一种靶向性的负载阿霉素的超分子水凝胶及其制备方法与流程
本发明属于超分子水凝胶载药领域,具体涉及一种靶向性的负载抗癌药物水凝胶的制备方法。
背景技术:
阿霉素又名多柔比星,是一种广谱强效的蒽醌类抗肿瘤药物,广泛用于治疗各种癌症,如乳腺癌、卵巢癌、膀胱癌、甲状腺癌以及非小细胞性肺癌等,其单独使用或联合其他抗肿瘤药物使用,均被认为是治疗实体瘤的一种强有力化疗方式,几乎对所有恶性增殖的肿瘤有一定效果。其作用机理是通过与dna结合引发拓扑异构酶ii裂解dna,破坏dna的三级结构,从而阻滞细胞增殖、促进细胞凋亡。通过对临床现象的观察与研究,研究人员发现阿霉素在发挥治疗效果的同时,也会出现较大的毒副作用,如心脏毒性、胃肠道反应、脱发、骨髓抑制等,阻碍了其在临床上的应用。
叶酸是一中水溶性的b族维生素,亦称作维生素b9,叶酸分子由蝶啶、对氨基苯甲酸和谷氨酸共同构成。在传统的肿瘤化疗中,多数抗肿瘤药物随血液分布于全身各处,因为抗癌药物对肿瘤细胞和人体的正常细胞都具有杀伤效应,所以对人体产生了很大的毒副作用,使化疗药物缺乏一定的选择性。科研人员发现叶酸受体是普遍存在于恶性肿瘤细胞中的一种受体,它是能与糖基化磷脂酸肌醇连接的一种膜糖蛋白,可以介导细胞内化,是一种能对叶酸产生极高亲和力的受体,其在正常组织中表达高度保守,而在卵巢癌、乳腺癌、子宫内膜癌、肺癌、肾癌、结肠癌和鼻咽癌等癌细胞中均高度表达,因此,叶酸作为一种很好的靶向药物,在临床试验中被广泛使用。
技术实现要素:
本发明的目的是提供一种靶向性的负载抗癌药物水凝胶及其制备方法。本发明所用的原料易得,操作步骤简单,且制备的水凝胶载药体系以共价键接枝的方式对药物进行负载,使得载药体系更加稳定。
为实现上述目的,本发明采用如下技术方案:
一种靶向性的负载阿霉素的超分子水凝胶,其特征在于,由α-环糊精(α-cd)、f127(普朗尼克f127、pluronicf127)、阿霉素修饰的环糊精衍生物(dox-2n-β-cd)和叶酸修饰的环糊精衍生物(fa-2n-β-cd)反应得到。
进一步地,阿霉素通过化学键接枝的方式载入水凝胶网络。
本发明还提供了上述的靶向性的负载阿霉素的超分子水凝胶的制备方法,其特征在于,包括以下步骤:称取dox-2n-β-cd、f127和fa-2n-β-cd于反应器中,加入去离子水,搅拌反应;加入α-cd,继续搅拌反应,反应结束后,使反应体系静置10-15h后得到靶向性的负载阿霉素的超分子水凝胶。所述的静置时间优选12h。
进一步地,所述的f127、dox-2n-β-cd与fa-2n-β-cd的质量体积之比分别为5%、2%-8%、5%,搅拌反应的温度为50-60℃,时间为4-5h,继续搅拌反应的温度为50-60℃,时间为4-5h,加入的α-cd的质量体积之比为10%-20%。
更进一步地,所述的f127、dox-2n-β-cd与fa-2n-β-cd的质量体积之比分别为5%、2%、5%,搅拌反应的温度为60℃,时间为4h,继续搅拌反应的温度为60℃,时间为4h,加入的α-cd的质量体积之比为20%。
进一步地,所述的dox-2n-β-cd的合成方法包括:
步骤a:将干燥的β-环糊精、对甲苯磺酰氯和氢氧化钠在去离子水中进行反应,停止反应后进行过滤,将得到的滤液调节ph后,在2-5℃低温下放置一昼夜以析出白色沉淀,过滤并重结晶后可得到中间化合物单(6-氧-对甲苯磺酰基)-β-环糊精(6-ots-β-cd);
步骤b:取单(6-氧-对甲苯磺酰基)-β-环糊精(6-ots-β-cd)与1,3-丙二胺在n-甲基吡咯烷酮(nmp)中反应,反应停止后将其滴入丙酮中析出沉淀,过滤收集沉淀即得产物单-6-丙二胺-β-环糊精(2n-β-cd);
步骤c:将药物阿霉素、单-6-丙二胺-β-环糊精和三氟乙酸在水和乙醇的混合溶剂中进行反应,停止反应后将其滴入丙酮中析出沉淀,过滤收集沉淀并在丙酮中洗涤,即可得到前药分子dox-2n-β-cd。
更进一步地,所述步骤a中的反应温度为10-20℃,β-环糊精与对甲苯磺酰氯的摩尔比为:1:0.8-0.9,所述溶剂水用量按照每克β-环糊精5-10ml确定,反应时间控制在2-2.5h,ph控制在7-8。优选地,所述步骤a中的反应温度为10-20℃,β-环糊精与对甲苯磺酰氯的摩尔比为:1:0.84,所述溶剂水用量按照每克β-环糊精7ml确定,反应时间控制在2-2.5h,ph控制在7-8。
更进一步地,所述的步骤b中的反应在在氮气氛围下进行,反应温度为70-80℃,反应时间为10-12h,1,3-丙二胺、6-ots-β-cd与nmp的摩尔比为:1:3-6:20-30。优选地,所述的步骤b中的反应在在氮气氛围下进行,反应温度为80℃,反应时间为10-12h,1,3-丙二胺、6-ots-β-cd与nmp的摩尔比为:1:5:25。
更进一步地,所述的步骤c中的反应在避光条件下进行,反应温度为室温,反应时间为48-60h,阿霉素与2n-β-cd的摩尔比为1.3-1.8:1。优选地,所述的步骤c中的反应在避光条件下进行,反应温度为室温,反应时间为48h,阿霉素与2n-β-cd的摩尔比为1.5:1。
进一步地,所述的fa-2n-β-cd的合成方法包括:将叶酸、2n-β-cd、二环己基碳二亚胺(dcc)和n-羟基琥珀酰亚胺(nhs)在dmso溶剂中避光反应,停止反应后将其滴入丙酮中析出沉淀,过滤收集并在丙酮中洗涤,即可得到黄色固体fa-2n-β-cd。
进一步地,所述的2n-β-cd、叶酸、dcc、nhs的摩尔比为:1-1.1:1.1-1.8:4-4.6:4-4.6,反应温度为室温,反应时间为48-60h。优选地,所述的2n-β-cd、叶酸、dcc、nhs的摩尔比为:1:1.5:4:4。反应温度为室温,反应时间为48h。
本发明首先对β-环糊精进行化学修饰,制备出含有氨基环糊精衍生物2n-β-cd;然后将靶向基团叶酸分子与2n-β-cd进行缩合反应,合成出前药体fa-2n-β-cd,与此同时,将抗癌药物阿霉素与2n-β-cd进行缩合反应,合成出另一前药体dox-2n-β-cd;最后以α-环糊精、f127、dox-2n-β-cd、fa-2n-β-cd为构筑基元构建超分子水凝胶材料。
与现有技术相比,本发明的有益效果是:
本发明水凝胶载药体系的原料易得,制作过程简单,性质易于调控,阿霉素和叶酸分别以共价键形式负载于水凝胶网络中,不仅能够降低药物阿霉素的毒副作用,而且还有效的提高了水凝胶的靶向性。
本发明制作过程简单,通过环糊精的空腔与f127分子链之间的相互作用制得负载阿霉素和叶酸的准聚轮烷,然后利用环糊精分子中羟基之间的氢键作用,进一步拓展为三维的超分子水凝胶材料。药物分子叶酸和阿霉素分别以共价键形式负载于水凝胶网络中,不仅能够降低药物阿霉素的毒副作用,而且还能有效的提高水凝胶的靶向性。
附图说明
图1是中间体6-ots-β-cd的核磁氢谱图。
图2是中间体2n-β-cd的核磁氢谱图。
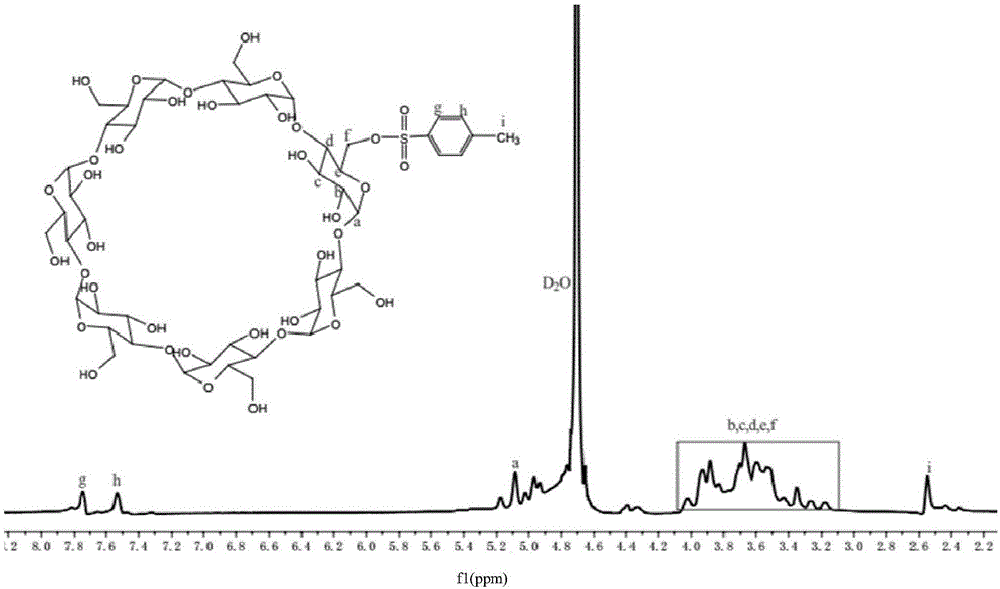
图3是中间体dox-2n-β-cd的核磁氢谱图。
图4是中间体fa-2n-β-cd的核磁氢谱图。
图5是实施例5制得的载药水凝胶体系扫描电镜图。
图6是四种超分子水凝胶的模量随频率变化曲线图。
图7是载药水凝胶与原料的x射线衍射对比图;
具体实施方式
下面结合具体实施例,进一步阐述本发明。应理解,这些实施例仅用于说明本发明而不用于限制本发明的范围。此外应理解,在阅读了本发明讲授的内容之后,本领域技术人员可以对本发明作各种改动或修改,这些等价形式同样落于本申请所附权利要求书所限定的范围。
实施例1:dox-2n-β-cd的合成:
通过1,3-丙二胺对β-环糊精进行化学修饰,制备出新型环糊精衍生物,通过阿霉素与胺基修饰的环糊精衍生物进行缩合反应,进一步合成前药分子dox-2n-β-cd,具体步骤为:
a、将0.19mol干燥的β-环糊精、0.16mol对甲苯磺酰氯、6.16mol氢氧化钠在1500ml去离子水中进行反应,反应温度为15℃,反应时间为2h,停止反应后进行过滤,得到的滤液调节ph为7后,在2℃温度下放置一昼夜以析出大量白色沉淀,过滤并重结晶后可得到中间化合物单(6-氧-对甲苯磺酰基)-β-环糊精(6-ots-β-cd)。通过核磁技术进行表征,如图1所示。
b、取1.6mmol单(6-氧-对甲苯磺酰基)-β-环糊精(6-ots-β-cd)与8mmol1,3-丙二胺在41.5mmoln-甲基吡咯烷酮中在氮气氛围下进行反应,反应温度为80℃,反应时间为10h,反应停止后将其滴入丙酮中析出沉淀。过滤收集沉淀即得产物单-6-丙二胺-β-环糊精(2n-β-cd)。通过核磁技术进行表征,如图2所示。
c、将0.7mmol药物阿霉素、0.4mmol2n-β-cd、0.001mmol三氟乙酸在20ml体积比为1:1的水和乙醇混合溶剂中在避光条件下室温进行反应,反应时间为48h,停止反应后将其滴入丙酮中析出沉淀,过滤收集沉淀并在丙酮中多次洗涤,即可得到前药体dox-2n-β-cd。通过核磁技术进行表征,如图3所示。
实施例2:fa-2n-β-cd的合成
通过叶酸与胺基修饰的环糊精衍生物进行缩合反应,进一步合成中间体fa-2n-β-cd,具体步骤为:
将1.1mmol叶酸、1mmol2n-β-cd、4.6mmoldcc和4.6mmolnhs在15mldmso溶剂中避光反应,反应温度为室温,反应时间为48h,停止反应后将其滴入丙酮中析出沉淀,过滤收集并在丙酮中多次洗涤,即可得到黄色固体fa-2n-β-cd,并通过核磁技术进行表征,如图4所示。
实施例3
一种靶向性的负载阿霉素的超分子水凝胶,由α-环糊精、f127、dox-2n-β-cd和fa-2n-β-cd反应得到,阿霉素通过化学键接枝的方式载入水凝胶网络。所述的靶向性的负载阿霉素的超分子水凝胶的制备方法为:
取0.12gdox-2n-β-cd、0.15gf127、0.15gfa-2n-β-cd于反应器中,加入3ml的去离子水,在60℃的温度下搅拌反应4h。然后将0.6gα-cd加入到反应体系中,在60℃的条件下继续搅拌反应4h,整个反应在避光条件下进行。反应结束后,使反应体系缓慢冷却到室温,静置12h后,冷冻干燥12h,即得干燥的靶向性的负载阿霉素的超分子水凝胶。f127、dox-2n-β-cd、fa-2n-β-cd与α-cd的质量/体积含量分别为5%、4%、5%、20%。
实施例4
一种靶向性的负载阿霉素的超分子水凝胶,由α-cd、f127、dox-2n-β-cd和fa-2n-β-cd反应得到,阿霉素通过化学键接枝的方式载入水凝胶网络。所述的靶向性的负载阿霉素的超分子水凝胶的制备方法为:
称取0.18gdox-2n-β-cd、0.15gf127、0.15gfa-2n-β-cd于反应器中,加入3ml的去离子水,在60℃的温度下搅拌反应4h。然后将0.3gα-cd加入到反应体系中,在60℃的条件下继续搅拌反应4h,整个反应在避光条件下进行。反应结束后,使反应体系缓慢冷却到室温,静置12h后,冷冻干燥12h,即得干燥的靶向性的负载阿霉素的超分子水凝胶。f127、dox-2n-β-cd、fa-2n-β-cd与α-cd的质量/体积含量分别为5%、6%、5%、10%。
实施例5
一种靶向性的负载阿霉素的超分子水凝胶,由α-cd、f127、dox-2n-β-cd和fa-2n-β-cd反应得到,阿霉素通过化学键接枝的方式载入水凝胶网络。所述的靶向性的负载阿霉素的超分子水凝胶的制备方法为:
称取0.24gdox-2n-β-cd、0.15gf127、0.15gfa-2n-β-cd于反应器中,加入3ml的去离子水,在60℃的温度下搅拌反应4h。然后将0.3gα-cd加入到反应体系中,在60℃的条件下继续搅拌反应4h,整个反应在避光条件下进行。反应结束后,使反应体系缓慢冷却到室温,静置12h后,冷冻干燥12h,即得干燥的靶向性的负载阿霉素的超分子水凝胶。f127、dox-2n-β-cd、fa-2n-β-cd与α-cd的质量/体积含量分别为5%、8%、5%、10%。
实施例6
一种靶向性的负载阿霉素的超分子水凝胶,由α-cd、f127、dox-2n-β-cd和fa-2n-β-cd反应得到,阿霉素通过化学键接枝的方式载入水凝胶网络。所述的靶向性的负载阿霉素的超分子水凝胶的制备方法为:
首先称取0.15gf127、0.15gfa-2n-β-cd与0.06gdox-2n-β-cd于反应器中,加入3ml的去离子水,在60℃的条件下搅拌反应4h。然后将0.6gα-cd加入到反应体系中,在60℃的条件下继续搅拌反应4h,整个反应在避光条件下进行。反应结束后,使反应体系缓慢冷却到室温,静置12h后,冷冻干燥12h,即得干燥的超分子水凝胶材料。f127、dox-2n-β-cd、fa-2n-β-cd与α-cd的质量/体积含量分别为5%、2%、5%、20%。
如图5所示,为实施例5制得的载药水凝胶体系扫描电镜图。如图6所示,是实施例3-6制得的四种超分子水凝胶的模量随频率变化曲线图,从图中可以看出,凝胶的弹性模量基本不随频率变化,表现出一种“类固体”的性质,且随着α-cd和f127浓度的增加,超分子水凝胶的弹性模量也随之提高。如图7所示,为实施例3-6制得的载药水凝胶的x射线衍射图,由于四种载药水凝胶均为多聚准轮烷结构,所以在图中统一称为hydrogel。
- 还没有人留言评论。精彩留言会获得点赞!