一种用于长链分子的数学序列重建方法与流程
本发明涉及一种长链分子序列数学重建算法,尤指一种用于基因dna序列测定、蛋白质氨基酸序列测定或其他长链结构化学物质检测的一种用于长链分子的数学序列重建方法。
背景技术:
在生物学和材料学中都涉及到序列的检测,即对某一链的各种基团排列进行测定,如蛋白质的序列测定,dna的序列测定,多糖的序列测定等等,以dna测序为例,来阐述序列测定中面对的瓶颈,在分子生物学研究中,dna的序列分析是进一步研究和改造目的基因的基础。
目前用于测序的技术主要有sanger等(1977)发明的双脱氧链末端终止法和maxam和gilbert(1977)发明的化学降解法,这二种方法在原理上差异很大,但都是根据核苷酸在某一固定的点开始,随机在某一个特定的碱基处终止,产生a,t,c,g四组不同长度的一系列核苷酸,然后在尿素变性的page胶上电泳进行检测,从而获得dna序列;sanger测序法属第一代测序技术,是测序技术发展的源头,此技术是通过核苷酸在某一固定的点开始,随机在某一个特定的碱基处终止,并且在每个碱基后面进行荧光标记,产生以a、t、c、g结束的四组不同长度的一系列核苷酸,然后在尿素变性的page胶上电泳进行检测,从而获得可见dna碱基序列的一种方法;第一代测序技术的主要特点是测序读长可达1000bp,准确性高达99.999%,但其测序成本高,通量低等方面的缺点,严重影响了其真正大规模的应用。因而第一代测序技术并不是最理想的测序方法。
经过不断的技术开发和改进,以roche公司的454技术、illumina公司的solexa,hiseq技术和abi公司的solid技术为标记的第二代测序技术诞生了;第二代测序技术大大降低了测序成本的同时,还大幅提高了测序速度,并且保持了高准确性,以前完成一个人类基因组的测序需要3年时间,而使用二代测序技术则仅仅需要1周,但在序列读长方面比起第一代测序技术则要短很多;不同公司第二代基因检测技术的共同特点是dna待测文库的构建,即利用超声波把待测的dna样本打断成小片段,目前除了组装之外和一些其他的特殊要求之外,主要是打断成200-500bp长的序列片段,并在这些小片段的两端添加上不同的接头,构建出单链dna文库,然后通过不同的技术将此文库中的不同序列片段进行检测,最后对此文库中的序列片段进行拼接;通过对dna链的碎片化,实现由sanger测序法的单链检测转化为多链检测,进而实现多通量,快速测序。
然而测序技术在近两三年中又有新的里程碑,以pacbio公司的smrt和oxfordnanoporetechnologies纳米孔单分子测序技术,被称之为第三代测序技术,与前两代相比,他们最大的特点就是单分子测序,测序过程无需进行pcr扩增;其中pacbiosmrt技术其实也应用了边合成边测序的思想,并以smrt芯片为测序载体;基本原理是:dna聚合酶和模板结合,4色荧光标记4种碱基(即是dntp),在碱基配对阶段,不同碱基的加入,会发出不同光,根据光的波长与峰值可判断进入的碱基类型;同时这个dna聚合酶是实现超长读长的关键之一,读长主要跟酶的活性保持有关,它主要受激光对其造成的损伤所影响。pacbiosmrt技术的一个关键是怎样将反应信号与周围游离碱基的强大荧光背景区别出来,他们利用的是zmw(零模波导孔)原理:如同微波炉壁上可看到的很多密集小孔,小孔直径有考究,如果直径大于微波波长,能量就会在衍射效应的作用下穿透面板而泄露出来,从而与周围小孔相互干扰;如果孔径小于波长,能量不会辐射到周围,而是保持直线状态(光衍射的原理),从而可起保护作用;同理,在一个反应管(smrtcell:单分子实时反应孔)中有许多这样的圆形纳米小孔,即zmw(零模波导孔),外径100多纳米,比检测激光波长小(数百纳米),激光从底部打上去后不能穿透小孔进入上方溶液区,能量被限制在一个小范围(体积20x10-21l)里,正好足够覆盖需要检测的部分,使得信号仅来自这个小反应区域,孔外过多游离核苷酸单体依然留在黑暗中,从而实现将背景降到最低。另外,可以通过检测相邻两个碱基之间的测序时间,来检测一些碱基修饰情况,既如果碱基存在修饰,则通过聚合酶时的速度会减慢,相邻两峰之间的距离增大,可以通过这个来之间检测甲基化等信息,smrt技术的测序速度很快,每秒约10个dntp;但是,同时其测序错误率比较高(这几乎是目前单分子测序技术的通病),达到15%,其出错是随机的,并不会像第二代测序技术那样存在测序错误的偏向,因而需要通过多次测序来进行有效的纠错。
技术实现要素:
为解决上述问题,本发明旨在公开一种长链分子序列数学重建算法,尤指一种用于基因dna序列测定、蛋白质氨基酸序列测定或其他长链结构化学物质检测的一种用于长链分子的数学序列重建方法;通过本发明的数学测序方法,提高第二代基因测序中的准确性,且本发明建立在严谨的数学算法上准确度更高。
为实现上述目的,本发明采用的技术方案是:
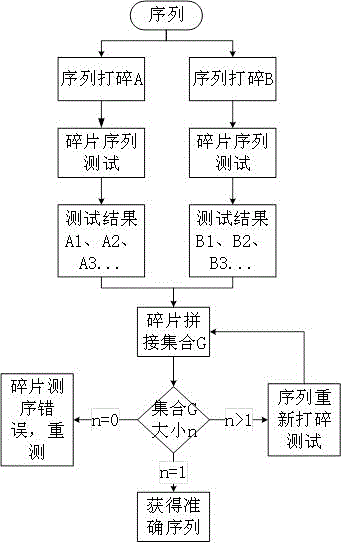
一种用于长链分子的数学序列重建方法,其特征在于,所述的测序方法主要包括以下步骤:
1)提供一个个体中至少两个待测dna分子链,或者对某个体的dna链用pcr仪进行增殖,设dna分子数量为x,x为≥2的自然数;
2)将所述x个dna分子打断成碎片序列,形成x个基因文库;
3)对x个基因文库的基因片段进行测序,获得x个基因文库的片段信息集合;
4)将x个基因文库的碎片进行全排列拼接,获取可能性集合;
5)对可能性集合求交集;
6)判断交集中的元素数量,通过精确的元素数量等式,筛选得到正确的基因序列图谱。
进一步地,当所述步骤5)的交集只有一个时,所得结果为所测试分子的序列结构;否则,重复所述步骤1)-5)进行测试和计算。
进一步地,所述步骤3)中,设x个基因文库分别为基因文库a、基因文库b、…、基因文库x,基因文库a的片段信息为{a1,a2,a3,…,am},基因文库b的片段信息为{b1,b2,b3,…,bn},基因文库x的片段信息为{x1,x2,x3,…,xn}。
进一步地,所述步骤4)中,设可能性集合分别为集合a、集合b、…、集合x,步骤5)的交集为交集g,g=a∩b∩…∩x;当g中元素数量=0,则步骤6)结果判断为x个基因文库中存在错误测序;若g中元素数量=1,则步骤6)结果判断交集g为正确的基因序列图谱;若g中元素数量>1,则需要重复步骤1)-5)的测试和计算,直到g中元素数量=1时,得到正确的基因序列图谱。
进一步地,所述步骤1)的dna分子链还可以替换为单一蛋白质分子链、单一多糖链,以测定重建蛋白质、多糖序列。
本发明的有益效果体现在:本发明属于数学算法技术,通过本发明的步骤方法可实现基因序列的测定重建,以及蛋白质、多糖或其他具有单一结构的聚合物的序列测定重建;采用本发明的步骤时,自动判断序列的正确性而进行重测,可提高目前的基因测序中的准确性,且本发明并非建立在基因库样本进行概率推测,而是建立在严谨的数学算法上,测定结构准确度高。
附图说明
图1是本发明的操作流程图。
具体实施方式
下面结合附图详细说明本发明的具体实施方式:
一种用于长链分子的数学序列重建方法,所述的测序方法主要包括以下步骤:
1)提供一个个体中至少两个待测dna分子链,或者对某个体的dna链用pcr仪进行增殖,设dna分子数量为x,x为≥2的自然数;dna分子链还可以替换为单一蛋白质分子链、单一多糖链或其他具有单一结构聚合物分子,以测定重建蛋白质、多糖序列;
2)将所述x个dna分子打断成碎片序列,形成x个基因文库;
3)对x个基因文库的基因片段进行测序,获得x个基因文库的片段信息集合;设x个基因文库分别为基因文库a、基因文库b、…、基因文库x,基因文库a的片段信息为{a1,a2,a3,…,am},基因文库b的片段信息为{b1,b2,b3,…,bn},基因文库x的片段信息为{x1,x2,x3,…,xn};
4)将x个基因文库的碎片进行全排列拼接,获取可能性集合;设可能性集合分别为集合a、集合b、…、集合x;
5)对可能性集合求交集;交集为交集g,g=a∩b∩…∩x;
6)判断交集中的元素数量,通过精确的元素数量等式,筛选得到正确的基因序列图谱;当所述步骤5)的交集只有一个时,所得结果为所测试分子的序列结构;否则,重复所述步骤1)-5)进行测试和计算;更具体地说,当g中元素数量=0,则步骤6)结果判断为x个基因文库中存在错误测序;若g中元素数量=1,则步骤6)结果判断交集g为正确的基因序列图谱;若g中元素数量>1,则需要重复步骤1)-5)的测试和计算,直到g中元素数量=1时,得到正确的基因序列图谱。
本发明与更新迭代的基因测序技术相比:第一代基因测序技术较其他现有技术较为准确,但速度太慢,第二代基因测试技术在第一代基因测序技术将单链基因打断成碎片,生成基因文库,同时对此碎片进行多通量分析,进而大大提高检测效率,带来的问题是如何将基因碎片进行重新拼接以得到完成基因图谱,目前的技术是blast,即利用现有基因库样本进行统计分析,获得最可能的定位,但此项技术存在测序错误的偏向;第三代测序技术采用单分子测序,测序错误率比较高,达到15%,本发明通过一种新型的基因碎片重新定位的技术和算法,从而提高第二代基因测序中的准确性,且本发明不是建立在基因库样本的概率推测,而是建立在严谨的数学算法上,准确度高。本发明把再测序得到的基因组序列在基因组上快速重建,通过blast进行序列比对,blast(basiclocalalignmentsearchtool)是一套在蛋白质数据库或dna数据库中进行相似性比较的分析工具,blast程序能迅速与公开数据库进行相似性序列比较。blast结果中的得分是对一种对相似性的统计说明,具体地说:采用本地blast(stand-aloneblast)把测出的基因组序列和人类基因组作批量比对(mega-blast),提交的序列片段在比对结束之后,可得到的是每一条序列在基因组染色体上的准确定位。
本发明不属于现有的算法逻辑,目前尚未有数学理论获得本发明的方法技术算法的原理,在本发明应用前,没有任何公开方法可达到本发明中准确率高的测定重建结果,因此本发明采用的方案并非显而易见的,对比所有的现有技术中,本发明不仅有效提高检测效率,准确度可达到100%,获得的结果是绝对准确,因此本发明的优点显而易见的,具有明显的进步性。
以人类基因组为例,人体基因组由23对染色体组成,其中包括22对体染色体、1条x染色体和1条y染色体,人类基因组含有约31.6亿个dna碱基对,碱基对是以氢键相结合的两个含氮碱基,以胸腺嘧啶(t)、腺嘌呤(a)、胞嘧啶(c)和鸟嘌呤(g)四种碱基排列成碱基序列;以100个碱基对为例,如果随机打断为20个碎片,那么20个碎片可以获得的拼接集合中将包含20!个元素,故不能够以全系列基因组来说明本发明的实施,本发明需要通过超级计算机和大规模存储器实现,通过算法设计可以大大减少本技术的计算量和存储量。
本发明文件以matlab计算来模拟短序列的重建算法,以说明本算法的可行性,详细如下:
例一:
对于需测定某基因序列tctaactg,对此基因序列进行随机打断,获取基因碎片tct、aac、tg,以此为基础建立起碎片文库a{'tct';'aac';'tg'};对此基因序列再进行随机打断,获取基因碎片tc、taac、tg,以此为基础建立起碎片文库b{'tc';'taac';'tg'}。对碎片文库a和碎片文库b分别进行全排列,获取两个集合a和b,求a和b的交集,c=a∩b={tctaactg},恰好是我们需重建的基因序列。
文库a{'tct';'aac';'tg'}如下:
{
'tctaactg'
'tcttgaac'
'aactcttg'
'aactgtct'
'tgtctaac'
'tgaactct'
}
文库b{'tc';'taac';'tg'}如下:
{
'tctaactg'
'tctgtaac'
'taactctg'
'taactgtc'
'tgtctaac'
'tgtaactc'
}
a和b的交集,c=a∩b={tctaactg}
例二:
对于需测定某基因序列tctaactggcgcctcgctgtggaaaa,对此基因序列进行随机打断,获取基因碎片tctaactgg、cgcctcgctg、tg和gaaaa,以此为基础建立起碎片文库a{'tctaactgg';'cgcctcgctg';'tg';'gaaaa'};对此基因序列再进行随机打断,获取基因碎片tctaact、g、gcg、cctcgc和tgtggaaaa,以此为基础建立起碎片文库b{'tctaact';'g';'gcg';'cctcgc';'tgtggaaaa'}。对碎片文库a和碎片文库b分别进行全排列,获取两个集合a和b,求a和b的交集,c=a∩b={tctaactggcgcctcgctgtggaaaa},恰好是我们需重建的基因序列,详细计算过程如下:
gene='tctaactggcgcctcgctgtggaaaa';//序列总数为24
//随机打断成1-10的小片段
breaks={'tctaactgg''cgcctcgctg''tg''gaaaa'};
//对此4个小片段全排列,可得24种不同序列
a={'gaaaatgcgcctcgctgtctaactgg'
'gaaaatgtctaactggcgcctcgctg'
'gaaaacgcctcgctgtgtctaactgg'
'gaaaacgcctcgctgtctaactggtg'
'gaaaatctaactggcgcctcgctgtg'
'gaaaatctaactggtgcgcctcgctg'
'tggaaaacgcctcgctgtctaactgg'
'tggaaaatctaactggcgcctcgctg'
'tgcgcctcgctggaaaatctaactgg'
'tgcgcctcgctgtctaactgggaaaa'
'tgtctaactggcgcctcgctggaaaa'
'tgtctaactgggaaaacgcctcgctg'
'cgcctcgctgtggaaaatctaactgg'
'cgcctcgctgtgtctaactgggaaaa'
'cgcctcgctggaaaatgtctaactgg'
'cgcctcgctggaaaatctaactggtg'
'cgcctcgctgtctaactgggaaaatg'
'cgcctcgctgtctaactggtggaaaa'
'tctaactggtgcgcctcgctggaaaa'
'tctaactggtggaaaacgcctcgctg'
'tctaactggcgcctcgctgtggaaaa'
'tctaactggcgcctcgctggaaaatg'
'tctaactgggaaaacgcctcgctgtg'
'tctaactgggaaaatgcgcctcgctg'}
//对之前的gene序列随机打断成1-10的小片段
breaks={'tctaact''g''gcg''cctcgc''tgtggaaaa'};
//对此5个小片段全排列,可得120种不同序列
b={'tgtggaaaacctcgcgcggtctaact'
'tgtggaaaacctcgcgcgtctaactg'
'tgtggaaaacctcgcggcgtctaact'
'tgtggaaaacctcgcgtctaactgcg'
'tgtggaaaacctcgctctaactggcg'
'tgtggaaaacctcgctctaactgcgg'
'tgtggaaaagcgcctcgcgtctaact'
'tgtggaaaagcgcctcgctctaactg'
'tgtggaaaagcggcctcgctctaact'
'tgtggaaaagcggtctaactcctcgc'
'tgtggaaaagcgtctaactgcctcgc'
'tgtggaaaagcgtctaactcctcgcg'
'tgtggaaaaggcgcctcgctctaact'
'tgtggaaaaggcgtctaactcctcgc'
'tgtggaaaagcctcgcgcgtctaact'
'tgtggaaaagcctcgctctaactgcg'
'tgtggaaaagtctaactcctcgcgcg'
'tgtggaaaagtctaactgcgcctcgc'
'tgtggaaaatctaactgcggcctcgc'
'tgtggaaaatctaactgcgcctcgcg'
'tgtggaaaatctaactggcgcctcgc'
'tgtggaaaatctaactgcctcgcgcg'
'tgtggaaaatctaactcctcgcggcg'
'tgtggaaaatctaactcctcgcgcgg'
'cctcgctgtggaaaagcggtctaact'
'cctcgctgtggaaaagcgtctaactg'
'cctcgctgtggaaaaggcgtctaact'
'cctcgctgtggaaaagtctaactgcg'
'cctcgctgtggaaaatctaactggcg'
'cctcgctgtggaaaatctaactgcgg'
'cctcgcgcgtgtggaaaagtctaact'
'cctcgcgcgtgtggaaaatctaactg'
'cctcgcgcggtgtggaaaatctaact'
'cctcgcgcggtctaacttgtggaaaa'
'cctcgcgcgtctaactgtgtggaaaa'
'cctcgcgcgtctaacttgtggaaaag'
'cctcgcggcgtgtggaaaatctaact'
'cctcgcggcgtctaacttgtggaaaa'
'cctcgcgtgtggaaaagcgtctaact'
'cctcgcgtgtggaaaatctaactgcg'
'cctcgcgtctaacttgtggaaaagcg'
'cctcgcgtctaactgcgtgtggaaaa'
'cctcgctctaactgcggtgtggaaaa'
'cctcgctctaactgcgtgtggaaaag'
'cctcgctctaactggcgtgtggaaaa'
'cctcgctctaactgtgtggaaaagcg'
'cctcgctctaacttgtggaaaaggcg'
'cctcgctctaacttgtggaaaagcgg'
'gcgcctcgctgtggaaaagtctaact'
'gcgcctcgctgtggaaaatctaactg'
'gcgcctcgcgtgtggaaaatctaact'
'gcgcctcgcgtctaacttgtggaaaa'
'gcgcctcgctctaactgtgtggaaaa'
'gcgcctcgctctaacttgtggaaaag'
'gcgtgtggaaaacctcgcgtctaact'
'gcgtgtggaaaacctcgctctaactg'
'gcgtgtggaaaagcctcgctctaact'
'gcgtgtggaaaagtctaactcctcgc'
'gcgtgtggaaaatctaactgcctcgc'
'gcgtgtggaaaatctaactcctcgcg'
'gcggtgtggaaaacctcgctctaact'
'gcggtgtggaaaatctaactcctcgc'
'gcggcctcgctgtggaaaatctaact'
'gcggcctcgctctaacttgtggaaaa'
'gcggtctaactcctcgctgtggaaaa'
'gcggtctaacttgtggaaaacctcgc'
'gcgtctaacttgtggaaaagcctcgc'
'gcgtctaacttgtggaaaacctcgcg'
'gcgtctaactgtgtggaaaacctcgc'
'gcgtctaactgcctcgctgtggaaaa'
'gcgtctaactcctcgcgtgtggaaaa'
'gcgtctaactcctcgctgtggaaaag'
'gcctcgcgcgtgtggaaaatctaact'
'gcctcgcgcgtctaacttgtggaaaa'
'gcctcgctgtggaaaagcgtctaact'
'gcctcgctgtggaaaatctaactgcg'
'gcctcgctctaacttgtggaaaagcg'
'gcctcgctctaactgcgtgtggaaaa'
'ggcgcctcgctgtggaaaatctaact'
'ggcgcctcgctctaacttgtggaaaa'
'ggcgtgtggaaaacctcgctctaact'
'ggcgtgtggaaaatctaactcctcgc'
'ggcgtctaacttgtggaaaacctcgc'
'ggcgtctaactcctcgctgtggaaaa'
'gtgtggaaaagcgcctcgctctaact'
'gtgtggaaaagcgtctaactcctcgc'
'gtgtggaaaacctcgcgcgtctaact'
'gtgtggaaaacctcgctctaactgcg'
'gtgtggaaaatctaactcctcgcgcg'
'gtgtggaaaatctaactgcgcctcgc'
'gtctaactgcgtgtggaaaacctcgc'
'gtctaactgcgcctcgctgtggaaaa'
'gtctaacttgtggaaaagcgcctcgc'
'gtctaacttgtggaaaacctcgcgcg'
'gtctaactcctcgctgtggaaaagcg'
'gtctaactcctcgcgcgtgtggaaaa'
'tctaactcctcgcgcggtgtggaaaa'
'tctaactcctcgcgcgtgtggaaaag'
'tctaactcctcgcggcgtgtggaaaa'
'tctaactcctcgcgtgtggaaaagcg'
'tctaactcctcgctgtggaaaaggcg'
'tctaactcctcgctgtggaaaagcgg'
'tctaactgcgcctcgcgtgtggaaaa'
'tctaactgcgcctcgctgtggaaaag'
'tctaactgcggcctcgctgtggaaaa'
'tctaactgcggtgtggaaaacctcgc'
'tctaactgcgtgtggaaaagcctcgc'
'tctaactgcgtgtggaaaacctcgcg'
'tctaactggcgcctcgctgtggaaaa'
'tctaactggcgtgtggaaaacctcgc'
'tctaactgcctcgcgcgtgtggaaaa'
'tctaactgcctcgctgtggaaaagcg'
'tctaactgtgtggaaaacctcgcgcg'
'tctaactgtgtggaaaagcgcctcgc'
'tctaacttgtggaaaagcggcctcgc'
'tctaacttgtggaaaagcgcctcgcg'
'tctaacttgtggaaaaggcgcctcgc'
'tctaacttgtggaaaagcctcgcgcg'
'tctaacttgtggaaaacctcgcggcg'
'tctaacttgtggaaaacctcgcgcgg'};
//求a集合和b集合的交集
c=intersect(a,b);
c='tctaactggcgcctcgctgtggaaaa'//c恰好等于gene
以上所述,仅是本发明的较佳实施例,并非对本发明的技术范围作任何限制,本行业的技术人员,在本技术方案的启迪下,可以做出一些变形与修改,凡是依据本发明的技术实质对以上的实施例所作的任何修改、等同变化与修饰,均仍属于本发明技术方案的范围内。
- 还没有人留言评论。精彩留言会获得点赞!