.5(c0.5,chcl3).
化合物36e
1-(2-((4r,6s)-6-((3-氯吡啶-2-基)甲基)-2,2-二甲基-1,3-二噁烷-4-基)乙基)-5-(4-氟苯基)-2-异丙基-n,4-二苯基-1h-吡咯-3-羧酰胺(36e)
向旋盖培养管中加入pd2(dba)3(1.9mg,0.0022mmol,0.1当量),对-茴香基二苯基膦(3.7mg,0.0132mmol,0.6当量)、1-氯-4-硝基苯(35mg,0.22mmol,10当量)和k3po4(47mg,0.22mmol,10当量)。然后将该管抽空并用氩气回填三次。然后通过注射器加入1,4-二噁烷(0.4ml),并将所得混合物在室温搅拌5分钟。依次添加36(15.0mg,0.022mmol)的二噁烷(0.6ml)溶液和脱气的去离子水(0.5ml)。将反应混合物在100℃加热15h,然后将其冷却至室温并用盐水(4ml)处理。将所得混合物用etoac萃取(2ml×3)。合并的有机层经无水na2so4干燥并真空浓缩。通过快速柱色谱法(硅胶,1:6至3:7的etoac:己烷)和ptlc(硅胶,1:3的etoac:己烷)纯化,得到36e(7.9mg,54%)。
物理状态:无色油状;
rf=0.38(硅胶,3:7的etoac∶己烷);
1hnmr(600mhz,丙酮-d6):δ8.45(dd,j=4.8hz,1.8hz,1h),8.30(brs,1h),7.79(dd,j=8.4hz,1.8hz,1h),7.45(d,j=7.8hz,2h),7.30-7.25(m,3h),7.20(t,j=7.8hz,2h),7.12–7.05(m,7h),6.99–6.96(m,1h),4.46–4.41(m,1h),4.11–4.06(m,1h),3.91–3.86(m,1h),3.85–3.81(m,1h),3.44–3.39(m,1h),3.13(dd,j=14.4hz,6.6hz,1h),2.88(dd,j=14.4hz,7.2hz,1h),1.76–1.64(m,2h),1.47(d,j=0.6hz,3h),1.45(d,j=0.6hz,3h),1.38(dt,j=12.6hz,2.4hz,1h),1.34(s,3h),1.24(s,3h),1.16–1.10(m,1h)ppm;
13cnmr(151mhz,丙酮-d6):δ166.4,163.1(d,j=245.5hz),156.2,148.3,140.6,139.3,137.6,136.2,134.5(d,j=8.3hz),132.2,130.8,129.9(d,j=3.3hz),129.3,128.9,128.6,126.7,123.8,123.8,122.4,120.2,118.0,116.0(d,j=21.6hz),99.2,68.7,67.3,42.3,41.3,39.2,37.0,30.5,26.9,22.4,22.3,20.1ppm;
19fnmr(376mhz,丙酮-d6):δ-114.92ppm;
hrms(esi-tof)计算值c40h42clfn3o3[m+h]+666.2893;实测值666.2884;
[α]d20=+26.2(c0.5,chcl3).
化合物36f
1-(2-((4r,6s)-2,2-二甲基-6-(4-硝基苄基)-1,3-二噁烷-4-基)乙基)-5-(4-氟苯基)-2-异丙基-n,4-二苯基-1h-吡咯-3-羧酰胺(36f)
向旋盖培养管中加入pd2(dba)3(1.9mg,0.0022mmol,0.1当量)、对-茴香基二苯基膦(3.7mg,0.0132mmol,0.6当量)、1-氯-4-硝基苯(35mg,0.22mmol,10当量)和k3po4(47mg,0.22mmol,10当量)。抽空该管,并用氩气回填三次。经由注射器添加1,4-二噁烷(0.4ml),并将所得混合物在室温搅拌5分钟。依次添加36(15.0mg,0.022mmol)的二噁烷(0.6ml)溶液和脱气的去离子水(0.5ml)。将反应混合物加热至100℃保持15h,然后将其冷却至室温并用盐水(4ml)处理。将所得混合物用etoac(2ml×3)萃取。合并的有机层经无水na2so4干燥并真空浓缩。通过快速柱色谱法(硅胶,1:9至1:3的etoac:己烷)和ptlc(硅胶,1:4的etoac:己烷)纯化,得到36f(10.5mg,72%)。
物理状态:黄色油状;
rf=0.45(硅胶,3:7的etoac∶己烷);
1hnmr(600mhz,丙酮-d6):δ8.28(brs,1h),8.15(dt,j=9.0hz,1.8hz,2h),7.51(dt,j=9.0hz,1.8hz,2h),7.45(d,j=8.4hz,2h),7.30–7.27(m,2h),7.20(t,j=7.8hz,2h),7.14–7.10(m,6h),7.09–7.05(m,1h),6.99–6.96(m,1h),4.17–4.13(m,1h),4.11–4.06(m,1h),3.92–3.87(m,1h),3.85–3.81(m,1h),3.44–3.37(m,1h),2.87(dd,j=13.8hz,7.2hz,1h),2.81(dd,j=13.8hz,7.2hz,1h),1.73–1.65(m,2h),1.46(d,j=0.6hz,3h),1.45(d,j=0.6hz,3h),1.36(dt,j=12.6hz,2.4hz,1h),1.32(s,3h),1.25(s,3h),1.09–1.03(m,1h)ppm;
13cnmr(151mhz,丙酮-d6):δ166.4,163.1(d,j=245.5hz),147.6,147.5,140.5,139.4,136.1,134.5(d,j=8.3hz),131.4,130.8,129.9(d,j=3.8hz),129.3,128.9,128.7,126.7,123.9,123.8,122.4,120.2,118.0,116.0(d,j=21.4hz),99.2,69.8,67.3,42.9,41.2,39.1,36.7,30.4,26.9,22.4,22.3,20.1ppm;
19fnmr(376mhz,丙酮-d6):δ-114.92ppm;
hrms(esi-tof)计算值c41h43fn3o5[m+h]+676.3181;实测值676.3182;
[α]d20=+10.8(c0.5,chcl3).
硼万古霉素类似物的合成
方案s2。42、43和44的合成
化合物s45
向在ch3cn(5.1ml)中的s44[根据文献报道(38,62)合成](600mg,0.43mmol,1.0当量)加入n-叔丁基二甲基甲硅烷基-n-甲基三氟乙酰胺(mtbstfa,2.4ml,10.2mmol,23.7当量),将所得混合物加热至50℃。30小时后,将反应混合物倒入饱和柠檬酸水溶液(50ml)/etoac(20ml)的混合物中并在室温下剧烈搅拌12小时。分离有机层,并用饱和nahco3水溶液(50ml)和盐水(50ml)洗涤。然后将水层用etoac(20ml×2)反萃取。合并的有机层经无水na2so4干燥并真空浓缩。通过快速柱色谱法(硅胶,1:1至4:1的etoac:己烷)和制备型tlc(7:93meoh/ch2cl2)纯化,得到所需产物s45(440mg,63%)。
物理状态:白色膜;
rf=0.31(硅胶,7:93meoh/ch2cl2);
1hnmr(600mhz,丙酮-d6):δ9.43(brs,1h),7.94(d,j=6.5hz,1h),7.57(dd,j=8.3,1.9hz,1h),7.53(brs,3h),7.49(s,1h),7.46(s,1h),7.45(s,1h),7.43–7.33(m,4h),7.26(d,j=8.3hz,1h),7.19(d,j=8.3hz,1h),7.08(s,1h),7.03(d,j=8.7hz,1h),6.77(d,j=9.9hz,1h),6.73(s,1h),6.67(d,j=2.3hz,1h),6.45(brs,1h),6.31(d,j=2.3hz,1h),5.94(brs,1h),5.85(s,1h),5.58(d,j=4.9hz,1h),5.54(s,1h),5.51(s,1h),5.39(d,j=12.3hz,1h),5.23(d,j=12.3hz,1h),5.20(brs,1h),5.10(brs,1h),4.96(d,j=6.5hz,1h),4.67(d,j=5.2hz,1h),4.63(t,j=7.2hz,1h),4.42(d,j=11.7hz,1h),4.18(s,3h),3.68(s,3h),3.67(s,3h),3.59(s,3h),2.83(s,3h),2.59(d,j=16.5hz,1h),2.42(d,j=16.3hz,1h),2.09(s,1h),1.66–1.57(m,2h),1.53(s,9h),1.54-1.48(m,2h),1.00(s,9h),0.92(s,9h),0.92(d,j=6.5hz,3h)0.86(d,j=6.5hz,3h),0.17(s,6h),0.13(s,3h),0.12(s,3h)ppm;
13cnmr(151mhz,丙酮-d6):δ172.3,171.7,171.3,171.3,171.1,170.8,168.9,168.0,161.1,159.9,158.1,156.9,154.5,153.0,151.5,151.5,141.5,140.0,138.4,137.0,136.9,136.2,135.7,130.0,129.3,129.3,129.0,128.3,127.9,127.6,126.1,125.4,124.7,124.1,122.1,113.8,106.5,106.1,105.4,99.6,80.3,74.6,74.0,67.2,64.3,61.5,60.0,57.5,56.5,56.2,56.1,55.7,55.4,55.2,52.0,38.1,37.2,28.9,28.6,26.5,26.3,26.3,25.7,23.7,23.3,22.8,19.1,19.1,–4.4,–4.6,–4.8,–4.8.
hrms(esi-tof,m/z):计算值c81h103cl2n8o19si2[m+h]+1617.6249;实测值1617.6248.
化合物42
向s45(600mg,0.37mmol)的etoh/etoac(4/1,50ml)溶液中加入pd/c(240mg,5%pd/c,50%湿粉)。将得到的黑色悬浮液在氢气氛下在室温搅拌12小时。然后将反应混合物通过硅藻土过滤,并用etoh/etoac(4∶1,150ml)洗涤。减压浓缩滤液。所得残留物通过制备型反相hplc(30分钟内85%–100%ch3cn/h2o,100%ch3cn持续30分钟,ch3cn和h2o均含有0.1%tfa)纯化,得到42(450mg,79%),为tfa盐。
注意:发现boc基团在纯化过程中裂解。
物理状态:淡黄色膜;
1hnmr(600mhz,meoh-d4):δ8.68(d,j=5.4hz,1h),7.60(dd,j=8.4hz,2.4hz,1h),7.48(brs,1h),7.42(brs,1h),7.37(d,j=8.4hz,1h)7.40–7.35(brm,1h),7.10(d,j=9.0hz,1h),7.04–7.02(m,2h),6.68(d,j=2.4hz,1h),6.58(d,j=2.4hz,1h),6.39(brs,2h),5.77(d,j=1.2hz,1h),5.65(brs,1h),5.46(s,1h),5.37(s,1h),5.30(brs,1h),4.80(s,1h),4.60(d,j=5.4hz,1h),4.23(s,3h),4.10(brs,1h),3.93–3.90(m,1h),3.87(s,3h),3.73(s,3h),3.67(s,3h),2.83(s,3h),2.83–2.78(m,1h),2.42(dd,j=16.8,5.4hz,1h),1.89–1.82(m,1h),1.79–1.74(m,2h),0.98–0.93(m,24h),0.15(s,3h),0.15(s,3h),0.13(s,3h),0.12(s,3h)ppm;
13cnmr(600mhz,meoh-d4):δ175.3,174.0,172.3,171.9,171.8,171.4,170.4,169.4,169.2,162.0,160.4,159.0,155.2,154.2,153.3,152.1,142.2,140.0,139.4,137.3,136.8,135.4,130.6,129.2,128.5,128.5,128.0,127.2,126.1,125.2,124.9,122.5,114.1,107.3,106.7,106.4,99.4,74.9,65.0,62.5,62.4,61.1,58.2,56.6,56.1,56.0,55.6,52.4,40.8,37.0,33.2,26.8,26.5,25.3,23.7,22.0,19.7,19.5,–4.3,–4.5,–4.7,–4.7ppm;
19fnmr(376mhz,meoh-d4):δ-77.2ppm;
hrms(esi-tof,m/z):计算值c69h89cl2n8o17si2[m+h]+1427.5256;实测值1427.5258.
化合物43
向42(15.0mg,0.0097mmol,1.0当量)在ch3cn(1.5ml)中的溶液中添加三(二甲基氨基)二氟三甲基硅酸锍(tasf)在dmf(120μl,1.0m,12.4当量)中的溶液。将所得混合物在室温下搅拌1.5小时,然后减压下将其浓缩至最终体积为约0.1ml。该残余物通过制备型反相hplc纯化(40分钟内30%-45%ch3cn/h2o,ch3cn和h2o均含有0.1%tfa),得到43(9.3mg,73%),为tfa盐。
物理状态:白色膜;
1hnmr(600mhz,meoh-d4)δ9.01(d,j=6.4hz,0.6h),8.73(d,j=5.8hz,0.4h),7.86(d,j=8.8hz,1h),7.75(d,j=2.1hz,1h),7.65(d,j=8.5hz,1h),7.64(d,j=2.1hz,1h)7.61(ddd,j=8.5,2.2,0.9hz,1h),7.21(d,j=8.5hz,1h),7.09(d,j=2.3hz,1h),6.85(d,j=8.8hz,1h),6.78(d,j=8.2hz,1h),6.68(d,j=2.3hz,1h),6.51(d,j=2.2hz,1h),6.13(brs,1h),6.06(s,1h),5.87(s,1h),5.40(dd,j=2.2,1.0hz,1h),5.37(s,1h),5.27(d,j=3.5hz,1h),4.78(s,1h),4.65(s,1h),4.27(dd,j=9.6,1.9hz,1h),4.18(s,1h),4.11(s,3h),4.02(t,j=7.2hz,1h),3.86(s,3h),3.66(s,3h),3.63(s,3h),3.03(d,j=15.7hz,1h),2.76(s,3h),2.03(dd,j=15.7,10.4hz,1h),1.90(dt,j=14.0,7.2hz,1h),1.69–1.57(m,2h),0.88(d,j=6.4hz,3h),0.85(d,j=6.4hz,3h).
13cnmr(600mhz,meoh-d4):δ175.8,174.6,172.8,171.7,170.0,169.9,169.4,169.0,161.9,160.4,158.7,154.2,153.0,152.3,151.1,142.7,141.7,138.1,136.8,136.7,136.6,130.2,129.0,129.0,128.9,128.5,127.6,127.3,125.3,125.3,124.8,122.4,113.8,109.8,106.7,106.3,99.2,74.3,73.4,63.9,62.1,61.9,59.5,58.5,56.6,56.3,56.2,56.0,55.2,53.0,52.9,40.2,38.7,36.4,33.0,25.5,23.2,22.8ppm;
19fnmr(376mhz,meoh-d4):δ-76.9ppm;
hrms(esi-tof,m/z):计算值c57h61cl2n8o17[m+h]+1199.3526;实测值1199.3521.
化合物s46
向42(45mg,0.029mmol,1.0当量)、n-羟基邻苯二甲酰亚胺(26mg,0.16mmol,5.5当量)和n,n-二甲基吡啶-4-胺(0.4mg,0.0033mmol,0.11当量)在ch2cl2(0.5ml)的悬浮液中添加n,n'-二异丙基碳二亚胺(25μl,0.16mmol,5.5当量)。将反应混合物在室温搅拌1h,然后添加acoh(10μl)。将所得混合物再搅拌2小时,然后直接进行快速柱色谱纯化(硅胶,柱:直径(d)1.6cm×长度(l)7.5cm,3:2的etoac:己烷(200ml)至1:19的meoh:ch2cl2(120)ml))。将用meoh-ch2cl2洗脱的合并的级分在减压下浓缩,并且将s46残余物(31mg)无需进一步纯化即可用于下一步。
注意:
(1)lc/ms表明所需的氧化还原活性酯(s46)仅用meoh/ch2cl2洗脱。发现非极性杂质(例如1,3-二异丙基脲)用etoac:己烷洗脱。
(2)dmap额外的量或更长的反应时间对反应收率有害。
(3)该氧化还原活性酯(s46)相当不稳定,应在纯化后3小时内用于下一步。替代地,它可以储存在-20℃下。
化合物s47
将含有s46(31mg)、mgbr2·oet2粉末(38mg,0.15mmol)的旋盖培养管抽空,并用氩气回填3次。接下来加入悬浮液c(0.4ml,在thf中的nicl2·6h2o/di-tbubipy),并将混合物在室温下剧烈搅拌15分钟(或超声处理直至观察不到颗粒状的mgbr2·oet2)。将得到的悬浮液冷却至0℃,并一次性加入[b2pin2me]li在thf(0.55ml)中的悬浮液。搅拌1小时后,将反应混合物用ch2cl2(5ml)稀释,通过硅胶和硅藻土的短垫过滤,用5%meoh/ch2cl2(50ml)洗涤。将滤液在减压下浓缩,并将残余物直接进行快速柱色谱法(硅胶,柱:d1.6cm×l7.5cm,1:1的etoac:己烷(200ml)至1:19的meoh:ch2cl2(120ml))。在减压下浓缩meoh-ch2cl2洗脱液,并且s47残留物(16mg)无需进一步纯化即可用于下一步。
注意:
(1)基于lc/ms分析,发现频哪醇酯在反应过程中水解。
(2)lc/ms表明,根据lc/ms分析,s47仅用meoh/ch2cl2洗脱。发现非极性杂质(例如b2pin2)已用etoac:己烷洗脱。
(3)在该步骤中,并非所有杂质都能通过快速色谱法去除;取而代之的是,不纯的物料被带入下一步骤。
化合物44
向s47(16mg)在ch3cn(1.3ml)中的溶液中添加三(二甲基氨基)二氟三甲基硅酸锍(tasf)在dmf(120μl,1.0m)中的溶液。将混合物在室温下搅拌1.5小时,并在减压下浓缩至最终体积为约0.3ml。残留物通过制备型反相hplc(30分钟内20%–50%ch3cn/h2o,ch3cn和h2o均包含0.1%tfa)纯化,得到44(4.1mg,3个步骤中11%),其为tfa盐。
注意:由于该化合物易于聚合,因而在纯净条件下不稳定。因此,纯化的化合物立即溶解。使用在meoh中的溶液用于hrms;meoh-d4中的溶液用于nmr研究;dmso中的溶液用于生物学研究。
物理状态:白色膜;
1hnmr(600mhz,meoh-d4):δ9.05(d,j=6.6hz,1h),7.65–7.58(m,4h),7.31(d,j=9.0hz,1h),7.30(d,j=9.6hz,1h),7.15(dd,j=9.0hz,1.8hz,1h),6.97–6.91(m,2h),6.81(s,1h),6.52(d,j=2.4hz,1h),5.81(d,j=5.4hz,1h),5.69(s,1h),5.65(s,1h),5.54(s,1h),5.35(d,j=3.6hz,1h),5.07(brs,1h),5.04(d,j=6.6hz,1h),4.43(s,1h),4.32(d,j=5.4hz,1h),4.14(s,3h),4.03(t,j=7.2hz,1h),3.87(s,3h),3.68(s,3h),3.65(s,3h),2.96(d,j=15.6,1h),2.77(s,3h),2.34(dd,j=16.2hz,9.0hz,1h),1.86–1.82(m,1h),1.79–1.73(m,1h),1.71–1.86(m,1h),1.01(d,j=6.0hz,3h),0.98(d,j=6.0hz,3h)ppm;
11bnmr(500mhz,meoh-d4):δ-0.87(s)ppm;
hrms(esi-tof,m/z):计算值c56h62bcl2n8o17[m+h]+1199.3698;实测值1199.3698.
43、44、万古霉素和万古霉素糖苷配基抗生素评估的实验程序。
抗生素敏感性使用临床和实验室标准协会肉汤微稀释法测定(63)。简而言之,将抗生素以2倍稀释度的形式在含有阳离子调节mueller-hinton肉汤(金黄色葡萄球菌菌株)或脑心浸液(肠球菌菌株)的96孔板上制备。抗生素储备液在二甲基亚砜(dmso)中制得。从稀释至最终浓度为5×105cfu/ml的新鲜平板刮板接种孔,并在37℃下孵育。在20小时视觉观察生长。所有mic都是至少三个独立测定值的平均值。
a金黄色葡萄球菌(staphylococcusaureus)(atcc25923)
b金黄色葡萄球菌(staphylococcusaureus)(耐甲氧西林,atcc43300)
c屎肠球菌(enterococcusfaecium)(vana,atccbaa-2317)
d粪肠球菌(enterococcusfaecalis)(vana,bm4166)
e粪肠球菌(enterococcusfaecalis)(vanb,atcc51299)
注意:化合物44在37℃,在空气下,在h2o中不是很稳定。在这种情况下,lc/ms分析(254nmuv检测器)表明,在24小时后,发现44中的20%分解。
探测肽底物上的立体选择性
化合物s48
1,3-二氧代异二氢吲哚-2-基(叔丁氧羰基)-l-丙氨酰基-l-缬氨酸酯(s48)
在–10℃下,将n-(3-二甲基氨基丙基)-n'-乙基碳二亚胺盐酸盐(edc,422mg,2.2mmol,1.1当量)加入boc-l-ala-l-val-oh(2.0mmol,1.0当量)和nhpi(359mg,2.2mmol,1.1当量)的ch2cl2(30ml)溶液中。在室温下搅拌1小时后,将混合物用水洗涤,并将水相用ch2cl2萃取三次。合并的有机相经无水na2so4干燥并真空浓缩。通过快速柱色谱法(硅胶,3∶7的etoac∶己烷至etoac)纯化,得到s48(591mg,62%)。
物理状态:白色泡沫;
rf=0.36(硅胶,2:3的etoac∶己烷);
1hnmr(600mhz,cdcl3):δ7.88(dd,j=5.5,3.1hz,2h),7.79(dd,j=5.5,3.1hz,2h),6.93–6.79(br,1h),5.02–4.88(m,2h),4.28–4.14(br,s,1h),2.48–2.32(m,1h),1.44(s,9h),1.38(d,j=7.0hz,3h),1.10(d,j=6.9hz,3h),1.08(d,j=6.9hz,3h)ppm;
13cnmr(151mhz,cdcl3):δ172.6,168.4,161.6,155.9,135.0,129.0,124.2,80.5,55.6,50.0,31.8,28.4,18.9,17.5ppm;
hrms(esi-tof,m/z):计算值c21h28n3o7[m+h]+434.1922;实测值434.1930.
化合物45
((2s)-1-((2-甲基-1-(4,4,5,5-四甲基-1,3,2-二氧杂硼杂环戊烷-2-基)丙基)氨基)-1-氧代丙烷-2-基)氨基甲酸叔丁酯(45)
在0.2mmol的规模上,按照通用程序c加入悬浮液c(在thf中的nicl2·6h2o/di-tbubipy)。快速柱色谱法(硅胶,3:7的etoac:己烷),得到45,为不可分离的非对映异构体的混合物(50mg,dr=1:1,67%)
物理状态:无色油状;
rf=0.22(硅胶,3:7的etoac∶己烷);
1hnmr(600mhz,c6d6):δ6.77(s,1h),6.73(s,1h),5.56(s,1h),5.37(s,1h),4.23(s,1h),4.11(s,1h),3.05(s,2h),2.10–2.04(m,2h),1.41(s,9h),1.40(s,9h),1.25–0.92(m,42h)ppm;
13cnmr(151mhz,c6d6):δ174.7,174.2,156.0,155.9,82.8,82.6,79.4,74.7,49.1,49.0,37.0,30.3,28.4,25.3,25.3,25.2,25.0,20.7,20.7,20.0,19.9,17.9,17.7ppm;
hrms(esi-tof,m/z):计算值c18h36bn2o5[m+h]+371.2712;实测值371.2710.
方案s3。46的合成
在2.0mmol的规模上,按照通用程序a加入boc-l-val-l-val-oh(s49)。通过快速柱色谱法(硅胶,1∶3的etoac∶己烷)纯化,得到s50a(187mg,20%)和s50b(395mg,43%)。
化合物s50a
1,3-二氧代异二氢吲哚-2-基(叔丁氧羰基)-l-戊基-l-缬氨酸酯(s50a)
物理状态:白色泡沫;
rf=0.40(硅胶,1:2的etoac∶己烷);
1hnmr(600mhz,cdcl3):δ7.90–7.88(m,2h),7.81–7.79(m,2h),6.48(brd,j=8.8hz,1h),5.09(brd,j=8.4hz,1h),4.98(dd,j=8.8,5.1hz,1h),3.91(dd,j=8.7,6.8hz,1h),2.43-2.38(m,1h),2.14(brs,1h),1.43(s,9h),1.11(t,j=6.3hz,6h),0.97(dd,j=16.5,6.8hz,6h)ppm;
13cnmr(151mhz,cdcl3):δ171.9,168.4,161.6,156.1,135.0,129.0,124.2,80.2,60.4,55.6,31.7,30.6,28.4,19.4,18.9,18.2,17.7ppm;
hrms(esi-tof,m/z):计算值c18h24n3o5[m-boc+h]+362.1710;实测值362.1705;
[α]d20=-31.8(c0.96,chcl3).
化合物s50b
1,3-二氧代异二氢吲哚-2-基(叔丁氧羰基)-l-戊基-l-缬氨酸酯(s50b)
物理状态:白色泡沫;
rf=0.4(硅胶,1:2的etoac∶己烷);
1hnmr(600mhz,cdcl3):δ7.87–7.85(m,2h),7.79–7.77(m,2h),6.60(brd,j=8.8hz,1h),5.15(d,j=8.9hz,1h),4.96(dd,j=8.8,5.2hz,1h),3.92(dd,j=8.8,6.8hz,1h),2.41-2.36(m,1h),2.10(brs,1h),1.42(s,9h),1.09(dd,j=6.9,4.6hz,6h),0.97(d,j=6.8hz,3h),0.94(d,j=6.8hz,3h)ppm;
13cnmr(151mhz,cdcl3):δ172.0,168.4,161.6,156.1,134.9,128.9,124.1,80.1,60.3,55.6,31.6,30.6,28.4,19.4,18.9,18.2,17.7ppm;
hrms(esi-tof,m/z):计算值c18h24n3o5[m-boc+h]+362.1710;实测值362.1714;
[α]d20=-31.2(c1.0,chcl3).
化合物46
((s)-3-甲基-1-(((s)-2-甲基-1-(4,4,5,5-四甲基-1,3,2-二氧杂硼杂环戊烷-2-基)丙基)氨基)-1-氧丁烷-2-基)氨基甲酸叔丁酯(46)
从s50a:
在0.2mmol的规模上,按照通用程序c,从s50a开始(在这种情况下使用1.0当量的mgbr2·et2o),用悬浮液c(在thf中的nicl2·6h2o/di-tbubipy)。通过快速柱色谱法(硅胶,1:3的etoac:己烷)纯化,得到46,其为不可分离的非对映异构体的混合物(37.1mg,d.r.=1.7:1,47%)
从s50b:
在0.2mmol的规模上,按照通用程序c,从s50b开始(在这种情况下使用1.0当量的mgbr2·et2o),用悬浮液c(在thf中的nicl2·6h2o/di-tbubipy)。通过快速柱色谱法纯化(硅胶,1∶3的etoac∶己烷),得到46,其为不可分离的非对映异构体的混合物(36.5mg,d.r.=1.7∶1,46%)。
物理状态:无色油状;
rf=0.30(硅胶,1:2的etoac∶己烷);
1hnmr(600mhz,cdcl3):δ6.30(brd,j=5.5hz,0.78h),6.22(brs,0.22h),5.10(brd,j=8.7hz,1h),3.92-3.86(m,1h),3.03(brs,1h),2.10(brs,1h),1.96-1.90(m,1h),1.42(s,9h),1.28–1.16(m,12h),0.95(d,j=6.7hz,3h),0.93(d,j=6.7hz,3h),0.92(d,j=6.7hz,6h)ppm;
13cnmr(151mhz,cdcl3):δ172.48(次要),172.46,155.92,83.37,79.91,59.57(次要),59.25,44.96(br),31.10,31.02(次要),30.01,29.91(次要),28.51,28.44,25.18,25.12,25.10(次要),25.03(次要),24.97,20.42,20.37(次要),20.12,20.03,19.35,19.21(次要),18.13(次要),17.90ppm;
hrms(esi-tof,m/z):计算值c20h40bn2o5[m+h]+399.3025;实测值399.3028.
化合物s51
(r)-2-(((s)-1-((1,3-二氧代异二氢吲哚-2-基)氧基)-3-甲基-1-氧丁烷-2-基)氨甲酰基)吡咯烷-1-羧酸叔丁酯(s51)
在1.0mmol的规模(基于boc-l-pro-l-leu-oh)上,使用与合成s48的步骤相同的步骤。通过快速柱色谱法(硅胶,1∶2的etoac∶己烷)纯化,得到s51(308mg,67%)。
物理状态:白色泡沫;
rf=0.4(硅胶,1:1的etoac∶己烷);
1hnmr(600mhz,cdcl3):δ7.88-7.85(m,2h),7.87(brs,0.6h),7.79-7.77(m,2h),6.63(s,0.4h),4.99-4.88(m,1h),4.37-4.30(m,1h),3.61–3.21(m,2h),2.47(brs,0.4h),2.43-2.37(m,1h),2.16(brs,0.6h),2.03-1.76(m,3h),1.51-1.39(m,9h),1.12-1.03(m,6h);(由于混合了旋转异构体,观察到了复杂的光谱);
13cnmr(151mhz,cdcl3):δ172.7,172.0,168.4,161.6,156.3,154.9,140.9,137.2,134.9,130.1,129.0,124.1,115.6,110.4,81.4,80.7,61.3,59.5,55.7,55.1,47.0,31.5,28.5,27.1,24.8,19.0,17.5;(由于混合了旋转异构体,观察到了复杂的光谱);
hrms(esi-tof,m/z):计算值c18h22n3o5[m-boc+h]+360.1554;实测值360.1554.
化合物47
2-(((s)-2-甲基-1-(4,4,5,5-四甲基-1,3,2-二氧杂硼杂环戊烷-2-2-基)丙)氨基甲酰基)吡咯烷-1-羧酸叔丁酯(47)
在0.28mmol的规模上,按照通用程序c加入s51和悬浮液c(在thf中的nicl2·6h2o/di-tbubipy)。通过快速柱色谱法(硅胶,2∶1的etoac∶己烷)纯化,得到47,其为非对映异构体的混合物(70.5mg,d.r.=2.6∶1,63%)。通过1hnmr和noesy在dmso-d6中于65℃确定非对映异构体比率。
物理状态:无色油状;
rf=0.30(硅胶,2:1的etoac∶己烷);
1hnmr(500mhz,dmso-d6):δ8.37(s,0.72h),8.28(s,0.28h),4.25(dd,j=8.5,2.8hz,1h),3.44–3.35(m,1h),3.34–3.27(m,1h),2.46(t,j=5.3hz,0.28h),2.40(t,j=4.7hz,0.72h),2.19–2.05(m,1h),1.89–1.74(m,4h),1.39(s,9h),1.13(s,3.36h),1.12(s,8.64h),0.93–0.85(m,6h)ppm;
13cnmr(126mhz,dmso-d6):δ174.9,153.0,80.6(次要),80.4,78.5,57.5(次要),57.3,46.2,28.9(次要),28.7,27.8,27.7,24.9(次要),24.8,24.7,20.1,20.0(次要),19.2ppm;
hrms(esi-tof,m/z):计算值c20h38bn2o5[m+h]+397.2868;实测值397.2864.
硼替佐米(万珂(velcade))的立体选择性合成
合成方案s4。硼替佐米(万珂)的合成
化合物s53
1,3-二氧代异二氢吲哚-2-基(叔丁氧羰基)-l-苯丙氨酰亮氨酸(s53)
在3.0mmol的规模上,按照通用程序a加入boc-l-phe-l-leu-oh(64)(s62)。通过快速柱色谱法(失活的硅胶,1:5.6的etoac:己烷)纯化,得到s53,其为不可分离的非对映异构体的混合物(1.42g,d.r.=3:2,90%)。非对映体比率通过1hnmr和noesy确定。
物理状态:白色泡沫;
rf=0.50(硅胶,2:3的etoac∶己烷);
1hnmr(600mhz,meoh-d4):次要异构体:δ7.94–7.89(m,4h),7.29–7.15(m,5h),4.78(dd,j=9.5hz,5.8hz,1h),4.39–4.35(m,1h),3.05(dd,j=13.7hz,7.2hz,1h),2.90(dd,j=13.6hz,8.1hz,1h),1.76–1.70(m,2h),1.54–1.50(m,1h),1.38(s,9h),0.94(d,j=6.6hz,3h),0.89(d,j=6.6hz,3h)ppm;主要异构体:δ7.94–7.89(m,4h),7.29–7.15(m,5h),4.92(dd,j=9.6hz,6.0hz,1h),4.39–4.35(m,1h),3.13(dd,j=14.4hz,5.4hz,1h),2.84(dd,j=13.8hz,9.0hz,1h),1.89–1.83(m,3h),1.37(s,9h),1.02(d,j=6.0hz,3h),0.99(d,j=6.0hz,3h)ppm;
13cnmr(151mhz,meoh-d4):次要异构体:δ174.4,170.4,163.1,157.3,138.3,136.3,135.5,130.4,130.1,129.5,127.7,124.9,124.0,80.7,57.4,50.2,41.2,39.6,28.6,28.4,25.7,25.5,23.2,21.7ppm;主要异构体:δ174.6,170.4,163.1,157.6,138.4,136.4,135.5,130.4,130.1,129.4,127.6,124.9,124.0,80.6,57.1,50.2,41.5,39.1,28.6,28.4,25.7,25.5,23.2,21.8ppm;
hrms(esi-tof,m/z):计算值c23h26n3o5[m-boc+h]+424.1867;实测值424.1871.
化合物48
叔丁基((s)-1-(((r)-3-甲基-1-(4,4,5,5-四甲基-1,3,2-二氧杂硼杂环戊烷-2-基)丁基)氨基)-1-氧代-3-苯基丙烷-2-基)氨基甲酸酯(48)
在0.6mmol的规模上,按照通用程序c加入悬浮液c(在thf中的nicl2·6h2o/di-tbubipy)和s53。反应从约15℃开始,经过3小时温热至室温。快速柱色谱法(硅胶,1:9的etoac:己烷至1:4的etoac:己烷),得到48,将其溶解在己烷中并通过硅藻土过滤。真空浓缩滤液,得到作为不可分离的非对映异构体的混合物的48(151mg,d.r.=5.1∶1,55%)。
物理状态:淡黄色油状;
rf=0.50(硅胶,2:3的etoac∶己烷);
1hnmr(600mhz,cdcl3):主要异构体:δ7.31–7.26(m,2h),7.24–7.21(m,3h),6.19(brs,1h),5.00(brs,1h),4.35(q,j=7.3hz,1h),3.10–3.02(m,2h),2.98(ddd,j=8.8hz,6.3hz,4.4hz,1h),1.49–1.42(m,1h),1.39(s,9h),1.37–1.35(m,2h),1.24(s,6h),1.23(s,6h),0.86(d,j=6.6hz,3h),0.84(d,j=6.6hz,3h)ppm;
13cnmr(151mhz,cdcl3):主要异构体:δ172.6,155.5,134.4,129.6,128.8,127.1,83.0,80.3,54.8,39.9,38.3,28.4,25.6,25.1,25.0,23.3,22.0ppm;
hrms(esi-tof,m/z):计算值c25h42bn2o5[m+h]+461.3181;实测值461.3179.
化合物49
(3-甲基-1-((s)-3-苯基-2-(吡嗪-2-羧酰胺基)丙酰胺基)丁基)硼酸(49)
硼替佐米(49)是采用稍加修改的文献方法(19)由48合成的。
boc脱保护:在0℃下向装有48(151mg,0.33mmol)的旋盖培养管中添加hcl的etoac(14wt%)溶液,并将反应混合物在0℃搅拌3h,并在室温下再搅拌1小时。将反应混合物浓缩至干,并将所得固体用己烷洗涤。所需产物为白色固体,无需进一步纯化即可用于下一步。
酯化:将ch2cl2(1.2ml,0.5m)添加至包含从先前步骤获得的盐酸盐的旋盖培养管中。将混合物冷却至0℃。逐滴加入二异丙基乙胺(0.15ml,0.86mmol),并将反应混合物搅拌5分钟。然后将2-吡嗪羧酸(56mg,0.45mmol)一次性加入溶液中。然后将o-(苯并三唑-1-基)-n,n,n',n'-四甲基脲鎓四氟硼酸酯(tbtu,118mg,0.37mmol)加入反应混合物中,将其在0℃下搅拌2小时并在室温下再搅拌1小时。然后将反应混合物真空浓缩。将粗残余物溶于etoac(10ml)中,并转移至分液漏斗中。依次用去离子水(2×10ml)、1%磷酸(2×10ml)、2%k2co3(2×10ml)和盐水(2×10ml)洗涤有机层。每个水层用etoac(2×10ml)反萃取。合并的有机层经无水na2so4干燥,过滤并真空浓缩。所得浅黄色泡沫无需进一步纯化即可用于下一步骤。
硼酸酯交换:将戊烷(0.8ml)和meoh(0.8ml)添加到装有从上一步骤获得的频哪醇硼酸酯的旋盖培养管中。然后将2-甲基丙烷硼酸(125mg,1.2mmol)加入溶液中。将1n的hcl(0.6ml)水溶液加入到反应混合物中,并将得到的两相溶液剧烈搅拌16小时。然后停止搅拌,并使两相混合物分离。水层用戊烷(2×10ml)洗涤,然后真空浓缩。将得到的膜在ch2cl2和1n的naoh水溶液(10ml)之间分配。水层用ch2cl2(3×10ml)洗涤,并将有机相用1n的naoh水溶液(2×10ml)反萃取。当将所需产物用ch2cl2(3×10ml)萃取到有机层中时,将1n的hcl水溶液添加到合并的水层中直到ph=6。合并的有机相经无水na2so4干燥、过滤并真空浓缩。将得到的残余物溶于etoac(2ml),然后将溶液真空浓缩。然后向残余物中加入己烷(2ml),并将悬浮液真空浓缩,得到产物49(64mg,d.r.=5.1∶1,51%,经历3个步骤)。
物理状态:白色固体;
1hnmr(600mhz,cd3cn:d2o=4:1):主要异构体:δ9.10(d,j=1.8hz,1h),8.74(d,j=2.4hz,1h)),8.61(dd,j=2.4hz,1.8hz,1h),7.26–7.22(m,4h),7.20–7.17(m,1h),4.78(dd,j=8.4hz,6.0hz,1h),3.19(dd,j=13.8hz,6.0hz,1h),3.07(dd,j=13.8hz,8.2hz,1h),2.93(dd,j=10.2hz,5.4hz,1h),1.44–1.33(m,2h),1.26–1.21(m,1h),0.80(d,j=6.6hz,3h),0.78(d,j=6.6hz,3h)ppm;
13cnmr(151mhz,cd3cn:d2o=4:1):主要异构体:172.4,164.5,148.7,145.0,144.7,144.4,137.7,130.4,129.5,127.8,54.9,40.2,40.2(brs),38.5,25.9,23.6,22.0ppm;
hrms(esi-tof,m/z):计算值c19h24bn4o3[m–h2o+h]+367.1936;实测值367.1950.
弹性蛋白酶抑制剂50的合成
方案s5。50和50a的合成
化合物50a
(甲氧羰基)-l-戊基-l-脯氨酰-l-缬氨酸(50a)
cbz脱保护:在配有搅拌棒的100ml烧瓶中装入z-l-pro-l-val-otbu(65)(s54,2.55g,6.3mmol)、10%pd/c(128mg,5wt%)和meoh(30ml)。然后将烧瓶抽空,并用来自气球的h2回填三次。将混合物在室温搅拌6h,并通过硅藻土短垫过滤,然后将其用meoh(10ml)冲洗。真空浓缩滤液,得到相应的胺,其为无色油状物。
形成酰胺键:将上述胺依次用s55(1.1g,6.3mmol,1.0当量),hobt·h2o(96mg,0.07mmol,0.11当量)和ch2cl2(25ml)处理。将所得溶液冷却至0℃,然后加入dcc(1.43g,6.9mmol,1.1当量)。将反应混合物在0℃下搅拌30分钟,然后在室温下搅拌过夜。将反应混合物通过硅藻土垫过滤;将滤液重新溶解在etoac中,并依次用0.1n的hcl水溶液、0.1m的nh4oh水溶液和盐水洗涤。有机层经无水na2so4干燥并真空浓缩,得到无色油状的s56(2.2g),其无需进一步纯化即可用于下一步骤。
tbu脱保护:在配有搅拌棒的25ml烧瓶中,将s56(428mg,1.0mmol)溶于ch2cl2(3ml)。加入tfa(3ml),并将所得溶液在室温搅拌5h。真空除去挥发物后,将粗混合物通过快速柱色谱法(硅胶,2:1的etoac:己烷)纯化,得到50a(359mg,80%,历经3个步骤)。
物理状态:白色泡沫;
rf=0.35(硅胶,1:2己烷:etoac);
1hnmr(600mhz,cdcl3):δ7.43(brd,j=8.4hz,2h),6.17(d,j=9.0hz,1h),4.64(dd,j=7.8hz,3.0hz,1h),4.48(dd,j=8.4hz,4.0hz,1h),4.29(t,j=8.4hz,1h),3.84(dd,j=16.8hz,8.4hz,1h),3.69–3.63(m,4h),2.33–2.29(m,1h),2.20–2.10(m,2h),2.03–1.92(m,3h),0.97(d,j=6.6hz,3h),0.95(d,j=6.6hz,3h),0.91(d,j=7.2hz,3h),0.89(d,j=7.2hz,3h)ppm;
13cnmr(151mhz,cdcl3):δ174.5,173.5,171.1,157.8,60.6,58.1,57.8,52.5,48.3,31.4,31.2,27.7,25.2,19.4,19.0,18.1,17.8ppm;
hrms(esi-tof,m/z):计算值c17h30n3o6[m+h]+372.2129;实测值372.2126;
[α]d20=-62.9(c0.79,chcl3)
化合物s57
1,3-二氧代异二氢吲哚-2-基(甲氧羰基)-l-戊基-l-脯氨酰缬氨酸酯(s57)
在2.34mmol的规模上,按照通用程序a加入(甲氧基羰基)-l-戊基-l-脯氨酰-l-缬氨酸。通过快速柱色谱法纯化(硅胶,1:1的etoac:己烷),得到s57(640mg,53%)。
物理状态:白色泡沫;
rf=0.40(硅胶,1:2己烷:etoac);
1hnmr(600mhz,cdcl3)δ7.88–7.84(m,2h),7.78–7.75(m,2h),7.50(d,j=8.4hz,1h),5.61(d,j=9.2hz,1h),4.84(dd,j=8.5,5.0hz,1h),4.61(dd,j=8.1,3.0hz,1h),4.29(dd,j=9.3,6.9hz,1h),3.79–3.72(m,1h),3.63(s,3h),3.64–3.61(m,1h),2.41–2.30(m,2h),2.17(dt,j=12.3,9.1hz,1h),2.01–1.96(m,2h),1.95–1.89(m,1h),1.07(d,j=7.2hz,3h),1.06(d,j=6.6hz,3h),0.97(d,j=6.7hz,3h),0.93(d,j=6.7hz,3h)ppm;
13cnmr(151mhz,cdcl3)δ172.5,171.2,168.4,161.7,157.3,134.9,129.0,124.1,60.0,57.7,56.0,52.4,48.0,31.6,31.4,27.4,25.3,19.5,18.8,17.8,17.7ppm;
hrms(esi-tof,m/z):计算值c25h33n4o8[m+h]+517.2293;实测值517.2289;
[α]d20=-61.0(c1.0,chcl3).
化合物s58
((s)-3-甲基-1-((s)-2-(((r)-2-甲基-1-(4,4,5,5-四甲基-1,3,2-二氧杂硼杂环戊烷-2-基)丙基)氨基甲酰基)吡咯烷-1-基)-1-氧丁烷-2基)-氨基甲酸甲酯(s58)
在0.33mmol的规模上,按照通用程序c加入悬浮液c(在thf中的nicl2·6h2o/di-tbubipy)。在这种情况下,使用了mgbr2·et2o(1.0当量)。通过快速柱色谱法纯化(硅胶,2∶3的etoac∶己烷至20∶1的ch2cl2∶meoh),得到浅黄色油状的s58(72mg,48%)。
化合物50
((r)-1-((s)-1-((甲氧羰基)-l-戊基)吡咯烷-2-甲酰胺基)-2-甲基丙基)硼酸(50)
在氩气下,将氨基硼酸酯s58(24mg,0.053mmol)溶解于ch2cl2(2ml)中;当滴加bcl3(0.16ml,在ch2cl2中为1.0m,3.0当量)时,用干冰/丙酮浴将溶液冷却至-78℃,然后将混合物在-78℃搅拌1h。然后使反应物升温至室温,并真空除去挥发物。加入无水甲醇(4ml),将所得混合物搅拌10分钟,然后真空浓缩。所得残余物用甲醇(4ml)处理10分钟,并真空浓缩。将该过程重复三次。然后将所得粗产物通过制备型反相hplc(10-40%ch3cn/h2o,历时25分钟,ch3cn和h2o均包含0.1%的tfa)纯化,冻干,得到白色松散粉末状的50(15.0mg,76%)。
物理状态:白色粉末;
1hnmr(600mhz,meoh-d4):δ4.61(dd,j=8.4hz,6.0hz,1h),4.17(d,j=7.8hz,1h),3.97–3.93(m,1h),3.75–3.71(m,1h),3.64(s,3h),2.33–2.24(m,2h),2.19–2.13(m,1h),2.08–1.98(m,3h),1.80–1.74(m,1h),1.05(d,j=6.6hz,3h),1.00(d,j=6.6hz,3h),0.96(d,j=6.6hz,3h),0.92(d,j=6.6hz,3h)ppm;
13cnmr(151mhz,meoh-d4):δ179.3,173.5,159.4,59.7,57.9,52.7,31.7,31.0,29.8,26.2,21.4,21.2,19.6,18.8ppm;
hrms(esi-tof,m/z):计算值c16h29bn3o5[m-h2o+h]+354.2195;实测值354.2189;
[α]d20=-81.1(c0.44,meoh).
弹性蛋白酶抑制剂mcbk320(51)和mcbk323(52)的合成
方案s6。51和52的合成
化合物s60
(叔丁氧羰基)-l-戊基-l-脯氨酰-l-缬氨酸(s60)
cbz脱保护:向配有搅拌棒的100ml烧瓶中装入z-l-pro-l-val-ome(66)(s59,1.95g,5.4mmol)、10%pd/c(98mg,5wt%)和meoh(25ml)。然后将该烧瓶抽空,并用来自气球的h2回填3次。将反应混合物在室温搅拌6h,并通过硅藻土薄垫过滤,然后将其用meoh(10ml)冲洗。真空浓缩滤液,得到相应的胺,其为无色油状物。
形成酰胺键:将上述胺依次用boc-l-缬氨酸(1.17g,5.4mmol,1.0当量)、hobt·h2o(83mg,0.61mmol,0.11当量)和ch2cl2(25ml)处理。将所得溶液冷却至0℃,然后加入dcc(1.23g,6.0mmol,1.1当量)。将反应混合物在0℃下搅拌30分钟,然后在室温下搅拌过夜。通过硅藻土垫过滤所得混合物;将滤液真空浓缩,再溶解在etoac中,并依次用0.1n的hcl水溶液、0.1m的nh4oh水溶液和盐水洗涤。有机层经无水na2so4干燥,真空浓缩,并通过快速柱色谱法(硅胶,2:1的etoac:己烷)纯化,得到boc-l-val-l-pro-l-val-ome(1.32g),其为无色油状物。
酯的水解:在配有搅拌棒的25ml烧瓶中装入boc-l-val-l-pro-l-val-ome(1.32g)和thf(3ml)。添加lioh(4ml,1m水溶液),并将所得溶液在室温下剧烈搅拌12h。向反应混合物中加入1n的hcl,直到ph=2-3,然后将混合物用etoac萃取。合并的有机层经无水na2so4干燥并真空浓缩,得到白色泡沫状的s60(1.24g,53%,经历3个步骤),其无需进一步纯化即可用于下一步骤。
化合物s61
1,3-二氧代异二氢吲哚-2-基(叔丁氧基羰基)-l-戊基-l-脯氨酰缬氨酸酯(s61)
在3.0mmol的规模上,按照通用程序a加入boc-l-戊基-l-脯氨酰-l-缬氨酸(s60)。通过快速柱色谱法纯化(硅胶,1:1的etoac:己烷),得到s61(920mg,55%)。
物理状态:白色泡沫;
rf=0.50(硅胶,1:2己烷:etoac);
1hnmr(600mhz,cdcl3):δ7.88–7.85(m,2h),7.79–7.76(m,2h),7.50(d,j=8.4hz,1h),5.28(d,j=9.6hz,1h),4.84(dd,j=8.4,4.8hz,1h),4.62(dd,j=7.8,3.0hz,1h),4.28(dd,j=9.6,6.6hz,1h),3.71–3.77(m,1h),3.60(dt,j=8.4,3.6hz,1h),2.43–2.39(m,1h),2.37–2.31(m,1h),2.11–2.19(m,1h),1.88-2.02(m,3h),1.41(s,9h),1.08(d,j=6.6hz,3h),1.07(d,j=6.6hz,3h),0.98(d,j=7.2hz,3h),0.92(d,j=7.2hz,3h)ppm;
13cnmr(151mhz,cdcl3):δ172.9,171.1,168.3,161.7,156.0,134.9,129.0,124.1,79.7,60.0,57.0,56.1,47.9,31.6,31.6,28.5,27.1,25.4,19.7,18.9,17.8,17.6ppm;
hrms(esi-tof,m/z):计算值c28h39n4o8[m+h]+559.2762;实测值559.2757.
[α]d20=-86.2(c1.0,chcl3).
化合物s62
((s)-3-甲基-1-((s)-2-(((r)-2-甲基-1-(4,4,5,5-四甲基-1,3,2-二氧杂硼杂环戊烷-2-基)丙基)氨基甲酰基)吡咯烷-1-基)-1-氧代丁-2-基)氨基甲酸叔丁酯(s62)
在1.1mmol的规模上,按照通用程序c用s61和悬浮液c(在thf中的nicl2·6h2o/di-tbubipy),在这种情况下,使用1.0当量的mgbr2·et2o。快速柱色谱(硅胶,2:3的etoac:己烷至3:1的etoac:己烷),得到浅黄色油状的s62(257mg,47%)。
物理状态:浅黄色油状;
rf=0.65(硅胶,1:2己烷:etoac);
1hnmr(600mhz,cdcl3):δ7.08(brs,1h),5.22(d,j=9.3hz,1h),4.66(dd,j=8.2,2.6hz,1h),4.28(dd,j=9.3,6.0hz,1h),3.70(q,j=8.7hz,1h),3.56(ddd,j=9.7,8.1,3.7hz,1h),2.97–2.86(m,1h),2.41–2.38(m,1h),2.19–2.11(m,1h),2.01–1.94(m,2h),1.94–1.80(m,2h),1.43(s,9h),1.25(d,j=5.4hz,12h),0.97(d,j=6.8hz,3h),0.95(d,j=6.8hz,3h),0.93(d,j=6.8hz,3h),0.91(d,j=6.7hz,3h)ppm;
13cnmr(151mhz,cdcl3):δ172.8,171.8,156.0,83.3,79.8,59.0,56.9,47.7,31.6,29.8,28.5,27.0,25.3,25.2,25.1,20.6,20.3,19.7,17.5ppm;
hrms(esi-tof,m/z):计算值c25h47bn3o6[m+h]+496.3552;实测值496.3550.
[α]d20=-73.6(c1.0,chcl3).
化合物51
(1-((s)-1-((4-(((4-氯苯基)磺酰基)氨基甲酰基)苯甲酰基)-l-戊基)吡咯烷-2-甲酰胺基)-2-甲基丙基)硼酸(51)
boc脱保护:在配有搅拌棒的培养管中,将s62(55mg,0.11mmol)溶于ch2cl2(1ml)中。在0℃下加入tfa(1ml),并将得到的溶液在0℃下搅拌2小时。使用旋转蒸发仪(水浴温度<25℃)在真空中除去挥发物,残留物无需纯化即可用于下一步。
酯化:然后加入苯甲酸s63(45mg,0.13mmol,1.2当量)和pybop(69mg,0.13mmol,1.2当量),并将混合物溶于dmf(2.0ml)。加入n-甲基吗啉(49μl,0.45mmol,4.0当量),并将反应在室温下搅拌3小时。然后将混合物用etoac稀释,用盐水洗涤,用无水na2so4干燥,在真空中浓缩,并通过快速柱色谱法(硅胶,10:1的ch2cl2:meoh)纯化,得到被一些三吡咯烷膦氧化物污染的频哪醇硼酸酯(69mg)。该混合物无需进一步纯化即可用于下一步骤。
硼酸酯交换:在配有搅拌棒的培养管中,将上述混合物(53mg)和phb(oh)2(14mg)溶于et2o(3ml)。加入2n的hcl(3ml),然后将所得两相混合物在室温下剧烈搅拌36h,然后将其用etoac(5ml×3)萃取。合并的有机层经无水na2so4干燥,真空浓缩。所得残留物通过制备型反相hplc(20-80%ch3cn/h2o,经历35分钟内,ch3cn和h2o均含有0.1%tfa)纯化,冻干,得到51(14.0mg,26%,经历3个步骤)。
物理状态:白色粉末;
1hnmr(600mhz,meoh-d4):δ8.10(d,j=8.4hz,2h),7.94–7.90(m,4h),7.66(d,j=9.0hz,2h),4.64(dd,j=8.4hz,3.6hz,1h),4.61(d,j=9.6hz,1h),4.12(dt,j=9.6hz,6.6hz,1h),3.82(dt,j=9.6hz,6.6hz,1h),2.38–2.31(m,2h),2.25-2.19(m,2h),2.14-2.02(m,2h),1.82–1.77(m,1h),1.16(d,j=6.6hz,3h),1.10(d,j=7.2hz,3h),0.98(d,j=6.6hz,3h),0.94(d,j=6.6hz,3h)ppm;
13cnmr(151mhz,meoh-d4):δ179.2,173.0,169.1,166.9,141.3,139.5,139.4,135.9,131.2,130.3,129.5,128.9,59.0,57.9,31.8,31.1,29.8,26.3,21.4,21.2,19.5,19.5ppm;
hrms(esi-tof,m/z):计算值c28h35bcln4o7s[m-h2o+h]+617.2003;实测值617.2002.
[α]d20=-72.2(c0.36,meoh).
化合物52
(4-(((2s)-1-((2s)-2-((1-硼烷-2-甲基丙基)氨基甲酰基)吡咯烷-1-基)-3-甲基-1-氧丁烷-2-基)氨基甲酰基)苯甲酰基)甘氨酸(52)
boc脱保护:在配有搅拌棒的培养管中,将s62(55mg,0.11mmol)溶于ch2cl2(1ml)中。在0℃下加入tfa(1ml),并将得到的溶液在0℃下搅拌2小时。使用旋转蒸发仪(水浴温度<25℃)在真空中除去挥发物,残留物无需纯化即可用于下一步骤。
酯化:然后加入苯甲酸s64(37mg,0.13mmol,1.2当量)和pybop(69mg,0.13mmol,1.2当量),并将混合物溶于dmf(2.0ml)。加入n-甲基吗啉(49μl,0.45mmol,4.0当量),并将反应在室温下搅拌3小时。然后将混合物用etoac稀释,用盐水洗涤,用无水na2so4干燥,真空浓缩,并通过快速柱色谱法(硅胶,10:1的ch2cl2:meoh)纯化,得到频哪醇硼酸酯(63mg,86%),其不经进一步纯化用于下一步骤。
整体脱保护:在配有搅拌棒的培养管中,将上述频哪醇硼酸酯(32mg)溶于ch2cl2(1ml)中。在0℃下加入tfa(1ml),并将所得溶液在室温下搅拌过夜。使用旋转蒸发仪(水浴温度<25℃)在真空中除去挥发物,并通过制备型反相hplc(20–80%ch3cn/h2o,历经40分钟,ch3cn和h2o均含有0.1%的tfa)纯化残留物,并冻干,得到52(13.0mg,52%,历经3个步骤)。
物理状态:白色粉末;
1hnmr(600mhz,甲醇-d4):δ7.96–7.90(m,4h),4.66–4.59(m,2h),4.16–4.07(m,3h),3.83(dt,j=10.1,6.8hz,1h),2.40–2.29(m,2h),2.26–2.16(m,2h),2.14–2.02(m,2h),1.82–1.72(m,1h),1.17(d,j=6.7hz,3h),1.12(d,j=6.7hz,3h),0.98(d,j=6.6hz,3h),0.95(d,j=6.6hz,3h)ppm;.
13cnmr(151mhz,甲醇-d4):δ179.3,173.1,173.0,169.50,169.48,138.1,138.0,128.8,128.6,59.0,58.0,42.3,31.8,31.1,29.8,26.3,21.4,21.2,19.6,19.5ppm;
hrms(esi-tof,m/z):计算值c24h34bn4o7[m-h2o+h]+501.2515;实测值501.2516;
[α]d20=-97.3(c0.26,meoh).
肽硼酸50、51和52的立体化学分配
方案s8。s62a和s62b的合成
频哪醇α-氨基硼酸酯s66a/s66b使用文献方法(67)进行了稍加修改后制备。
化合物s66a
(r)-2-甲基-n-((r)-2-甲基-1-(4,4,5,5-四甲基-1,3,2-二氧杂硼杂环戊烷-2-基)丙基)丙烷-2-亚磺酰胺(s66a)
依次向配有搅拌棒的培养管中加入pcy3·hbf4(12mg,0.033mmol,1.2mol%)、甲苯(0.55ml)、cuso4水溶液(1.1ml,0.03m,1.2mol%)和苄胺(15.3μl,0.14mmol,5mol%)。在室温下,当先加入醛亚胺s65(480mg,2.74mmol,1.0当量)的甲苯(5.0ml)溶液,然后加入b2pin2(1.39g,5.5mmol,2.0当量)时,搅拌混合物10分钟。将混合物剧烈搅拌14小时,用etoac稀释并通过硅胶塞过滤,用etoac洗脱。浓缩滤液,并通过快速柱色谱法(硅胶,1:3的etoac:己烷)纯化,得到被杂质污染的s66a(1.07g,d.r.>20:
1),该杂质源自可以在下一步骤中除去的b2pin2。
化合物s66b
(r)-2-甲基-n-((s)-2-甲基-1-(4,4,5,5-四甲基-1,3,2-二氧杂硼杂环戊烷-2-基)丙基)丙烷-2-亚磺酰胺(s66b)
向配有搅拌棒的培养管中依次加入p(oph)3的溶液(0.33ml,在甲苯中为0.1m,1.2mol%)、cuso4水溶液(1.1ml,0.03m,1.2mol%)和苄胺(15.3μl,0.14mmol,5mol%)。将混合物搅拌10分钟,然后依次加入醛亚胺s65(480mg,2.74mmol,1.0当量)的甲苯(5.0ml)溶液和b2pin2(1.39g,5.5mmol,2.0当量)。将混合物剧烈搅拌14小时,用etoac稀释,并通过硅胶塞过滤,用etoac洗脱。将滤液真空浓缩,并通过快速柱色谱法(硅胶,1:3的etoac:己烷)纯化,得到被杂质污染的s66b(857mg,d.r.=6.1:1),该杂质源自可以在下一步骤中除去的b2pin2。
使用文献程序(19)制备α-硼胺盐酸盐s67a/s67b
化合物s67a
(r)-2-甲基-1-(4,4,5,5-四甲基-1,3,2-二氧杂硼杂环戊烷-2-基)丙-1-胺盐酸盐(s67a)
在氩气下,将s66a(190mg,被b2pin2杂质污染)溶解在1,4-二噁烷(1.2ml)和甲醇(0.1ml)中。在室温下加入hcl(80μl,4.0m,在1,4-二噁烷中),并将所得混合物在相同温度下搅拌,然后真空除去挥发物。将所得固体用己烷和et2o的2∶1的混合物一起研磨,得到s67a(48mg,42%,历经2个步骤)。
物理状态:白色固体;
1hnmr(600mhz,cdcl3)δ8.23(s,3h),2.79(brs,1h),2.26(pd,j=6.9,4.8hz,1h),1.28(brs,12h),1.11(d,j=7.0hz,3h),1.10(d,j=7.0hz,3h).
13cnmr(151mhz,cdcl3)δ85.2,44.4(br),29.3,25.2,24.8,20.4,19.9.
hrms(esi-tof,m/z):计算值c10h23bno2[m+h]+200.1816;实测值200.1812.
[α]d20=-3.0(c1.0,chcl3).
化合物s67b
(s)-2-甲基-1-(4,4,5,5-四甲基-1,3,2-二氧杂硼杂环戊烷-2-基)丙-1-胺盐酸盐(s67b)
在氩气下,将s66b(350mg,被bpin杂质污染)溶解在1,4-二噁烷(2.4ml)和甲醇(0.2ml)中。在室温下加入hcl(0.16ml,4.0m,在1,4-二噁烷中),并在相同温度下搅拌所得混合物,然后真空除去挥发物。将所得固体用己烷和et2o的2∶1的混合物研磨,得到s67b(94mg,37%,历经2个步骤)。
物理状态:白色固体;
1hnmr(600mhz,cdcl3)δ8.25(s,3h),2.80(q,j=5.6hz,1h),2.26(pd,j=6.9,4.9hz,1h),1.28(brs,12h),1.12(d,j=7.2hz,3h),1.11(d,j=7.2hz,3h);
13cnmr(151mhz,cdcl3)δ85.2,44.5(br),29.3,25.2,24.8,20.4,19.9;
hrms(esi-tof,m/z):计算值forc10h23bno2[m+h]+200.1816;实测值200.1817;
[α]d20=+2.7(c1.0,chcl3).
化合物s62a
向装有boc-l-val-l-pro-oh(s68,34mg,0.11mmol,1.2当量)和1-[双(二甲基氨基)亚甲基]-1h-1,2,3-三唑并[4,5-b]3-氧化六氟磷酸吡啶鎓(hatu,44mg,0.12mmol,1.3当量)的培养管中加入dmf(0.5ml),然后加入二异丙基乙胺(45μl,0.26mmol,2.9当量)。在0℃下逐滴加入在dmf(1.0ml)中的s67a(21mg,0.089mmol)。加完后,将反应物在室温下搅拌1小时。混合物用et2o稀释,用盐水洗涤,经无水na2so4干燥,真空浓缩。通过快速柱色谱法(硅胶,1:1的etoac:己烷至3:1的etoac:己烷)纯化所得残余物,得到呈无色油状的s62a(32.3mg,73%)。
s62a的nmr光谱与通过脱羧硼化制备的s62的光谱一致。这确认了s62中硼的立体中心α的构型为r。
化合物s62b
向装有boc-l-val-l-pro-oh(s68,26mg,0.083mmol,1.2当量)和1-[双(二甲基氨基)亚甲基]-1h-1,2,3-三唑并[4,5-b]3-氧化六氟磷酸吡啶鎓(hatu,34mg,0.089mmol,1.3当量)的培养管中加入dmf(0.5ml),然后加入二异丙基乙胺(45μl,0.2mmol,2.9当量)。在0℃下逐滴加入在dmf(1.0ml)中的s67b(16mg,0.068mmol,1.0当量)。在加完后,将反应物在室温下搅拌1小时。将混合物用et2o稀释,用盐水洗涤,用无水na2so4干燥,真空浓缩,并通过快速柱色谱法(硅胶,1:1的etoac:己烷至etoac)纯化,得到无色油状的s62b(22mg,65%)。
物理状态:无色油状;
rf=0.60(硅胶,1:2的etoac∶己烷);
1hnmr(600mhz,cdcl3)δ7.08(brs,1h),5.21(d,j=9.3hz,1h),4.65(dd,j=8.2hz,2.3hz,1h),4.28(dd,j=9.4hz,6.1hz,1h),3.70(td,j=9.4hz,7.1hz,1h),3.56(ddd,j=9.6hz,8.1hz,3.4hz,1h),2.94(td,j=5.7hz,2.6hz,1h),2.39(ddd,j=12.8hz,6.1hz,2.6hz,1h),2.14–2.05(m,1h),1.98(dtd,j=12.3hz,6.8hz,3.5hz,2h),1.88(tdd,j=11.3hz,9.0hz,5.8hz,2h),1.42(s,9h),1.21(d,j=6.3hz,12h),1.00(d,j=6.8hz,3h),0.94(d,j=6.8hz,3h),0.91(d,j=6.8hz,6h)ppm;
13cnmr(151mhz,cdcl3)δ172.9,171.8,156.0,83.1,79.8,59.0,56.9,47.7,45.5,31.6,29.8,28.5,27.2,25.2,25.12,25.10,20.4,20.3,19.9,17.7ppm.
s62b的nmr光谱与s62的不同。
弹性蛋白酶抑制试验
测试的化合物:
有关化学结构,请参见图4a。有关药动学特征,请参见图4b。
材料和方法:
对化合物50-58、50a,50b和51a进行此测定。通过echo分配器将在dmso中的系列稀释的化合物分配到384孔黑色不透明板中。将0.1μg/ml人嗜中性粒细胞弹性蛋白酶(epc,catalog#se563,owensville,ms)或用测定缓冲液(100mmhepes,500mmnacl,0.02%tween20)稀释的人痰加入384孔板中,用不同浓度的不同化合物在室温下孵育30分钟。反应中的dmso的终浓度为0.1%。然后将100μm最终浓度的弹性蛋白酶底物meosuc-aapv-amc(bachem,catalog#i-1270,torrance,ca)添加到反应系统中,紧接着在pherastar读板器上以380nm的激发和460nm的发射光读取酶动力学特征,间隔3分钟,总共30分钟。用mars软件计算代表酶活性vmax的荧光强度对时间的斜率(slope)。相对抑制百分比计算如下:
使用prism软件使用log(激动剂)对响应(三个参数)方法,基于相对抑制%曲线计算ic50。所有实验按一式三份进行至少独立的三次。所有实验的ic50结果显示为一式三份的平均值,误差线表示标准偏差,如各图所示。对于化合物51、52和58,使用2.5μm、25μm、50μm和100μm弹性蛋白酶底物(meosuc-aapv-amc)重复上述测定,并使用混合模型基于这些结果计算ki/nm值(68)。
人体痰液中弹性蛋白酶浓度的定量:
人体痰液购自discoverylifesciences(lososos,ca)。用测定缓冲液(100mmhepes,500mmnacl,0.02%tween20)将人类痰液按1:10的体积稀释,然后剧烈涡旋。将1:10稀释的人类痰液进一步稀释为1:30、1:90、1:270、1:810和1:2430。如上所述,通过弹性蛋白酶抑制测定法测定弹性蛋白酶浓度。具体而言,在测定缓冲液中制备了一系列人类嗜中性粒细胞弹性蛋白酶标准品(起始浓度为2μg/ml,并进一步按1:2稀释)。将样品和标准品接种在384孔黑色实心底板中;然后,将最终浓度为100μm的底物meosuc-aapv-amc加入反应系统中,紧接着在如上所述用pherastar读板器上读取酶动力学特征。酶动力学读数的斜率通过mars软件计算。基于标准曲线计算人痰的弹性蛋白酶水平。
结果:
注意:平均值±sd,n=3,代表3个独立的一式三份的实验。使用非线性3参数对数抑制曲线来计算ic50值。曲线拟合统计:纯化的hne,r2≥0.95,cf患者痰,r2≥0.93,copd患者痰,r2≥0.93。
ki值:
注意:测量进行了3次重复,并记录了平均值。
弹性蛋白酶抑制的时间依赖性
方法:
弹性蛋白酶抑制测定的程序稍加修改进行。将0.1μg/ml人嗜中性粒细胞弹性蛋白酶用一系列浓度的抑制剂孵育5、15、30和60分钟,然后添加终浓度为100μm的底物meosuc-aapv-amc。在pherastar酶标仪上读取酶动力学特征,并用上述方法计算ic50。
结果:
注意:测量进行了3次重复,并记录了平均值。
血浆稳定性测定
材料:
1)测试化合物50、50a、51、51a和52。在该测定法中,使用丙烷作为对照化合物;使用前,所有储备液均储存在–40℃下。
2)测试系统:从bioreclamationivt(目录号:msepledta2-m;批次号:mse244515)获得至少20个雄性个体的cd-1小鼠血浆。edta-k2用作抗凝剂。
程序:实验前,将冷冻的血浆在37℃的水浴中解冻。将血浆以4000rpm离心5分钟,如有必要,去除血块。根据需要将ph调节至7.4±0.1。制备中间溶液(1mm),并用90μl45%meoh/h2o稀释10μl中间溶液,从而制备了100μm配料溶液。通过将98μl的空白血浆与2μl的配料溶液(100μm)混合以达到2μm的最终浓度,制成重复的测试样品。将样品在37℃下孵育。在每个时间点(0、10、30、60和120分钟),加入400μl终止溶液(由50%meoh/ch3cn中的200ng/ml的1-丁基-3-对甲苯磺酰基脲(tolbutamide)和20ng/ml丁螺环酮组成),以在彻底混合下沉淀蛋白质。然后将样品板以4,000rpm离心10分钟。从每个孔中转移等分的上清液(100μl),并与200μl超纯水混合。将样品以800rpm的速度振摇约10分钟,然后进行lc-ms/ms分析。
数据分析:使用以下公式计算血浆中孵育后测试化合物的剩余百分比:
剩余百分比=100(在tn时的par/在t0时的par)
其中par是分析物与内标(is)的峰面积比,指定的孵育时间点是t0(0分钟),tn(n=0、10、30、60、120分钟)。
lc-ms/ms条件:使用ace5-苯基50×2.1mm色谱柱(零件号ace-125-0502),使用0.1%甲酸的水溶液和0.1%甲酸的乙腈溶液作为流动相,通过lc/ms分析每种化合物。1-丁基-3-对甲苯磺酰基脲(tobutamide)用作内标。收集的数据由analyst1.6.2软件和multiquant3.0.2软件处理。结果:
小鼠肝微粒体代谢稳定性测定
材料:
1)在该测定中测试化合物50、50a、51、51a和52。睾丸激素、双氯芬酸和普罗帕酮用作对照。
2)缓冲液:
1.100mm磷酸钾缓冲液,ph7.4。
2.10mmmgcl2
3)化合物稀释:
通过用dmso(45μl)和1:1甲醇/水(450μl)(浓度=100μm,45%meoh)稀释5μl化合物或对照储备液(在dmso中为10mm)来制备中间溶液。通过用450μl的100mm磷酸钾缓冲液稀释50μl中间溶液来制备工作溶液,ph=7.4(浓度=10μm,4.5%meoh)。
4)nadph再生系统(孵化时最终等电脱氢酶浓度=1单位/ml)包括:购自sigma(产品编号n0505)的β-烟酰胺腺嘌呤二核苷酸磷酸,购自sigma(产品编号i1252)的异柠檬酸和购自sigma(产品编号i2002)的异柠檬酸脱氢酶。
5)使用得自xenotech的小鼠肝微粒体(目录号m1000,批号1310028)制备肝微粒体溶液(终浓度为0.5mg蛋白质/ml)。
6)终止溶液:含有100ng/ml的1-丁基-3-对甲苯磺酰基脲(tolbutamide)和100ng/ml柳胺心定作为内标(is)的冷乙腈
程序:除基质空白外,将10μl/孔的化合物工作溶液或对照工作溶液添加到所有板(t0、t5、t10、t20、t30、t60、ncf60)中。将80μl/孔的微粒体溶液添加到每个板中。将微粒体溶液和化合物的混合物在37℃下孵育约10分钟。然后将10μl/孔的nadph再生系统(预热至37℃)添加到每个板中以开始反应。将板温育指定的持续时间(基质空白:1h;t60:1h;t30:31min;t20:40min;t1050min;t5:55min)。对于ncf60(无辅因子的缩写),未添加nadph再生系统,但被10μl/孔的磷酸钾缓冲液(100mm,ph7.4)代替;将得到的混合物在37℃下孵育1小时。
然后用含有100ng/ml的1-丁基-3-对甲苯磺酰基脲(tolbutamide)和100ng/ml柳胺心定(300μl/孔)的终止溶液(在4℃下冷却)终止反应。将样品板振荡约10分钟,然后在4℃下以4000rpm离心20分钟。离心时,向8个新的96孔板中装入300μl的hplc级水。最后将100μl上清液添加到300μl的hplc级水中,并混合用于lc/ms/ms分析。
apricot移液机器人用于96孔板形式的上述所有添加、混合和转化。
数据分析
一级动力学方程用于计算t1/2和clint(mic):
当
结果:
注意:1)*ncf:无辅助因子的缩写。在60分钟的孵育过程中,没有将nadph再生系统添加到ncf样品中(用缓冲液代替),如果剩余的ncf小于60%,则会出现非nadph依赖性。
1)r2是用于确定动力学常数的线性回归的相关系数。
2)t1/2为半衰期,clint(mic)为固有清除率。
3)对于五个种类,
4)用于小鼠,
5)肝血清除率
肝提取率
由此,对于小鼠肝脏,qh(ml/min/kg肝脏)=90.0ml/min/kg
动力学溶解度测试
材料:测试了化合物50、50a、51、51a和52。
程序:将每种化合物的储备溶液(10μl;在dmso中为10mm)用磷酸盐缓冲液(490μl;50mm,ph6.8)稀释。将所得混合物摇动24小时。然后将样品过滤。然后通过uv光谱法[通过标准曲线(1、20和200μm)校准]确定动力学溶解度。
结果:
caco-2渗透性测定
材料:
1)caco-2培养:将从atcc购买的caco-2细胞以1×105细胞/cm2的密度接种到96孔bd插入板的聚乙烯膜(pet)上,每4~5天更新一次培养基,直到21日至第28天形成融合细胞单层。
2)化合物信息:对化合物51和51a进行测定。地高辛、非诺特罗和普萘洛尔分别用作标准品。
运输方式:
研究中使用的转运缓冲液是含ph值为7.40±0.05的10mmhepes的hbss。按一式两份,以2μm双向测试化合物。地高辛按一式两份以10μm双向测试,而非诺特罗和普萘洛尔按一式两份以2μm沿a(顶)至b(基底外侧)方向测试。将最终dmso浓度调节至小于1%。在37±1℃的co2培养箱中,在饱和湿度下,用5%co2,在没有摇动的情况下,将板孵育2小时。所有样品在与含有内标的乙腈混合后,均以4000rpm离心20分钟。随后,将100μl上清液用100μl蒸馏水稀释以用于lc/ms/ms分析。使用分析物/内标物的峰面积比,通过lc/ms/ms方法对起始溶液、供体溶液和接收器溶液中测试化合物和对照化合物的浓度进行定量。在转运测定后,应用萤光素黄排斥测定来确定caco-2细胞单层完整性。本文提供的所有数据均已通过此测试。
数据分析:表观渗透系数papp(cm/s)使用以下公式计算:
其中,
外排率的计算公式如下:
外排率=papp(ab)/papp(ab)
回收率使用以下公式计算:
%回收率=100×[(vr·cr)+(vd·cd)]/(vd·c0)
其中vd是供体室的体积(顶侧为0.075ml,基底外侧为0.25ml);cd和cr分别是供体室和接收器室中转运化合物的最终浓度。
lc/ms条件:使用ace5-苯基50×2.1mm色谱柱(零件号ace-125-0502)通过lc/ms分析每种化合物,其中0.1%的甲酸水溶液和0.1%的乙腈甲酸作为流动相。1-丁基-3-对甲苯磺酰基脲(tobutamide)用作内标。收集的数据由analyst1.6.2软件和multiquant3.0.2软件处理。
结果:
注意:1)对于地高辛和测试化合物,接收器样品中的信号响应低于定量的限度。为了方便计算papp值,取而代之的是使用50作为接收器样品中分析物的峰面积。
2)在37±1℃和5%co2和饱和湿度下孵育120分钟,评估渗透率。
与三氟甲基酮51a相比,硼酸酯51和52的adme数据摘要
实施例部分中引用的文献
69.a.suzuki,angew.chem.int.ed.50,6722(2011).
70.w.l.a.brooks,b.s.sumerlin,chem.rev.116,1375(2016).
71.s.d.bull,etal.acc.chem.res.46,312(2013).
72.p.c.trippier,c.mcguiganmed.chem.commun.1,183(2010).
73.a.draganov,d.wang,b.wang,top.med.chem.17,1(2016).
74.c.ballatore,d.m.huryn,a.b.smith,chemmedchem8,385(2013).
75.r.smoum,a.rubinstein,v.m.dembitsky,m.srebnik,chem.rev.112,4156(2012).
76.h.c.brown,hydroboration(benjamin/cummings,1980).
77.c.m.vogels,s.a.westcott,curr.org.chem.9,687(2005).
78.a.s.dudnik,g.c.fu,j.am.chem.soc.134,10693(2012).
79.t.c.atack,r.m.lecker,s.p.cook,j.am.chem.soc.136,9521(2014).
80.r.b.bedfordetal.,organometallics33,5940(2014).
81.c.-t.yangetal.,angew.chem.int.ed.51,528(2012).
82.h.ito,k.kubota,org.lett.14,890(2012).
83.h.c.brown,t.e.cole,organometallics2,1316(1983).
84.k.-s.lee,a.r.zhugralin,a.h.hoveyda,j.am.chem.soc.131,7253(2009).
85.j.a.schniffner,k.müther,m.oestreich,angew.chem.int.ed.49,1194(2010).
86.p.andrés,g.ballano,m.isabelcalaza,c.cativiela,chem.soc.rev.45,2291(2016).
87.m.a.beenen,c.an,j.a.ellman,j.am.chem.soc.130,6910(2008).
88.i.a.mkhalid,j.h.barnard,t.b.marder,j.m.murphy,j.f.hartwig,chem.rev.110,890(2010).e.j.olhava,m.d.danca,us7442830b1(2008).
89.t.qinetal.,science352,801(2016).
90.j.cornellaetal.,j.am.chem.soc.138,2174(2016).
91.j.wangetal.,angew.chem.int.ed.55,9676(2016).
92.f.toriyamaetal.,j.am.chem.soc.138,11132(2016).
93.t.qinetal.,angew.chem.int.ed.55,266(2016).
94.t.hatakeyamaetal.,j.am.chem.soc.132,10674(2010).
95.r.b.bedfordetal.,chem.eur.j.20,7935(2014).
96.r.a.hussainyetal.,j.med.chem.54,3480(2011).
97.p.lassalasetal.,acsmed.chem.lett.59,3183(2016).
98.j.schmidt,j.choi,a.liu,m.slusarczyk,g.c.fu,science354,1265(2016).
99.y.xi,j.hartwig,j.am.chem.soc.138,6703(2016).
100.g.a.molander,n.ellis,acc.chem.res.40,275(2007).
101.s.n.mlyanrski,a.s.karns,j.p.morken,j.am.chem.soc.134,16449(2012).
102.a.bonet,m.odachowski,d.leonori,s.essafi.v.k.aggarwal,nat.chem.6,584(2014).
103.s.laulhé,j.m.blackburn,j.l.roizen,org.lett.18,4440(2016).v.m.dembitsky,m.srebnik,tetrahedron59,579(2003).j.j.mcatee,s.l.castle,q.jin,d.l.boger,bioorg.med.chem.lett.12,1319(2002).
104.p.r.bernsteinetal.,j.med.chem.37,1259(1994).
105.c.a.vealeetal.,j.med.chem.40,3173(1997).
106.p.r.bernsteinetal.,j.med.chem.38,212(1995).
107.j.p.burkhartetal.,j.med.chem.37,223(1995).
108.p.d.edwardsetal.,j.med.chem.40,1876(1997).
109.k.hemmi,i.shima,k.imai,h.tanaka,ep0494071a2(1992).
110.t.kinoshita,i.nakanishi,a.sato,t.tada,bioorg.med.chem.lett.13,21(2003).
111.f.vonnussbaum,v.m.-j.li,bioorg.med.chem.lett.25,4370(2015).
112.f.vonnussbaum,etal.,chemmedchem10,1163(2015).
113.f.otto,etal.,wo2015096873(2015).
114.t.j.blench,etal.,wo2013037809a1(2013).
115.l.
116.m.d.schultz,bioorg.med.chem.lett.23,5992(2013).
117.a.zervosen,etal.,j.am.chem.soc.133,10839(2011).
118.m.groll,c.r.berkers,h.l.ploegh,h.ovaa,structure14,451(2006).
119.m.d.schultz,bioorg.med.chem.lett.23,5992(2013).
120.a.zervosen,etal.,j.am.chem.soc.133,10839(2011).
121.m.groll,c.r.berkers,h.l.ploegh,h.ovaa,structure14,451(2006).
本文所引用的所有专利和出版物都以相同的程度通过引用并入本文,就好像每个单独的出版物均被明确地和单独地表示通过引用整体并入。
- 还没有人留言评论。精彩留言会获得点赞!