基于定向进化的方法、装置和系统与流程
相关申请的交叉引用
本申请基于35u.s.c.§119(e)要求于2013年9月27日提交,标题为:基于结构的预测性建模的美国临时专利申请号61/883,919的权益,为了所有的目的通过引用将其以其全部并入本文。
本申请涉及但不限于基于结构的预测性建模。
背景技术:
蛋白设计长久以来被认为是艰巨的任务,只因为一个原因,构成可搜索的序列空间的可能分子的组合式激增。蛋白的序列空间是极大的,并且使用本领域目前已知的方法彻底地搜索是不可能的,本领域目前已知的方法通常被鉴定有用的多肽所需的时间和成本所限制。问题的一部分由必须要测序、筛选和测定的多肽变体的巨大的量而引起。定向进化方法提高了深入研究具有有益特征的候选生物分子的效率。如今,蛋白的定向进化由往往迭代进行的多种高通量筛选和重组方式主导。
用于搜索序列-活性空间的多种计算技术也已被提出。相对来说,这些技术处于其初期,并仍然需要重大进展。因此,用于提高筛选、测序和测定候选生物分子的效率的新方法是高度期望的。
技术实现要素:
本公开内容涉及分子生物学、分子进化、生物信息学和数字化系统的领域。
本公开内容的方法在优化用于工业和治疗用途的蛋白方面具备实用性。方法和系统对于设计和开发具有有益特性或活性的酶是尤其有用的。
本公开内容的某些方面涉及用于开发具有有益特性的蛋白和/或引导定向进化程序的方法。本公开内容展示了用于从复杂的生物分子文库或多组此类文库鉴定具有期望的特性的(或最适合于向此类特性定向进化的)生物分子的方法。本公开内容的一些实施方案提供了用于参考结构数据构建序列活性模型的方法,该模型可被用于引导具有有益特性的蛋白的定向进化。一些实施方案采用遗传算法和结构数据来过滤掉不提供信息的数据。一些实施方案使用支持向量机来训练序列活性模型。过滤方法和训练方法可产生具有比传统建模方法高的预测能力的序列活性模型。
本公开内容的一些实施方案提供了用于执行定向进化的方法。在一些实施方案中,该方法使用包括一个或更多个处理器和系统存储器的计算机系统来实施。该方法包括:(a)从分子的物理测量值接收具有信息的数据集,其中,所述数据集包括多个变体生物分子的每一个的以下信息:(i)变体生物分子对在所述变体生物分子的结合位点中的配体的活性;(ii)变体生物分子的序列;以及(iii)一个或更多个几何参数,所述一个或更多个几何参数表征在所述结合位点中的所述配体的几何结构;(b)过滤所述数据集以通过移出变体生物分子中的一个或更多个的信息来产生经过滤的数据子集,其中过滤包括测试用多个选择的数据子集训练的序列活性模型的预测能力,每个选择的数据子集具有从(a)的数据集移出的一组特定的变体生物分子的信息;以及(c)使用所述经过滤的数据子集来训练改进的序列活性模型。在一些实施方案中,所述多个变体生物分子的每一个的信息还包括(iv)表征所述配体在所述结合位点中的相互作用的相互作用能。在一些实施方案中,所述变体生物分子是酶。
在一些实施方案中,改进的序列活性模型通过支持向量机、多元线性回归、主成分回归、偏最小二乘回归或神经网络获得。
在一些实施方案中,过滤数据集包括从数据集移出几何参数中的至少一个。在一些实施方案中,用遗传算法进行数据集的过滤。在一些实施方案中,遗传算法改变阈值来将与一个或更多个变体生物分子的几何参数相关的信息移出。
在一些实施方案中,用于定向进化的方法还包括应用改进的序列活性模型来鉴定被所述改进的序列活性模型预测为具有满足某个标准的活性的一个或更多个新的生物分子变体。所述一个或更多个新的生物分子变体的每一个具有与为(a)的数据集提供信息的生物分子变体的序列不同的序列。在一些实施方案中,应用改进的序列活性模型鉴定一个或更多个新的生物分子变体包括进行遗传算法,在该遗传算法中,利用改进的序列活性模型作为适应度函数来评价潜在的新的生物分子变体。
在一些实施方案中,用于定向进化的方法还包括测定新的生物分子变体的活性。在一些实施方案中,该方法还包括通过体外测定来测量变体生物分子的活性。
在一些实施方案中,该方法还包括针对新的生物分子变体中的每一个生成结构模型。该方法还使用该结构模型来生成新的生物分子变体的结合位点的几何参数。该几何参数表征了在新的生物分子变体的结合位点中的配体的几何结构。在一些实施方案中,该方法还包括接收生物分子变体的结构模型和使用该结构模型来确定一个或更多个几何参数。在一些实施方案中,结构模型是同源模型。在一些实施方案中,该同源模型使用生物分子的物理结构测量细节来制作。生物分子的物理结构测量细节可包括通过nmr或x射线晶体学获得的原子的三维位置。
在一些实施方案中,该方法还包括使用对接器来确定一个或更多个几何参数。在一些实施方案中,该方法还使用对接器来确定相互作用能。
在一些实施方案中,所处理的变体生物分子是多个酶。在一些实施方案中,变体生物分子对配体的活性是酶对底物的活性。在一些实施方案中,酶对底物的活性包括底物被酶酶促转化的一个或更多个特征。
在一些实施方案中,用于定向进化的方法还包括使用改进的序列活性模型来鉴定具有期望的活性的一个或更多个生物分子。在一些实施方案中,该方法还包括合成具有期望的活性的生物分子。
在一些实施方案中,还提供了实施用于生物分子的定向进化的方法的计算机程序产品和计算机系统。
在一些实施方案中,本申请提供了一种计算机程序产品,所述计算机程序产品包括一个或更多个计算机可读的非瞬时储存介质,所述非瞬时储存介质具有计算机可执行指令存储于其上,所述计算机可执行指令当被计算机系统的一个或更多个处理器执行时,导致所述计算机系统执行用于进行定向进化的方法,所述方法包括:
(a)通过所述计算机系统接收具有来自分子的物理测量值的信息的数据集,其中所述数据集包括多个变体生物分子的每一个的以下信息:(i)所述变体生物分子对在所述变体生物分子的结合位点中的配体的活性;(ii)所述变体生物分子的序列;以及(iii)一个或更多个几何参数,所述一个或更多个几何参数表征在所述结合位点中的配体的几何结构;
(b)通过所述计算机系统过滤所述数据集,以通过移出一个或更多个所述变体生物分子的信息来产生经过滤的数据子集,其中所述过滤包括测试用多个选择的数据子集训练的序列活性模型的预测力,每一个选择的数据子集具有从(a)的所述数据集移出的一组特定的变体生物分子的信息;以及
(c)通过所述计算机系统使用所述经过滤的数据子集训练改进的序列活性模型。
在一些实施方案中,本申请提供了一种计算机系统,包括:
一个或更多个处理器;
系统存储器;以及
一个或更多个计算机可读存储介质,所述一个或更多个计算机可读存储介质具有计算机可执行指令存储于其上,所述计算机可执行指令当被所述一个或更多个处理器执行时,导致所述计算机系统执行用于进行定向进化的方法,所述方法包括:
(a)接收具有来自分子的物理测量值的信息的数据集,其中所述数据集包括多个变体生物分子的每一个的以下信息:(i)所述变体生物分子对在所述变体生物分子的结合位点中的配体的活性;(ii)所述变体生物分子的序列;以及(iii)一个或更多个几何参数,所述一个或更多个几何参数表表征在所述结合位点中的配体的几何结构;
(b)过滤所述数据集以通过移出所述变体生物分子中的一个或更多个的信息来产生经过滤的子集,其中所述过滤包括测试用多个选择的数据子集训练的序列活性模型的预测力,每一个选择的数据子集具有从(a)的所述数据集移出的一组特定的变体生物分子的信息;以及
(c)使用所述经过滤的数据子集来训练改进的序列活性模型。
本申请提供了以下内容:
项目1.一种进行定向进化的方法,所述方法包括:
(a)从分子的物理测量值接收具有信息的数据集,其中所述数据集包括多个变体生物分子的每一个的以下信息:(i)所述变体生物分子对在所述变体生物分子的结合位点中的配体的活性;(ii)所述变体生物分子的序列;以及(iii)一个或更多个几何参数,所述一个或更多个几何参数表征在所述结合位点中的所述配体的几何结构;
(b)过滤所述数据集,以通过移出一个或更多个所述变体生物分子的信息来产生经过滤的数据子集,其中,所述过滤包括测试用多个选择的数据子集训练的序列活性模型的预测力,每一个选择的数据子集具有从(a)的所述数据集移出的一组特定的变体生物分子的信息;以及
(c)使用所述经过滤的数据子集来训练改进的序列活性模型。
项目2.根据项目1所述的方法,其中过滤所述数据集包括从所述数据集移出所述一个或更多个几何参数的至少一个。
项目3.根据前述项目中任一项所述的方法,其中所述过滤所述数据集用几何算法来进行。
项目4.根据项目3所述的方法,其中所述遗传算法改变阈值来将与一个或更多个所述变体生物分子的遗传参数有关的信息移出。
项目5.根据前述项目中任一项所述的方法,还包括应用改进的序列活性模型来鉴定被所述改进的序列活性模型预测为具有满足一个或更多个标准的活性的一个或更多个新的生物分子变体,其中所述一个或更多个新的生物分子变体的每一个具有与为(a)的所述数据集提供信息的生物分子变体的序列不同的序列。
项目6.根据项目5所述的方法,其中应用所述改进的序列活性模型鉴定一个或更多个新的生物分子变体包括执行遗传算法,在所述遗传算法中,利用所述改进的序列活性模型作为适应度函数来评价潜在的新的生物分子变体。
项目7.根据项目5所述的方法,还包括测定所述新的生物分子变体的活性。
项目8.根据项目5所述的方法,还包括;
针对所述新的生物分子变体中的每一个生成结构模型;以及
使用所述结构模型来生成所述新的生物分子变体的结合位点的几何参数,其中所述几何参数表征在所述新的生物分子变体的结合位点中的配体的几何结构。
项目9.根据前述项目中任一项所述的方法,还包括通过体外测定测量所述变体生物分子的活性。
项目10.根据前述项目中任一项所述的方法,还包括接收生物分子变体的结构模型并使用所述生物分子变体的结构模型来确定所述一个或更多个几何参数。
项目11.根据项目10所述的方法,其中所述生物分子变体的结构模型是同源模型。
项目12.根据项目11所述的方法,其中所述同源模型使用生物分子的物理结构测量细节来制作。
项目13.根据项目12所述的方法,其中生物分子的所述物理结构测量细节包括通过nmr或x射线晶体学获取的原子的三维位置。
项目14.根据项目10所述的方法,还包括使用对接器来确定所述一个或更多个几何参数。
项目15.根据前述项目中任一项所述的方法,其中多个变体生物分子中的每一个的信息还包括(iv)表征所述配体在所述结合位点中的相互作用的相互作用能。
项目16.根据项目15所述的方法,还包括使用对接器来确定所述相互作用能。
项目17.根据前述项目中任一项所述的方法,其中所述改进的序列活性模型通过支持向量机、多元线性回归、主成分回归、偏最小二乘回归或神经网络获得。
项目18.根据项目17所述的方法,其中所述改进的序列活性模型通过支持向量机获得。
项目19.根据前述项目中任一项所述的方法,其中所述多个变体生物分子包括多个酶。
项目20.根据项目19所述的方法,其中所述变体生物分子对配体的活性酶对底物的活性。
项目21.根据项目20所述的方法,其中酶对底物的活性包括所述底物被所述酶催化转化的一个或更多个特征。
项目22.根据前述项目中任一项所述的方法,还包括使用所述改进的序列活性模型来鉴定具有期望的活性的一个或更多个生物分子。
项目23.根据项目22所述的方法,还包括合成具有期望的活性的生物分子。
项目24.一种计算机程序产品,所述计算机程序产品包括一个或更多个计算机可读的非瞬时储存介质,所述非瞬时储存介质具有计算机可执行指令存储于其上,所述计算机可执行指令当被计算机系统的一个或更多个处理器执行时,导致所述计算机系统执行用于进行定向进化的方法,所述方法包括:
(a)通过计算机系统接收具有来自分子的物理测量值的信息的数据集,其中所述数据集包括多个变体生物分子的每一个的以下信息:(i)所述变体生物分子对在所述变体生物分子的结合位点中的配体的活性;(ii)所述变体生物分子的序列;以及(iii)一个或更多个几何参数,所述一个或更多个几何参数表表征在所述结合位点中的配体的几何结构;
(b)通过所述计算机系统过滤所述数据集,以通过移出一个或更多个所述变体生物分子的信息来产生经过滤的数据子集,其中所述过滤包括测试用多个选择的数据子集训练的序列活性模型的预测力,每一个选择的数据子集具有从(a)的所述数据集移出的一组特定的变体生物分子的信息;以及
(c)通过所述计算机系统使用所述经过滤的数据子集训练改进的序列活性模型。
25.一种计算机系统,包括:
一个或更多个处理器;
系统存储器;以及
一个或更多个计算机可读存储介质,所述一个或更多个计算机可读存储介质具有计算机可执行指令存储于其上,所述计算机可执行指令当被所述一个或更多个处理器执行时,导致所述计算机系统执行用于进行定向进化的方法,所述方法包括:
(a)接收具有来自分子的物理测量值的信息的数据集,其中所述数据集包括多个变体生物分子的每一个的以下信息:(i)所述变体生物分子对在所述变体生物分子的结合位点中的配体的活性;(ii)所述变体生物分子的序列;以及(iii)一个或更多个几何参数,所述一个或更多个几何参数表表征在所述结合位点中的配体的几何结构;
(b)过滤所述数据集以通过移出所述变体生物分子中的一个或更多个的信息来产生经过滤的子集,其中所述过滤包括测试用多个选择的数据子集训练的序列活性模型的预测力,每一个选择的数据子集具有从(a)的所述数据集移出的一组特定的变体生物分子的信息;以及
(c)使用所述经过滤的数据子集来训练改进的序列活性模型。
下文将参考相关附图展示这些特征以及其他特征。
附图说明
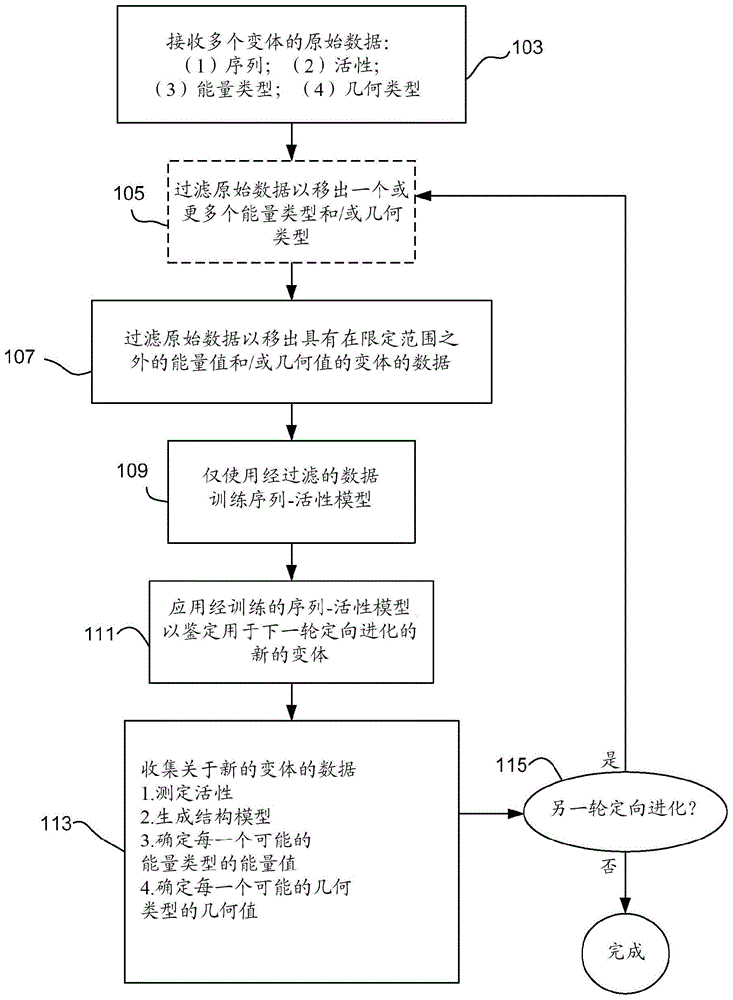
图1a是图示根据本公开内容的一些实施方案的定向进化工作流的流程图。
图1b是图示根据本公开内容的一些实施方案的过滤原始数据的一个方法的流程图。
图1c是展示根据一些实施方案的数据过滤过程的流程图,其中特征选择阶段不被执行或被与距离选择阶段结合。
图2a至图2c示出了序列活性数据集的三个表格表示,以说明根据本公开内容的一些实施方案过滤数据的实例。
图3a是显示根据本公开内容的一些实施方案的用于过滤原始数据以移出一个或更多个能量类型和/或几何类型的遗传算法的流程图。
图3b是显示根据本公开内容的一些实施方案的用于过滤原始数据以移出具有在所定义的范围之外的能量值和/或几何值的变体的数据的遗传算法的流程图。
图3c是显示根据本公开内容的一些实施方案的用于使用高预测力的序列活性模型鉴定新的生物分子变体的遗传算法的流程图。
图4示出了根据一些实施方案的可被实施的示例性数字设备。
详细描述
本文公开了用于参考结构数据开发序列活性模型的方法。序列活性模型可被用于引导具有有益特性的蛋白的定向进化。一些实施方案可有助于搜索巨大的序列空间并快速地深入研究具有有益特性的分子。在寻找或开发具有期望特性的蛋白的过程中还可节省材料和/或资源。一些实施方案对于设计和开发对涉及特定底物的催化反应具有期望的活性和/或选择性的酶是尤其有用的。
i.定义
除非本文另外定义,本文使用的所有技术和科学术语具有如本领域普通技术人员通常理解的相同含义。包含本文包括的术语的多本科学词典是本领域技术人员熟知并可获得的。与本文描述的那些方法和材料相似或等同的任何方法和材料在本文公开的实施方案的实践中具备实用性。
通过整体地参考说明书,下文紧接着定义的术语可被更充分地理解。只是为了描述特定的实施方案并帮助理解本说明书中描述的复杂概念的目的而提供定义。它们并非意图限制本公开内容的完整范围。特别地,要理解,本公开内容不限于本文所描述的特定序列、组合物、算法、系统、方法学、方案和/或试剂,因为这些可根据本领域技术人员使用它们的背景而变化。
如本说明书和所附的权利要求书中使用的,单数形式“一(a)”、“一(an)”和“该(the)”包括复数的指代对象,除非该内容和上下文另外明确地指明。因此,例如,提及“一个装置(adevice)”包括两个或更多个此类装置的组合,诸如此类。除非另外指明,“或”连接意图以其作为布尔逻辑算符的正确含义使用,包括择一性的特征选择(a或b,其中选择a是与b互相排斥的)和合取性的特征选择(a或b,其中a和b两者均被选择)两者。
支持向量机(svm)是具有用于分类和回归分析的相关学习算法的机器学习工具。基础svm取用一组输入数据和预测值,对于每个给出的输入值,两个可能的类别的所述每个给出的输入值形成输出。假定一组训练样例,每一个训练样例被标记为属于两个分类中的一个,svm训练算法构建将新的样例分配到一个分类或另一个分类的模型。svm是作为空间中的点的样例被映射以使得不同分类的样例被清晰的即尽量宽的间距分开的表述,其通过最大化数据点之间的距离和分隔开两个分类的超平面来实施。除了进行线性分类之外,svm可使用核技巧有效地进行非线性分类,以隐含式地将输入值映射到高维特征空间中。
当用于优化序列活性模型时,svm将已基于活性分类成两个或更多个组的训练组序列作为输入值。支持向量机通过根据训练组的不同成员如何接近超平面界面来不同地加权训练组的不同成员,所述超平面界面将训练组的“活性”和“非活性”成员分隔开。该技术需要科学家先决定将哪些训练组成员置于活性组以及将哪些训练组成员置于非活性组。这可通过选择活性的适当数值充当训练组的活性成员和非活性成员之间的分界线来完成。支持向量机将根据该分类生成向量w,其可为限定训练组中的活性组成员和非活性组成员的序列的自变量中的个体自变量提供系数值。这些系数可被用于如本文其他处描述的对个体残基“排名”。该技术试图鉴定超平面,所述超平面使在该平面的相对侧上的最靠近的训练组成员之间的距离最大化。在另一个变体中,进行支持向量回归建模。在该情形中,因变量为连续的活性值的向量。支持向量回归模型将生成系数向量w,其可被用来对个体残基排名。
svm已在很多研究中被用来用于大数据集,并且在dna微阵列领域中已相当普及。其潜在优势包括(通过加权)细微地辨别哪些因素将样本彼此分开的能力。就svm能够精确地梳理出哪些残基对功能有贡献来说,其可以是根据本发明对残基排名特别有用的工具。svm被描述于s.gunn(1998)“supportvectormachinesforclassificationandregressions”技术报告,南安普敦大学工程与应用科学学院电子和计算机科学系,其为了所有目的被通过引用并入本文。
对接器(对接软件或对接程序)—“对接器”是在计算上预测配体是否将与蛋白或其他的生物分子中的感兴趣的结合位点结合或对接的计算机程序。通过其配体与结合位点接近并最终结合的过程有时候被称为“对接”。对接的概念可被理解为导致配体与生物分子结合的相互作用,以此方式配体不被容易地移开。在成功的对接中,配体和生物分子形成稳定的复合体。对接的配体可充当激动剂或拮抗剂。对接器可模拟和/或表征对接。
对接器通常被实现为可与诸如一个或更多个处理器的硬件相关联被暂时性地或永久性地存储的软件。市售可得的对接程序包含cdocker(accelrys)、dock(加利福尼亚大学,旧金山)、autodock(斯克里普斯研究所)、flexx(tripos.com)、gold(ccdc.cam.ac.uk)和glide(schrodinger.com)。
多种对接器输出配体和生物分子之间的结合的对接得分或其他测量值。对于一些配体-生物分子结合,对接程序将确定结合不可能发生。在此类情况中,对接程序将输出配体不与生物分子结合的结论。
对接器可生成配体相对于结合位点的“位姿(pose)”。这些位姿中的一些可被用于生成对接得分或另外评价对接。在一些实施方案中,对接器允许使用者指定很多位姿(n)来用于评价对接。在评价对接中,仅具有最佳对接得分的前“n”个位姿被考虑。
对接器可被编程以输出配体将与生物分子的结合位点对接的可能性或此类对接(万一其发生)的质量的评价。在一个层面上,对接器确定配体是否可能与生物分子结合位点结合。如果对接器逻辑性地总结出结合是不可能的或者是高度不利的,则其可输出“未找到精确位姿”的结果。当对接程序生成的所有构象与结合位点具有不利的范德瓦尔斯冲突和/或静电排斥时,这可能发生。在对接程序的以上实例中,如果第二个操作未能找到具有小于阈值的软能量的位姿,对接器可返回诸如“未找到精确位姿”的结果。因为软能量首先考虑包括范德瓦尔斯力和静电力的非键相互作用,因此未找到精确位姿的结果意味着对于给定数量的位姿配体与生物分子受体具有严重的空间冲突和/或静电排斥。
在某些实施方式案中,对接器输出代表配体和生物分子结合位点之间的相互作用的对接得分。对接器可计算配体-生物分子相互作用的多种特征。在一个实例中,输出仅仅是配体和生物分子之间的相互作用能。在另一个实施方案中,总能量是输出。总能量可被理解为配体-生物分子相互作用能和配体张力的组合。在某些实施方案中,可使用诸如charmm的力场来计算此类能量。
在多种实施方案中,对接程序通过考虑配体在生物分子的结合位点中的多个位姿来生成此类输出。每个位姿将具有其自身的相关能量值。在一些实施方案中,对接程序对位姿排名并考虑与高排名的位姿中的一个或更多个相关的能量。在一些情况中,它可对某些高排名位姿的能量求平均值或者否则对排名靠前的位姿进行统计分析。在其他实施方案中,它仅仅选择与排名靠前的位姿相关的值并将其输出为得到的能量,用于对接。
“位姿”是配体相对于生物分子的结合位点的位置或方位。在位姿中,配体的一些或所有原子的三维位置相对于结合位点中的原子的位置中的一些或所有是特定的。尽管配体的构象不为其位姿-因为构象不考虑结合位点-构象可被用于确定位姿。在一些实施方案中,配体的方位和构象共同定义位姿。在一些实施方案中,只有当配体的方位/构象组合满足参考结合位点中的定义的阈值能量水平时,位姿才存在。
可采用多种计算机制来生成用于对接的位姿。实例包含关于可旋转键的系统的或随机的扭转搜索、分子动力学模拟和以“开发”新的低能构象的遗传算法。这些技术被用于修改配体和/或结合位点的计算表示,以搜索“位姿空间”。
对接器评价位姿以确定配体如何与结合位点相互作用。在一些实施方案中,它们通过基于以上提到的相互作用类型中的一种或更多种(例如,范德瓦尔斯力)计算相互作用的能量来完成这。该信息被用来表征对接,并且在一些情况中产生对接得分。在一些实施方案中,对接器基于对接得分来对位姿排名。在一些实施方案中,对接器将具有不良对接得分的位姿排除在考虑之外。
在某些实施方案中,虚拟蛋白筛选系统评价位姿以确定该位姿是否是活性的。如果位姿满足限定的约束,则其被视为是活性的,所述限定的约束已知对于在考虑中的期望的活性是重要的。例如,虚拟蛋白筛选系统可确定位姿是否支持配体在结合位点中的催化转化。
“配体”是与生物分子的结合位点相互作用以形成包含至少配体和生物分子的稳定的复合物的分子或复合物。除了配体和生物分子之外,稳定的复合物可包含(有时候需要)其他化学实体,诸如有机和无机辅因子(例如,辅酶和辅基)、金属离子等。配体可以是激动剂或拮抗剂。
当生物分子是酶时,结合位点是催化位点,且配体是底物、底物的反应中间体或底物的过渡态。“反应中间体”是在从底物到反应产物的转化中从底物产生的化学实体。底物的“过渡态”是对应于沿着反应途径最高势能的状态的底物。在趋于具有短暂存在的过渡态,碰撞反应物分子继续形成产物。在本公开内容中,有时候当在过程中描述底物时,中间体和过渡态也可适用于该过程。在此类情况中,底物、中间体和过渡态可被统一称为“配体”。在一些情况中,在底物的催化转化中生成多种中间体。在某些实施方案中,被选择用于分析的配体种类(底物或中间体或过渡态)是已知与催化转化中的限速步骤有关的配体种类。例如,在限速步骤中可以在化学上修饰与酶辅因子共价结合的底物。在此类情况中,底物-辅因子种类被用于为相互作用建模。
应清楚的是,配体的概念比“底物”的概念更广义。一些配体与结合位点结合,但是不经历催化转化。实例包含在药物设计领域中评价的配体。此类配体可以是为了药理学目的针对其与靶生物分子非共价结合的能力而选择的小分子。在一些情况中,配体被评价其加强、活化或抑制生物分子的天然行为的能力。
如本文所使用的,“生物分子(biomolecule)”和“生物分子(biologicalmolecule)”指通常在生物有机体中发现的分子。在一些实施方案中,生物分子包括具有多个亚单位的聚合生物大分子(即,“生物聚合物”)。典型的生物分子包括但不限于与天然存在的聚合物诸如rna(由核苷酸亚单位形成)、dna(由核苷酸亚单位形成)和肽或多肽(由氨基酸亚单位形成)共有一些结构特征的分子,包括例如rna、rna类似物、dna、dna类似物、多肽、多肽类似物、肽核酸(pna)、rna和dna的组合(例如,嵌合体(chimeraplast))等。不意图生物分子被限制为任何特定的分子,因为任何合适的生物分子在本公开内容中具备实用性,包括但不限于,例如,脂质、碳水化合物或通过一种或更多种遗传上可编码的分子(例如,一种或更多种酶或酶通路)制备的其他有机分子等。本公开内容的一些方面特别感兴趣的是具有与配体相互作用以影响化学或生物转化(例如,底物的催化、生物分子的活化或生物分子的失活)的结合位点的生物分子。
在一些实施方案中,“有益特性”或“活性”是以下中的一个或更多个的增加或降低:催化速率(kcat)、底物结合亲和力(km)、催化效率(kcat/km)、底物特异性、化学选择性、区域选择性、立体选择性、立体特异性、配体特异性、受体激动、受体拮抗、辅因子的转化、氧气稳定性、蛋白表达水平、溶解度、热活性、热稳定性、ph活性、ph稳定性(例如,在碱性ph或酸性ph)、葡萄糖抑制和/或对抑制剂(例如,乙酸、凝集素、单宁酸和酚类化合物)和蛋白酶的抗性。其他期望的活性可包括响应于特定刺激的改变的特征;例如,改变的温度和/或ph特征。在理性配体设计的背景中,靶向的共价抑制(tci)的优化是一种类型的活性。在一些实施方案中,如本文描述的筛选的一种或更多种变体作用于同一个底物,但是对于以下活性中的一种或更多种不同:产物形成的速率、底物到产物的转化百分比、选择性和/或辅因子的转化百分比。不意图本公开内容限于任何特定的有益特性和/或期望的活性。
在一些实施方案中,“活性”被用于描述酶催化底物到产物的转变的能力的更受限的概念。相关的酶特征是其对特定产物的“选择性”,所述特定产物诸如对映体或区域选择性产物。本文提出的“活性”的广义定义包括选择性,尽管传统上选择性有时候被视为与酶活性不同。
术语“蛋白”、“多肽”和“肽”可互换地使用来表示通过酰胺键共价连接的至少两个氨基酸的聚合物,而不管长度或翻译后修饰(例如,糖基化、磷酸化、脂质化、豆蔻酰化、泛素化等)如何。在一些情况中,聚合物具有至少约30个氨基酸残基,并且通常具有至少约50个氨基酸残基。更通常地,它们含有至少约100个氨基酸残基。不意图将本发明限于任何特定长度的氨基酸序列。这些术语包括常规被认为是全长蛋白或肽的片段的组分。该定义包括d-氨基酸和l-氨基酸、以及d-氨基酸和l-氨基酸的混合物。本文描述的多肽不局限于遗传上编码的氨基酸。事实上,除了遗传上编码的氨基酸,本文描述的多肽可以全部或部分地由天然存在的和/或合成的非编码氨基酸组成。在一些实施方案中,多肽为全长的原型(ancestral)多肽或亲本多肽的一部分,与全长亲本多肽的氨基酸序列相比包含氨基酸添加或缺失(例如,空位)和/或取代,同时仍然保持功能活性(例如,催化活性)。
如本文所使用的,术语“野生型(wild-type)”或“野生型(wildtype)”(wt)指天然存在的有机体、酶和/或其他蛋白(例如,非重组酶)。与野生型生物分子相互作用的底物或配体有时候被视为“天然”底物或配体。
如本文所使用的,术语“变体”、“突变体”、“突变体序列”和“变体序列”指在一些方面与标准序列或参考序列(例如,在一些实施方案中,亲本序列)不同的生物序列。该不同可被称为“突变”。在一些实施方案中,突变体是已通过至少一个取代、插入、交换(cross-over)、缺失和/或其他遗传操作改变的多肽序列或多核苷酸序列。为了本公开内容的目的,突变体和变体不限于特定的产生所述突变体和变体的方法。在一些实施方案中,突变体或变体序列与亲本序列相比具有增加的、减少的或基本上相似的活性或特性。在一些实施方案中,变体多肽与野生型多肽(例如亲本多肽)的氨基酸序列相比包含已突变的一个或更多个氨基酸残基。在一些实施方案中,在组成多种多肽的变体多肽中,与亲本多肽相比,多肽的一个或更多个氨基酸残基被保持恒定、是不变的、或未被突变。在一些实施方案中,亲本多肽被用作用于生成具有改进的稳定性、活性或其他期望的特性的变体的基础。
如本文使用的,术语“酶变体”和“变体酶”被用来指与参考酶相似(尤其在它们的功能上),但是在其氨基酸序列中具有使其在序列上不同于野生型或另一种参考酶的突变的酶。可以通过本领域技术人员熟知的很多种不同的诱变技术制备酶变体。另外,诱变试剂盒也是从很多商业的分子生物学供应商可得的。对于在限定的氨基酸处形成特定的取代(定点)、在基因的局部区域中形成特定的或随机的突变(区域特定的)或在整个基因上形成随机诱变(例如,饱和诱变),方法是可得的。本领域的技术人员已知产生酶变体的许多合适的方法,包含但不限于利用pcr的单链dna或双链dna的定点诱变、盒式诱变、基因合成、易错pcr、重排、和化学饱和诱变或本领域已知的任何其他合适方法。在产生变体之后,可针对期望的特性(例如,高的或增长的或者低的或降低的活性、增加的热稳定性和/或碱稳定性)对其筛选。
“酶组(apanelofenzymes)”是经选择以使得该组的每个成员催化相同的化学反应的一组酶。在一些实施方案中,组的成员可全体转化多个底物,每个底物经历相同的反应。通常选择组成员来有效转化多个底物。在一些情况中,组是市售可得的。在其他情况中,它们是实体专用的。例如,组可包括在筛选程序中被鉴定命中(hits)的多种酶。在某些实施方案中,组的一个或更多个成员只作为计算表示而存在。换言之,酶是虚拟酶。
“模型”是生物分子或配体的结构的表示。其有时候被提供为用于被表示的实体的原子或部分的三维位置的集合。模型常常包含酶变体的结合位点或其他方面的计算上产生的表示。与本文的实施方案相关的模型的实例从使用诸如rosetta(rosettacommons.org/software/)或moleculardynamics模拟的程序的同源建模、蛋白线程或从头开始的蛋白建模产生。
“同源模型”是至少包含在考虑中的配体的结合位点的蛋白或蛋白的一部分的三维模型。同源建模依赖于以下观察:在同源蛋白中蛋白结构趋于被保留。同源模型提供了包含主链和侧链的残基的三维位置。该模型从可能相似于建模的序列的结构的同源蛋白的结构模版生成。在一些实施方案中,结构模版被用于两个步骤中:“将序列与模板对齐”和“建立同源模型”。
“将序列与模板对齐”步骤将模型序列与一个或更多个结构模版序列对齐并准备用于建立同源模型的输入序列对齐。对齐鉴定模型序列和结构模板序列之间空位和其他差异区域。
“建立同源模型”使用结构模板的结构特征得到空间约束,该空间约束继而被用来使用共轭梯度和模拟退火优化程序产生例如模型蛋白结构。可从诸如nmr或x射线晶体学的技术获得模板的结构特征。此类技术的实例可在综述文章,“aguidetotemplatebasedstructureprediction”,qux,swansonr、dayr、tsaij.currproteinpeptsci.2009年6月;10(3):270-85中找到。
术语“活性构象”被用来指允许蛋白(例如,酶)导致底物经历化学转化(例如,催化反应)的该蛋白的构象。
“活性位姿”是其中配体可能经历催化转化或执行诸如与结合位点共价结合的一些期望的作用的位姿。
术语“序列”在本文中被用来指任何生物序列的顺序和身份,所述任何生物序列包含但不限于全基因组、全染色体、染色体片段、用于与基因相互作用的基因序列的集合、基因、核算序列、蛋白、肽、多肽、多糖等。在一些上下文中,“序列”指蛋白中的氨基酸残基的顺序和身份(即,蛋白序列或蛋白字符串)或指核酸中的核苷酸的顺序和身份(即,核酸序列或核酸字符串)。序列可由字符串表示。“核酸序列”指构成核酸的核苷酸的顺序和身份。“蛋白序列”指构成蛋白或肽的氨基酸的顺序和身份。
“密码子”指三个连续核苷酸的特定序列,其为遗传密码的一部分并限定蛋白中的特定氨基酸或者起始或终止蛋白合成。
术语“基因”被广泛地用来指dna或与生物功能相关的其他核酸的任何片段。因此,基因包括编码序列以及任选地其表达所需的调控序列。基因还任选地包括例如形成其他蛋白的识别序列的不表达的核酸片段。基因可从多种来源获得,包括从感兴趣的来源克隆或从已知的或预测的序列信息合成,并且基因可包含被设计为具有期望的参数的序列。
“部分”是分子的一部分,其可包含整个功能组或功能组的部分诸如亚结构,而功能组是促成那些分子的特征性化学反应的分子内的原子或键的组。
“筛选”指其中确定一个或更多个生物分子的一个或更多个特性的方法。例如,典型的筛选方法包括其中确定一个或更多个文库的一个或更多个成员的一个或更多个特性的那些筛选方法。可利用生物分子和生物分子的虚拟环境的计算模型来在计算上进行筛选。在一些实施方案中,针对选择的酶的期望的活性和选择性提供了虚拟蛋白筛选系统。
“表达系统”是用于表达由基因或其它核酸编码的蛋白或肽的系统。
“定向进化”、“导向进化”或“人工进化”指通过人工选择、突变、重组或其他操作来人工地改变一个或更多个生物分子序列(或表示该序列的字符串)的计算机模拟的、体外或体内过程。在一些实施方案中,定向进化发生在繁殖性群体中,其中(1)存在多种个体;(2)一些种类具有可遗传的遗传信息;并且(3)一些种类在适应度(fitness)方面不同。繁殖成功通过预先确定的特性诸如有益的特性的选择的结果来确定。繁殖性群体可以是例如,体外过程中的物理群体或计算机模拟过程中的计算机系统中的虚拟群体。
定向进化方法可被容易地应用于多核苷酸,以产生可被表达、筛选和测定的变体文库。诱变和定向进化方法在本领域中是众所周知的(参见,例如,美国专利第5,605,793、5,830,721、6,132,970、6,420,175、6,277,638、6,365,408、6,602,986、7,288,375、6,287,861、6,297,053、6,576,467、6,444,468、5,811238、6,117,679、6,165,793、6,180,406、6,291,242、6,995,017、6,395,547、6,506,602、6,519,065、6,506,603、6,413,774、6,573,098、6,323,030、6,344,356、6,372,497、7,868,138、5,834,252、5,928,905、6,489,146、6,096,548、6,387,702、6,391,552、6,358,742、6,482,647、6,335,160、6,653,072、6,355,484、6,03,344、6,319,713、6,613,514、6,455,253、6,579,678、6,586,182、6,406,855、6,946,296、7,534,564、7,776,598、5,837,458、6,391,640、6,309,883、7,105,297、7,795,030、6,326,204、6,251,674、6,716,631、6,528,311、6,287,862、6,335,198、6,352,859、6,379,964、7,148,054、7,629,170、7,620,500、6,365,377、6,358,740、6,406,910、6,413,745、6,436,675、6,961,664、7,430,477、7,873,499、7,702,464、7,783,428、7,747,391、7,747,393、7,751,986、6,376,246、6,426,224、6,423,542、6,479,652、6,319,714、6,521,453、6,368,861、7,421,347、7,058,515、7,024,312、7,620,502、7,853,410、7,957,912、7,904,249号,和所有相关的非美国副本;ling等人,analbiochem,254(2):157-78[1997];dale等人,meth.mol.biol.,57:369-74[1996];smith,ann.rev.genet.,19:423-462[1985];botstein等人,science,229:1193-1201[1985];carter,biochem.j.,237:1-7[1986];kramer等人,cell,38:879-887[1984];wells等人,gene,34:315-323[1985];minshull等人,curr.op.chem.biol.,3:284-290[1999];christians等人,nat.biotechnol.,17:259-264[1999];crameri等人,nature,391:288-291[1998];crameri等人,nat.biotechnol.,15:436-438[1997];zhang等人,proc.nat.acad.sci.u.s.a.,94:4504-4509[1997];crameri等人,nat.biotechnol.,14:315-319[1996];stemmer,nature,370:389-391[1994];stemmer,proc.nat.acad.sci.usa,91:10747-10751[1994];wo95/22625;wo97/0078;wo97/35966;wo98/27230;wo00/42651;wo01/75767;和wo2009/152336,其全部被通过引用并入本文。)
在某些实施方案中,定向进化方法通过重组编码从亲本蛋白开发的变体的基因以及通过重组编码亲本蛋白变体文库中的变体的基因而产生蛋白变体文库。该方法可使用包含编码亲代变体文库中的至少一种蛋白的序列或子序列的寡核苷酸。亲代变体文库的一些寡核苷酸可以是密切相关的,只在选择替代性氨基酸的密码子方面不同,所述替代氨基酸被选择为通过与其他变体重组而被改变。所述方法可被进行一个或多个循环,直到实现期望的结果。如果使用多个循环,则每个循环通常包括筛选步骤以鉴定具有可接受的或改进的性能和待用于至少一个随后的重组循环的那些变体。在一些实施方案中,筛选步骤涉及用于确定酶对期望的底物的催化活性和选择性的虚拟蛋白筛选系统。
在一些实施方案中,定向进化方法通过在特定残基处定点定向诱变来产生蛋白变体。通常通过结合位点的结构分析、量子化学分析、序列同源性分析、序列活性模型等来鉴定这些特定残基。一些实施方案采用饱和诱变,其中技术人员试图在特定位点或基因的窄区域处产生所有可能的(或尽可能接近于所有可能的)突变。
“重排”和“基因重排”是通过一系列链延伸循环来重组亲本多核苷酸的片段的集合的定向进化方法类型。在某些实施方案中,一个或更多个链延伸循环是自引发的;即,除了片段自身之外不添加引物而进行。每个循环包括:通过杂交使单链片段退火,随后通过链延伸延长退火的片段,以及变性。在重排的过程中,生长的核酸链通常在有时被称作“模板转换”的过程中被暴露于多个不同的退火伴侣,所述“模板转换”包括将来自一个核酸的一个核酸区域与来自第二核酸的第二区域转换(即,第一和第二核酸在重排过程中充当模板)。
模板转换经常产生嵌合序列,所述嵌合序列由在不同起源的片段之间引入交叉造成。交叉通过转换的模板在多个循环的退火、延伸和变性期间的重组产生。因此,重排通常导致变体多核苷酸序列的产生。在一些实施方案中,变体序列包括变体的“文库”(即,包括多个变体的组)。在这些文库的一些实施方案中,变体包含来自两个或更多个亲本多核苷酸的序列片段。
当采用两个或更多个亲本多核苷酸时,个体亲本多核苷酸足够同源,以使得来自不同亲本的片段在重排循环中使用的退火条件下杂交。在一些实施方案中,重排允许具有相对有限/低同源性水平的亲本多核苷酸重组。经常地,个体亲本多核苷酸具有不同和/或独特的区域和/或其他感兴趣的序列特征。当使用具有不同的序列特征的亲本多核苷酸时,重排可产生高度多样性的变体多核苷酸。
多种重排技术在本领域是已知的。参见,例如,美国专利第6,917,882、7,776,598、8,029,988、7,024,312和7,795,030号,其所有被通过引用以其全文并入本文。
一些定向进化技术采用“重叠延伸基因拼接法”或“基因soeing”,其为不依赖于限制位点重组dna序列并直接体内生成突变的dna片段的基于pcr的方法。在技术的一些实现中,初始pcr产生被用作第二pcr的模板dna的重叠基因片段,以产生全长产物。内部pcr引物在中间片段上生成重叠、互补的3’末端,并引入核苷酸取代、插入或删除用于基因剪接。这些中间片段的重叠链在第二pcr的3'区域杂交并被延伸以产生全长产物。在多种应用中,全长产物通过侧翼引物扩增,所述侧翼引物可包含用于为了克隆目的将产物插入表达载体的限制酶位点。参见,例如,horton等人,528-35[1990]。“诱变”是将至少一个突变引入标准或参考序列诸如亲本核酸或亲本多肽的过程。
定点诱变用于引入突变的有用技术的一个实例,尽管任何合适的方法具备实用性。因此,可选地或另外地,突变体可通过以下来提供:基因合成、饱和随机诱变、残基的半合成组合文库、递归序列重组(“rsr”)(参见,例如,美国专利申请公布号2006/0223143,通过引用以其整体并入本文)、基因重排、易错pcr和/或任何其他合适的方法。
合适的饱和诱变程序的一个实例被描述于美国专利申请公布号2010/0093560中,其被通过引用以其全文并入本文。
“片段”为核苷酸或氨基酸序列的任何部分。片段可利用本领域已知的任何合适的方法产生,包括但不限于,裂解多肽或多核苷酸序列。在一些实施方案中,片段通过使用裂解多核苷酸的核酸酶来产生。在一些另外的实施方案中,片段利用化学技术和/或生物合成技术生成。在一些实施方案中,片段包含至少一个亲本序列的子序列,所述子序列利用互补核酸的部分链延长生成。在涉及计算机模拟的技术的一些实施方案中,计算上产生虚拟片段以模拟通过化学和/或生物技术产生的片段的结果。在一些实施方案中,多肽片段表现出全长多肽的活性,而在一些其他的实施方案中,多肽片段不具有由全长多肽表现出的活性。
“亲本多肽”、“亲本多核苷酸”、“亲本核酸”和“亲本”通常被用来指在多样性生成程序诸如定向进化中被用作起点的野生型多肽、野生型多核苷酸或变体。在一些实施方案中,亲本自身经由重排或其他多样性生成程序产生。在一些实施方案中,定向进化中使用的突变体与亲本多肽直接相关。在一些实施方案中,亲本多肽在暴露于极端的温度、ph和/或溶剂条件时是稳定的并可充当用于生成用于重排的变体的基础。在一些实施方案中,亲本多肽对于极端的温度、ph和/或溶剂条件是不稳定的,并且亲本多肽被演变以制备稳健的变体。
“亲本核酸”编码亲本多肽。
“文库”或“群体”指至少两个不同的分子、字符串和/或模型,诸如核酸序列(例如,基因、寡核苷酸等)或来自其的表达产物(例如,酶或其他蛋白)的集合。文库或群体通常包括很多不同的分子。例如,文库或群体通常包括至少约10个不同的分子。大的文库通常包括至少约100个不同的分子、更通常地至少约1000个不同的分子。对于一些应用,文库包括至少约10000或更多个不同的分子。然而,不意图本发明被限制于特定数目的不同分子。在某些实施方案中,文库包括通过定向进化程序产生的很多变异或嵌合的核酸或蛋白。
当来自两种核酸的每一种的序列被组合以产生子代核酸时,所述两种核酸被“重组”。当两种核酸均是用于重组的底物时,所述两种核酸被“直接”重组。
术语“选择”指其中一种或更多种生物分子被鉴定为具有一种或更多种感兴趣的特性的过程。因此,例如,技术人员可筛选文库以确定一个或更多个文库成员的一种或更多种特性。如果一个或更多个该文库的成员被鉴定为拥有感兴趣的特性,则其被选择。选择可包括分离文库成员,但这不是必需的。另外,选择和筛选可以并且经常是同时的。本文公开的一些实施方案提供了用于筛选并选择具有期望的活性和/或选择性的酶的系统和方法。
“下一代测序”或“高通量测序”是使测序过程并行化的测序技术,一次产生数千计或数百万计的序列。合适的下一代测序方法的实例包括但不限于,单分子实时测序(例如,pacificbiosciences,menlopark,california)、离子半导体测序(例如,iontorrent,southsanfrancisco,california)、焦磷酸测序(例如,454,branford,connecticut)、连接测序(例如,solidsequencingoflifetechnologies,carlsbad,california)、通过合成和可逆终止物的测序(例如,illumina,sandiego,california)、诸如透射电子显微术的核酸成像技术等。
“因变量”(“dv”)表示输出或结果,或者被测试以查看其是否是所述结果。“自变量”(“iv”)表示输入或原因,或被测试以查看其是否是所述原因。因变量可被研究以查看其是否随着自变量变化而变化以及变化多少。
在以下的简单的随机线性模型中:
yi=a+bxi+ei
其中,项yi是因变量的第i个值,且xi是自变量(iv)的第i个值。项ei被称为“误差”且包含不由自变量解释的因变量的变异性。
自变量(iv)也被称为“预测变量”、“回归量”、“控制变量”、“操纵变量”、“解释变量”或“输入变量”。
术语“系数”指因变量或含有因变量的表达式乘以的标量值。
术语“正交的”和“正交性”指与模型中的其他自变量或其他关系不相关的自变量。
术语“序列活性模型”指描述一方面的生物分子的活性、特征或特性与另一方面的多种生物序列之间的关系的任何数学模型。
术语“字符串”指生物分子的表示,该表示保存了关于该分子的序列/结构信息。在一些实施方案中,字符串包含关于变体文库中的序列突变的信息。生物分子的字符串和生物分子的活性信息可被用作序列活性模型的训练集。生物分子的非序列特性可被储存或否则与针对生物分子的字符串相关。
“参考序列”为从其产生序列的变异的序列。在一些情形中,“参考序列”被用来限定变异。此类序列可以是被模型预测为具有期望的活性的最高值(或最高值中的一个)的序列。在另一种情形中,参考序列可以是原始蛋白变体文库的成员的序列。在某些实施方案中,参考序列为亲本蛋白或亲本核酸的序列。
词语“训练集”指一个或更多个模型与其拟合并基于其建立的一组序列活性数据或观察值。例如,对于蛋白序列活性模型,训练集包括原始的或改进的蛋白变体文库的残基序列。通常,这些数据包括完整的或部分的残基序列信息,以及文库中每个蛋白的活性值。在一些情况中,多种类型的活性(例如,速率常数数据和热稳定性数据)被共同提供在训练集中。活性有时是有益的特性。
术语“观察值”是关于蛋白或其他生物实体的信息,所述信息可被用于训练集来生成诸如序列活性模型的模型。术语“观察值”可指任何测序和/或测定的生物分子,包括蛋白变体。在某些实施方案中,每个观察值是文库中的变体的活性值和相关序列。通常,生成序列-活性模型采用的观察值越多,该序列活性模型的预测力越好。
词语“预测力”指模型在多种条件下正确地预测数据的因变量的值的能力。例如,序列活性模型的预测力指模型由序列信息预测活性的能力。
词语“交叉验证”指用于测试模型预测因变量的值的能力的普遍性的方法。所述方法利用一组数据制作模型,并利用不同的一组数据测试模型误差。第一组数据被视为训练集,而第二组数据为验证集。
词语“系统方差”指以不同的组合被改变的一个项或一组项的不同描述符。
词语“系统地变化的数据”指由以不同的组合被改变的一个项或一组项的不同描述符产生、推导或得到的数据。很多不同的描述符可同时但以不同的组合被改变。例如,从其中氨基酸的组合已被改变的多肽收集的活性数据是系统地变化的数据。
词语“系统地变异的序列”指其中每个残基见于多种背景的一组序列。原则上,系统变异的水平可通过这些序列彼此正交的程度(即,与平均值相比最大程度地不同)来定量。
术语“切换(toggling)”指将多种氨基酸残基类型引入优化的文库中的蛋白变体的序列中的特定位置。
术语“回归”和“回归分析”指用来理解自变量中与因变量有关的那些自变量,并被用来探索这些关系的形式的技术。在有限的情形中,回归分析可被用来推导自变量和因变量之间的因果关系。它是用于评价变量之间的关系的统计技术。当焦点是关于因变量和一个或更多个自变量之间的关系时,其包括很多用于对若干个变量建模和分析的技术。更特别地,回归分析帮助技术人员理解当任一个自变量变化而其他自变量保持固定时,因变量的典型值(typicalvalue)如何改变。回归技术可被用于从包括多个观察值的训练集生成序列活性模型,所述多个观察值可包括序列和活性信息。
“偏最小二乘法”(“pls”)是通过将预测变量(例如,活性)和可观察变量(例如,序列)投射到新的空间而发现线性回归模型的方法族。pls也被称为“潜在结构投射法”。x(自变量)和y(因变量)数据两者均被投射至新的空间。pls被用来找出两个矩阵(x和y)之间的基本关系。潜在变量模型被用来对x和y空间中的协方差结构建模。pls模型将试图找出在x空间中的多维方向,所述多维方向解释y空间中的最大多维变化的方向。当预测器(predictor)的矩阵具有比观察值更多的变量时,并且当在x值中存在多重共线性时,pls回归是特别有用的。
潜在变量(与可观察变量相对)是未直接被观察到但从观察到的或直接测量的变量推测的变量。旨在以潜在变量的形式解释观察到的变量的数学模型被称为潜在变量模型。
“描述符”指用来描述或标识项的事物。例如,字符串中的字符可以是由该字符串表示的多肽中的氨基酸的描述符。
在回归模型中,因变量通过项的和与自变量相关。每个项包括自变量和相关回归系数的乘积。在纯粹的线性回归模型的情况中,回归系数由以下表达形式中的β给出:
yi=β1xi1+...+βpxip+εi=xitβ+εi
其中yi是因变量,xi是自变量,εi是误差变量,并且t表示转置矩阵(transpose),即向量xi和β的内积。
词语“主成分回归”(“pcr”)指当评估回归系数时使用主成分分析的回归分析。使用自变量的主成分,而不是直接将因变量对自变量回归。pcr在回归分析中通常只使用主成分的子集。
词语“主成分分析”(“pca”)指使用正交变换将可能相关的变量的一组观察值转换成称为“主成分”的线性不相关的变量的一组值的数学程序。主成分的数目小于或等于最初变量的数目。该转换以使得第一主成分具有最大的可能方差(即,在数据中占尽可能多的变化性),并且每个之后的成分在其与之前的成分正交(即,与之不相关)的约束下转而具有最高的可能方差的方式被定义。
“神经网络”为含有互相连接的处理元件或“神经元”的组的模型,所述处理元件或“神经元”使用联结法(connectionistapproach)计算处理信息。神经网络被用来对输入和输出之间的复杂关系建模和/或被用来找出数据中的模式。大多数神经网络以非线性、分散式、平行的方式处理数据。在大多数情况中,神经网络是在学习阶段期间改变其结构的自适应系统。由处理元件统一且平行地执行多种功能,而不利用对被分配了多个单元的子任务的清晰描述。
通常,神经网络包括简单处理元件的网络,所述网络呈现出通过处理元件和元件参数之间的联系确定的复杂性整体行为。神经网络与被设计为改变网络中的联系的强度的算法一起使用以产生期望的信号流。所述强度在训练或学习期间被改变。
“遗传算法”(“ga”)是模仿进化过程的过程。遗传算法(ga)被用于很多领域来解决未被完全表征或太复杂以致不允许被完全表征的问题,但是对于所述问题一些分析评价是可获得的。即,ga被用来解决可通过对解的相对值(或至少一个可能的解相对于另一个解的相对值)的一些定量测量来评价的问题。在本公开内容的上下文中,遗传算法是用于在计算机中选择或操作字符串的过程,通常其中该字符串对应于一个或更多个生物分子(例如,核酸、蛋白等)或者被用于训练诸如序列活性模型或支持向量机的模型的数据。
在一个实例中,在算法的第一代中,遗传算法提供并评价模型的群体。每个模型包含描述在至少一个自变量(iv)和因变量(dv)之间的关系的多个参数。“适应度函数”评价群体的成员模型并基于一个或更多个标准来将它们排名,所述一个或更多个标准诸如高的期望的活性或低的模型预测误差。群体的成员模型在遗传算法的上下文中有时候也被称为个体或染色体。在一些实施方案中,使用赤池信息准则(aic)或贝叶斯信息准则(bic)来评价模型适应度,其中,具有最小aic或bic值的个体被选作最适应的个体。选择高排名的模型用于升级到第二代和/或交配以产生“子代模型”的群体用于算法的第二代。第二代中的群体通过适应度函数来相似地评价,并将高排名的成员升级和/或与第一代交配。遗传算法继续以该方式用于后续的代数,直到满足“收敛准则”,在该点处,算法以一个或更多个高排名的个体(模型)结束。
在另一个实例中,“个体”是变体肽序列,并且适应度函数是这些个体的预测的活性。每一代包含个体肽序列的群体,对其进行适应度的评价。在一代中最适应的被选择用于升级和/或交配,以产生下一代群体。多代之后,遗传算法可收敛至高性能的肽序列的群体。
如在以上实例中的,遗传算法通常运行经过多次迭代以搜寻参数空间中的最佳参数。遗传算法的每次迭代也被称为遗传算法的一“代”。遗传算法的一代中的模型形成用于该代的“群体”。在遗传算法的上下文中,术语“染色体”和“个体”有时候被用作群体中的模型或一组模型参数的别称。其被如此使用是因为,来自亲代的模型将其参数(或“基因”)传递至子代模型,这类似亲本染色体将其基因传递至子代染色体的生物过程。
术语“遗传操作”(“go”)指生物的和/或计算的遗传操作,其中任何类型的字符串的任何群体中(以及由此而来的由此类字符编码的物理对象的任何物理特性中)的所有改变可被描述为随机和/或预先确定地应用有限组的逻辑代数函数的结果。go的实例包括但不限于扩增、交换、重组、突变、连接、片段化等。
“赤池信息量准则”(“aic”)是对统计模型的相对拟合优度的测量,并且其经常被用作在有限组的模型中选择模型的标准。aic建立在信息熵的概念上,事实上当给定的模型被用来描述真实性时提供对信息丢失的相对测量。其可被说成是描述模型结构中偏差和方差之间的权衡,或不严格地讲,模型的准确性和复杂性之间的权衡。aic可如下计算:
aic=-2logel+2k,
其中,l是函数的最大似然性,且k是待评价的模型的自由参数的数目。
“贝叶斯信息准则”(“bic”)为在有限组的模型中选择模型的准则,且与aic紧密相关。bic可如下计算:bic=-2logel+kloge(n),其中,n是数据观察值的数目。由于观察值的数目增加,bic往往比aic对额外数目的自由参数罚分更重。
模型的“似然性函数”或“似然性”为统计模型的参数的函数。给出一些观察结果的一组参数值的似然性等于给出那些参数值的那些观察结果的概率,即l(θ|x)=p(x|θ)。
“集成模型”为其项包括一组模型的所有项的模型,其中,集成模型的项的系数基于该组中的个体模型的相应项的加权的系数。系数的加权基于个体模型的预测力和/或适应度。
“蒙特卡罗模拟”为依赖大量的随机抽样以获得模仿真实现象的数值结果的模拟。例如,从区间(0,1]抽取大量的伪随机均匀变量,并将小于或等于0.50的值指定为正面并将大于0.05的值指定为反面,是对重复掷硬币行为的蒙特卡罗模拟。
ii.工作流的一般说明
a.一轮定向进化的工作流
在某些实施方案中,整个工作流利用体外技术和计算技术两者用于控制定向进化过程。该过程的计算方面采用结构模型和序列活性模型。
每轮定向进化采用一组新的结构模型和新的序列活性模型。另外,在每一轮中,经鉴定用于另外的分析的生物分子变体利用变体的三维结构模型来评价。将来自结构模型的信息与变体的序列和测定数据(活性)组合,以产生大的未经过滤的数据集。通常,将数据集的一部分用作训练集。对于当前轮次的定向进化,训练集训练序列活性模型,然后序列活性模型鉴定用于下一轮定向进化的生物分子变体。
在某些实施方案中,采用一种或更多种遗传算法(ga)来评价在每一轮定向进化的开始提供的未经过滤的组合数据。ga鉴定未经过滤的数据集中所包含的信息的子集,该子集被用作用于训练新的序列活性模型的自变量。活性是因变量;序列活性模型将活性提供为在过滤期间鉴定的自变量的函数。在多种实施方案中,序列活性模型是非线性模型。在某些实施方案中,序列活性模型是n维空间中的超平面,所述n维空间可通过支持向量机产生。
在图1a中示出的实例中,定向进化工作流如下展开。最初,针对多个生物分子变体收集信息。这些变体的每一个可以已经在前一轮的定向进化中被鉴定。如果该项目刚刚开始(即,无之前轮次的定向进化),从不同的来源获得变体,所述不同的来源诸如已知具有潜在的感兴趣的特性的一组生物分子。有时候,选择第一轮的变体以跨越相对宽的范围的序列和/或活性空间。
在鉴定完变体之后,评价系统获得每一个变体的多种类型的信息。尤其,确定每个变体的至少一个感兴趣的活性和序列。在一些实施方案中,序列被表示为来自野生型序列或其他参考序列的突变的集合。在一些实施方案中,活性被存储为具有限定的单位的数值。在一些实施方案中,活性值被归一化。如果给定的变体的序列不是已知的,可通过对变体的物理样本测序来获得。
除了序列活性数据之外,对每一个变体生物分子生成结构模型。在某些实施方案中,结构模型是同源模型。计算上评价结构模型,以获得与每一个变体的序列和活性数据组合的另外的数据。在一些实现中,每一个变体的结构模型被用来鉴定配体与生物分子的受体位点的相互作用能和/或描述配体在受体位点中的几何结构的一个或更多个参数。此类几何结构可包括在配体的原子和结合位点中的残基部分的原子和/或结合位点中的辅因子部分的原子之间的距离。下文展示了某些实例。
未经过滤的数据集包括每一个变体的序列和活性数据,并且通常包括每一个变体的多种另外类型的信息。如本文描述的,这些另外类型的信息源自于每一个变体的结构模型。另外,这些另外的数据通常包含(i)被考虑的配体和每一个变体的结合位点之间的相互作用或结合能和/或(ii)表征配体与受体的相互作用的结构/几何描述符。参见图1a的模块103。
已经发现,原始的未经过滤的数据集对于训练新的序列活性模型并不总是最佳的。而是,组合的原始数据集的经过滤的子集通常提供更有用的序列活性模型。因此,如模块105和107中所示的,对来自模块103的原始数据集进行过滤。
过滤可通过任何合适的技术来完成。如以下更详细地描述的,一种任选的技术将从变体的结构模型获取的某些类型的参数(例如,某些底物原子到残基原子的距离)移出。模块105。例如,未经过滤的数据集可包含受体结合位点中的配体的十种可用的几何特征,但是过滤消除了这些中的三个,以使得只有七种此类参数的子集被用于训练集。这些参数连同序列一起充当针对训练集训练的序列活性模型中的自变量。可选地或另外地,过滤可移出具有落在被确定可用于产生序列活性模型的范围以外或阈值以下的一个或更多个自变量的值的变体。模块107。在某些实施方案中,以该方式过滤的自变量源自结构模型。
如在模块109所示的,在适当地过滤原始数据集之后,其被用于产生序列活性模型。如所提到的,序列活性模型可以是非线性模型,诸如通过支持向量机确定的n维空间中的超平面。在产生序列活性模型之后,它被用于鉴定用于下一轮定向进化的高性能变体。参见模块111。在一个实施方案中,经训练的序列活性模型与遗传算法(ga)一起被用来选择可能具有有益特性的多个变体序列。所选择的变体被用于下一轮的定向进化。在此类下一轮中,如以上描述的(模块103,任选地105、107和109)处理用序列活性模型选择的变体。然而,它们首先被分析以产生新的原始数据集。参见模块113。在某些实施方案中,物理地产生变体并测定活性。这提供了原始数据中的一些。还在结构上对变体建模,以确定在较前轮次的定向进化中使用的能量类型和几何结构类型中的每一种的相互作用能量值和结合配体的几何值。可采用对接器来产生这些数据类型的值。如果有必要,对变体中一个或更多个测序以完善原始数据。
以该方式继续多个轮次的定向进化,直到一个或更多个轮次显示出有限的改进或达到其他的收敛准则。然后结束定向进化项目。在图1a中,收敛准则检验由决策模块115示出。
b.模型产生工作流
如以上所示的,一些实现在训练序列活性模型之前过滤原始数据集。过滤可将某些变量类型从原始数据移出。每个变量类型为用于序列活性模型的潜在自变量。可选地,或另外地,过滤可移出具有在所限定的范围之外的参数值的某些变体。已经发现,此类过滤减少由使用数据训练的模型产生的噪声。在一些实现中,使用一个或更多个ga来完成过滤。在某些实施方案中,从原始数据过滤的数据的类型被限制于配体和生物分子之间的相互作用能和/或配体在生物分子结合位点中的几何特征。
图1b展示了过滤原始数据的一种方法。在示出的实施方案中,来自三个来源的数据被组合以形成原始数据集153。每一个变体从所有三个来源给出(contributes)其自身的数据。组合数据包含配体-变体相互作用的活性数据。可利用诸如液相色谱法、气相色谱法等的标准测定手段来产生由模块141表示的活性数据。另外,对具有期望的活性数据的个体变体提供序列数据。由模块143表示的序列数据可以是提前已知的或可通过对变体的氨基酸或编码核酸测序来确定。可利用许多可用的测序技术中的任一种来进行测序。在一些实施方案中利用大量平行测序。最后,可从变体的结构模型产生结构数据。可使用不只是结构模型还有评价配体在考虑中的变体的结构模型的结合位点中的位姿的对接程序(对接器)来获取此类信息。原始结构数据包含许多类型的参数的数据,所述许多类型的参数包括特定相互作用能类型以及在配体和辅因子和/或结合位点残基之间原子到原子的距离。原始结构数据由图1b中的模块145表示。
数据的所有三个来源如图1b中所示地组合,以提供组合的原始数据153。在某些实施方案中,组合的原始数据以计算机可读的文件或文件组的形式来提供,所述文件或文件组可用于通过过滤工具或计算机实现的算法进一步处理。
在示出的实施方案中,显示了两个单独的过滤阶段:阶段155中的特征选择和阶段157中的距离选择。在示出的实施方案中,这些过滤操作中的每一个利用其自身的遗传算法采用其自身的序列活性模型作为目标函数来完成。在特定的实施方案中,如在图1b中示出的,利用支持向量机159和161来产生序列活性模型。特征选择过滤器鉴定特定相互作用能类型和/或原子到原子的距离用于从组合的原始数据集的移出。在该实施方案中,“距离”的概念包括其他的几何参数,诸如配体原子相对于生物分子和/或辅因子原子的角度特征、扭转特征和整体的位置特征。对于促成数据集的所有变体,移出经鉴定的数据类型。当利用几何算法时,移出过程可以是不固定的。换言之,在进行特征选择遗传算法期间,被移出的数据中的一个或更多个可以只是暂时性地被移出,持续一代或更多代。以下描述了用于完成这的合适技术的实例。距离选择过滤器移出促成原始数据的某些变体的数据。该过滤器选择在指定的数值范围之外的某些能量值和/或距离值。具有在这些范围之外的能量值和/或距离值的任何变体其数据被整体地从原始数据集移出。当利用遗传算法实现过滤时,在随后执行遗传算法期间,如果合适,在过程中的一个点移出的变体数据可以被重新引入。例如,在遗传算法的一代期间移出的变体数据可在随后的世代被重新引入。以下更加详细地描述了该过程。
在如关于模块155和157描述的过滤结束之后,使用经过滤的数据训练序列活性模型。在一些实现中,利用支持向量机进行训练。得到的序列活性模型被示为模块165。在基于预测的活性值考虑并对变体序列排名的不同的遗传算法中,其被用作目标函数。在讨论中的遗传算法被示于图1b的模块167中。
在一些其他的实施方案中,不进行特征选择阶段155。因此,无特征被过滤掉。换言之,对于预测遗传算法167,所有可用的特征被用于训练序列活性模型165。过滤仅仅移出具有在鉴定的范围之外的能量值或几何值的变体。在一些其他的实施方案中,特征选择阶段155和距离选择阶段157被合并成单个选择阶段,其可利用遗传算法来实现。在这些实施方案中,在利用遗传算法评价的训练集数据中,特征类型和特征值两者均是变化的。
图1c展示了其中不进行特征选择阶段或使其与距离选择阶段157合并的过程。如示出的,利用单一遗传算法(singlegeneticalgorithm)173来过滤原始数据171,所述单次遗传算法173选择具有约束在选定的范围之内的一个或更多个几何参数的变体。在一个实例中,几何参数是底物的原子和结合位点中的残基或辅因子的原子之间的距离。例如,一个参数可以是结合位点中辅因子上的氮原子和酪氨酸残基上的氧原子之间的距离,另一个参数可以是底物上的羰基炭和辅因子上的磷原子之间的距离,等等。这些距离的每一个可被设置在任意阈值内(例如,第一距离可能需要小于5埃,且第二距离可能需要小于7.5埃)。
算法173的适应度函数是利用参数限制的不同组合来训练的序列活性模型175的预测准确性。以此方式,针对约束的几何参数的多种组合训练准确的序列活性模型175的能力来评价该约束的几何参数的多种组合。在某些实施方案中,利用支持向量机来训练序列活性模型。
将未被遗传算法173选择的变体从考虑移出,以产生变体过滤的数据集177。换言之,通过单一遗传算法173过滤的结果是原始数据171的子集,所述子集仅包括数据171中变体的子集的数据。该子集被用来训练高度精确的序列活性模型,其反过来被用于另一种遗传算法预测算法179中。在某些实施方案中,预测算法179鉴定被预测具有高活性的新的变体序列。它可通过将可选的氨基酸(或核苷酸)序列应用于经训练的序列活性模型并确定哪些序列可能具有高的有益特征(例如,序列活性模型的活性)的值来完成这。遗传算法179产生可选的序列,经训练的序列活性模型对其评价适应度。最终,鉴定高性能变体序列,用于另外的调查和/或产生。
iii.遗传算法应用的一般说明
一些实施方案提供了利用遗传算法来产生用于训练序列活性模型的经过滤的数据集的方法(例如,以下描述的第一遗传算法和第二遗传算法),所述序列活性模型诸如通过支持向量机优化的模型。其他的实施方案提供了利用遗传算法来调整序列活性模型的系数的值以使模型适应经过滤的训练数据集的方法。又其他的实施方案利用遗传算法来探索序列空间并鉴定具有有利特性的蛋白变体(例如,以下描述的第三遗传算法)。
在遗传算法中,合适的适应度函数和合适的交配程序(matingprocedure)被定义。适应度函数提供了用于确定哪些“个体”(在一些实施方案中为模型)对于观察的数据“最适应”或具有最高的预测能力(即,模型可能提供最佳结果)的准则。在一些实施方案中,模型通过由一个或更多个自变量(iv)和因变量(dv)之间的关系来限定,且该关系通过一个或更多个参数来描述。遗传算法提供了搜遍参数空间来找出产生最成功的模型的参数或参数值的范围的组合的机制。
遗传算法中的许多过程受生物遗传操作的启示。同样地,在遗传算法中使用的术语借用关于遗传操作的生物术语。在这些实施方案中,群体中的“个体”(有时候被称为成员或染色体)中的每一个包括代表为模型被测试的所有参数的“基因”,和对于该参数具有在限定的范围内的选择的值的基因。例如,染色体可具有代表在位置131处gly的存在的基因。
在一些实施方案中,遗传算法可被用于选择模型的合适的iv(例如,以下描述的用于列过滤的第一遗传算法)。此类算法的一个实例包含二进制值1和0的基因/参数,每个参数与一个iv关联。如果在运算结束时,最适应的个体中一个iv的参数收敛至0,则将该iv从模型剔除。相反地,该项被保留。
在一些实施方案中,模型的适应度由模型的预测力来测量。在一些实施方案中,适应度通过基于以下描述的混淆矩阵的命中率来测量。在一些实施方案中,适应度通过aic或bic来测量。该实例中的模型在一些情况中事实上可以是用于产生那些模型的基础数据集。
评估特定世代中的每个“模型”的预测力之后,检查遗传算法的收敛性或其他准则(诸如,固定的代数)以确定该过程是否应继续用于另外的世代。假定该遗传算法尚未满足终止的标准,则对当前世代的模型排名。具有最高预测力的那些模型可被保留并用于下一代。例如,可采用10%的精选率(elitismrate)。换言之,前10%的模型(如使用拟合函数确定并通过例如准确性或aic测量的)被保留以成为下一代的成员。下一代中其余90%的成员通过与来自前一代的“亲本”交配来获得。
如所指出的,“亲本”为选自前一代的模型。通常,虽然选择倾向于前一代的更合适成员,虽然在其选择中可能存在随机的成份。例如,亲本模型可使用线性加权(例如,比另一个模型表现好1.2倍的模型多20%的可能被选择)或几何加权(即,模型中的预测差异被乘幂数以获得选择的概率)来选择。在一些实施方案中,通过从前一代中的模型排名中简单地选择表现最佳的两个或更多个模型而不选择其他模型来选择亲本。在这些实施方案中,对选自前一代的所有模型交配。在其他实施方案中,来自前一代的一些模型在不交配的情况下被选择纳入下一代中,并且来自前一代的表现较差的模型被随机地选择作为亲本。这些亲本可彼此交配和/或与如此被选择待纳入下一代的表现较好的模型交配。
已选择一组亲本模型之后,通过提供来自一个亲本的一些基因(参数值)和来自另一亲本的其他基因(参数值),将成对的此类模型交配以产生子代模型。在一个方法中,比对两个亲本的系数并接连地考虑每个值以确定该子代应采用来自亲本a的项还是来自亲本b的项。在一个实现中,交配程序从亲本a开始并且随机地确定“交换(crossover)”事件是否应发生在所遇到的第一个项处。如果是这样,从亲本b采用该项。如果不是,从亲本a采用该项。接连地考虑下一项用于交换等。这些项继续来自在考虑中的提供先前项的亲本,直到交换事件发生。在那个点,下一项由另一亲本提供,并且所有的后续项均由该亲本提供直至另一个交换事件发生。为了保证在子代模型中两个不同位置处不选择同一项,可采用多种技术,例如,部分匹配交换技术。在一些实施方案中,子代染色体可采用基因的值的平均值,而不使用来自亲本之一的基因的值。
在一些实施方案中,遗传算法还采用一个或更多个突变机制以生成模型的另外的多样性,这有助于探索未被亲代中的任何现存基因覆盖的参数空间的区域。另一方面,突变机制影响收敛性,以使得突变率越高或突变范围越大,收敛(如果有的话)所用的时间越长。在一些实施方案中,突变通过随机选择染色体/模型和随机选择所述染色体的参数/基因来实现,然后所述参数/基因被随机改变。在一些实施方案中,参数/基因的随机改变的值从具有限定范围的随机均匀分布抽取。在其他实施方案中,参数/基因的随机改变的值从具有限定范围的随机正态分布抽取。
已考虑每个参数之后,子代“模型”被确定用于下一代。选择另两个亲本以产生另一个子代模型,等等。最终,新一代中的子代群体准备好以以上描述的方式通过适应度函数评价。
过程一代接一代地继续,直到满足中止准则,诸如值的收敛。在那个点,从当前世代选择至少一个排名靠前的模型作为整体最佳模型。收敛性可通过很多常规技术测试。在一些实施方案中,其包括确定来自很多后续世代的最佳模型的性能没有明显地改变。中止准则的实例包括但不限于到目前为止生成的世代的数目、来自当前文库的最佳蛋白的活性、期望的活性的量级和在最后一代模型中观察到的改进的水平。
iv.利用遗传算法进行数据过滤的实施方案
在一些实施方案中,对于从可用信息获得并使用序列活性模型,有两个或三个阶段。这些步骤的每一个使用遗传算法。在三阶段过程中,第一遗传算法针对来自原始数据集的数据操作来选择用于序列活性模型的自变量。这些自变量选自可用自变量(有时候被称为参数)的池。不是所有的可用自变量被用于最终模型。在一个实施方案中,序列或突变信息总是被用作自变量,但是其他类型的自变量通过遗传算法来选择。很好地完成(或者在一些实施方案中最佳地完成)准确地预测活性的自变量的特定组合被选择。例如,除了序列信息之外可使用五到十个可用自变量,但是只有这些非序列变量中的三个被选择用于序列活性模型。遗传算法鉴定自变量的许多可选的组合中的哪一个最佳地完成训练序列活性模型以预测活性。
另一个遗传算法鉴定数据集中的一些或所有的非序列自变量的合适范围。对于这些自变量,可通过阈值或截断值来限定范围。在两阶段过程或三阶段过程两者中均使用该遗传算法。
最后的遗传算法鉴定应经受选择或另外的分析的生物分子(例如,蛋白变体)序列。该遗传算法提供了多种序列并利用使用经过滤的数据训练的序列活性模型来测试其适应度,所述经过滤的数据是利用一个或两个前述遗传算法选择的。该遗传算法和本文讨论的其他遗传算法之间的区别值得注意。该算法将核酸、氨基酸或其他生物分子序列提供为群体中的个体。相对地,在本文讨论的其他遗传算法中,个体为模型或模型参数的集。
在一些实施方案中,序列活性模型是非线性模型。在其他实施方案中,其为线性模型。
如在图2a至图2c中示出的,序列活性模型训练集可用的数据包括被用于准备训练集的多个变体生物分子中的每一个的信息。每一个变体的信息包括其序列和其活性。在本文展示的多个实例中,活性是酶在转变底物方面的速率和/或立体选择性。其他类型的活性或有益特性可被采用,且这些类型中的一些在本文的其他地方被描述。活性数据从体外分析和/或计算技术来确定,所述计算技术诸如在与本发明同日提交的美国专利申请第61/883,838号中描述的虚拟筛选,并且其被通过引用以其全部并入本文。
在某些实施方案中,序列信息可被提供为对起始骨架的一组突变,该骨架可以是野生型序列或一些其他序列诸如共有序列。关于突变的序列信息可以以在给定位置处的起始残基和取代残基的形式提供。另一个备选简单地鉴定特定位置处的最终残基。在多种实施方案中,序列信息通过遗传算法或其他计算技术提供,并因此不需要对核酸或其他成分测序而得知。如果需要测序,可采用许多测序类型中的任一种。这些类型中的一些在本文其他地方被描述。例如,在一些实施方案中,使用了高通量技术来测序核酸。
除了序列和活性数据之外,原始数据包括多种类型的另外的信息,所述多种类型的另外的信息可被并入或不并入序列活性模型的最终训练集。该另外的信息可以为许多不同的类型。每种类型潜在地充当序列活性模型的自变量。如本文所说明的,遗传算法或其他技术评价每种类型的信息的有用性。
在多种实施方案中,另外的信息描述配体-受体结合的特征。此类信息可源自测量和/或计算。如所提到的,变体的结构模型可鉴定这些其他类型的信息的值。在一个实例中,结构模型是同源模型。对接器或相似的工具可被用来从结构模型获得另外的信息。从对接器生成的信息实例包括如通过诸如accelryscdocker程序的对接程序计算的相互作用能和/或总能量。其他的实例涉及表征配体或其部分或原子相对于辅因子、结合位点残基的相对位置和/或与在考虑中的变体的结合位点有关的其他特征的几何参数。如所提到的,一些该信息可涉及关于底物或中间体和结合位点中的辅因子或残基的相对位置的距离、角度和/或扭转信息。例如,相互作用能量值可基于范德瓦尔斯力和/或静电相互作用。配体的内能也可被考虑。
图2a-图2c示出了根据本公开的一些实施方案过滤原始序列活性数据集的实例。图2a示出了转氨酶家族的n个变体的原始序列活性数据集。每个变体与活性数据、序列数据、能量数据和几何数据相关联。在一些实施方案中,活性数据可以是催化速率、对映体专一性等,其可通过本文其他处描述的多种方法来测定。每种变体的三个序列位置p1、p2和p3被提供于用于包括在序列活性模型中的原始数据集中。此外,提供了如本文其他处通过虚拟对接系统确定的两种能量值总能量和相互作用能用于潜在地包括在模型中。最终,通过虚拟对接系统提供了五种几何值用于潜在地包括在模型中。在该涉及配体的实例中,这些几何值中的每一个是当被对接到酶变体时与当被对接到野生型酶时配体的关键原子之间的距离。特别地,n1表示氮原子,p是磷酸基团的磷,c(o)是羧基的碳原子,c(h3)是甲基的碳原子并且o(h)是羟基的氧原子。
根据一些实施方案,原始序列活性数据可通过遗传算法过滤,以排除对训练高预测力的序列活性模型不提供信息的数据列。图2b示出了通过遗传算法过滤的数据列的实例。在该实现中,遗传算法产生个体的群体,每个个体具有表明能量和几何值是否应被包括在序列活性模型中的一组二进制值的“基因”或系数(例如,0和1)。图2b中的实例示出了ga群体的个体的效应,所述个体具有以下参数:e总=1,e相互作用=1,n1=1,p=1,c(o)=0,c(h3)=1,o(h)=0。当参数取0值时,与该参数相关的特征被有效地从模型排除。该ga个体过滤掉几何数据c(o)和o(h),从而提供用于训练序列活性模型的数据子集。在一些实施方案中,使用包括三个序列iv、两个能量iv和三个几何iv的数据子集来训练序列活性模型。注意,ga的二进制值的系数或基因可独立于序列活性模型来实现,以使得序列活性模型不包含系数值。在一些实施方案中,利用svm来优化序列活性模型,其输出预测的活性的命中和未命中。对每个个体确定的ga的适应度函数基于预测的准确性。以以上描述的相同的方式测试ga的一代群体中的多个个体。每个个体具有一组值为0或1的参数,其中,0值的参数有效过滤掉一组特征,从而产生用于训练序列活性模型的数据子集。基于个体的适应度函数来比较并对所述个体排名。然后,如在本文其他地方所描述的,利用至少一个多样性机制来选择一个或更多个“最适合”的个体作为用于下一代群体的亲本。在一些实施方案中,利用赤池信息准则(aic)或贝叶斯信息准则(bic)来实现适应度的比较,其中,具有最小aic或bic的个体被选为最适合的个体。通常,重复ga,持续两代或三代,直到满足收敛准则。
注意,在一些实施方案中,列过滤是任选的。根据一些实施方案,原始序列活性数据可通过遗传算法过滤,以排除数据行而不是数据列,或除了列过滤之外排除数据行。图2c示出了通过遗传算法过滤掉的数据行(酶变体)的实例。在该实现中,遗传算法提供个体的群体,每个个体具有指示排除阈值的一组连续值的“基因”或系数。如果能量值和几何值大于变体的阈值,则将该变体从序列活性模型排除。图2c中的实例示出了具有以下阈值的ga个体:e总>1.5,e相互作用>1.5,n1>3.3,p>2.8,c(o)>3.6,c(h3)>6且o(h)>6。这些阈值仅仅是为了说明的目的,并且不表示实际实现的最佳阈值。在该实例中,该ga个体过滤掉变体1和变体5,提供了数据的子集以训练序列活性模型。注意,ga的阈值可独立于序列活性模型来实现,以使得序列活性模型不包含阈值。在一些实施方案中,如在列过滤中的,序列活性模型利用svm来优化,其输出预测的活性的命中和未命中。个体的适应度函数基于预测的准确性。以在以上实例中描述的相同的方式测试ga的多个个体。基于个体的适应度函数来比较并将所述个体排名。然后,如在本文其他地方所描述的,利用至少一种多样性机制来选择一个或更多个最适合的个体以产生下一代群体。
在一些实施方案中,源自图2a至图2c的实例中所示的ga的最适合的个体提供了数据的子集并训练支持向量机,以限定具有高预测力的序列活性模型的参数。在一些实施方案中,如以下进一步描述的,该序列活性模型可引导用于新一轮的定向进化的新变体的设计。在获得一个或更多个“最佳序列活性模型”之后,一些实施方案利用这些模型来引导实际蛋白的合成,其可通过定向进化进一步开发。如在本文其他处描述的,一些实施方案提供了用于通过修改模型预测的序列来设计具有期望的活性的蛋白的方法。
a.第一遗传算法-参数的选择
在某些实施方案,诸如在图3a中示出的实施方案中,遗传算法从可用参数的池选择特定参数,以及选择多个变体的活性信息。图3a中示出的实施方案是实现图1a中示出的过程中的过滤原始数据以移出一个或更多个能量类型和/或几何类型的步骤105的一种方式。这些参数的数据被提供在未经过滤的数据集中。参见图3a的模块303。在第一遗传算法的执行期间,可将所有数据合并在一个或更多个计算机可读文件中,以方便访问。
为了实现第一遗传算法,来自可用参数池的随机选择的参数集被用来提供第一代数据子集。参见模块305。充当自变量集合的每个参数集合限定了独特的数据子集。不同的随机选择的多组自变量(即,多个个体数据子集)被用来训练序列活性模型。在一些实施方案中,使用相同数量的自变量来产生每个数据子集。在许多实现中,序列或突变信息被用作各个和每个数据子集中的另外的自变量。数据子集共同组成遗传算法的一代群体中的“个体”。
在遗传算法的第一代中,从每个数据子集提供序列活性模型,每个模型与自变量的不同的随机选择的组合相关联。然后使用这些模型来预测活性。参见模块307。在某些实施方案中,预测针对并不被用来实际训练模型的序列进行,通过交叉验证测试模型的预测力。例如,未经过滤的数据可以是对100个变体可用的,但是这些变体中的仅70个的数据被用来训练序列活性模型。将剩余的30个变体或更确切地这些剩余的30个变体的数据用作测试集以测试序列活性模型的有效性,提供模型的预测力的交叉验证。
使在第一遗传算法的第一代期间获得的数据子集基于其训练准确预测活性的模型的能力排名。参见模块311。利用可被看作是所训练的模型的性能的适应度函数来进行排名。换言之,该过程从以被不同方式过滤移出不同的变量组合的原始数据推导出模型。模型评价用来训练其的数据子集(即,个体)的适应度。
最低排名的数据子集反映了最低排名的自变量集合,并在转移至遗传算法的第二代之前被拒绝。用通过与来自第一代的性能靠前的模型类型交配获得的数据子集替换被拒绝的数据子集。参见模块313。
可通过多种技术来进行数据子集的交配。基本上,来自两个亲本数据子集的每一个的所选自变量的一些被用于交配,因此它们可向前转到子代数据子集。在一个实例中,两个亲本数据子集被表示为1和0的序列,以表示来自可用自变量的池的特定参数是否被用作数据子集中的自变量。数据子集的这些二进制表示在交叉点处被截断,并且产生的片段与来自另一个亲本的数据子集的互补片段连接。
可以多种方式实现适应度函数、或更精确地,评价特定序列活性模型的准确性的方法。在一个方法中,适应度函数使用混淆矩阵来评价模型准确性。在此类技术中,在测试集中使用的变体的每一个根据其测量的活性是否大于或小于限定的阈值而被认为是活性的或非活性的。相似地,序列活性模型的特征为基于其预测活性值是否大于或小于限定的阈值来预测来自测试集的变体是活性的或非活性的。对于测试集的每个成员,比较成员的实际的活性状态和预测的活性状态。当序列活性模型正确地将测试变体表征为活性的或非活性的时,序列活性模型获取信用。当在测试变体被测量为活性的时,序列活性模型预测该测试变体为非活性的时候,或者当在测试变体被测量为非活性的时,序列活性模型预测该测试变体为活性的时候,序列活性模型丢失信用。这四个可选方案组成混淆矩阵。特定模型正确地预测活性或非活性的频率被用来对用于训练模型的数据子集排名。用于表征模型的准确性的另一个选项依赖于在其预测的活性(或其量级)和实际测量的活性之间的误差或差异。对于测试集的所有成员,该距离可被加和或平均。
在第一代遗传算法结束时,序列活性模型的几个自变量组(即,数据子集)被选择。如所提到的,高排名的数据集被选择用于与下一代交配和/或升级至下一代。这些子集包括选择的结构(例如,距离)和/或除了序列自变量之外能量自变量。
针对使用第二代数据子集训练的模型的预测能力评价第二代数据子集。重复该过程,持续多代,直到自变量的选择收敛。参见收敛模块309。在某些实施方案中,收敛准则确定当前代与前一代相比的改进是否小于一个或更多个连续代的阈值水平。在一些实施方案中,测试收敛的其他方式包括但不限于测试最大或最小适应度值如100%适应度、运行固定的代数、在固定的时间限制内运行或以上的组合。在某些实施方案中,在每一代产生并评价约5-100个数据子集。在某些实施方案中,在每一代产生并评价约30-70个数据子集。不意图本发明限于任何特定的数据子集数和/或代数。
b.第二遗传算法
在如在图3b中示例的第二遗传算法中,提供了实现图1a的步骤107的过程,以过滤原始数据,从而移出具有在限定的范围之外的能量值和/或几何值的变体的数据。在图3b中,在第一遗传算法中鉴定的自变量是固定的。未被选择的自变量不再被视为是有意义的,且第二遗传算法始于接收通过第一遗传算法过滤的数据集。参见模块323。可假设,通过第一遗传算法选择的自变量是可能在准确地预测活性方面(至少使用考虑中的序列活性模型的形式(例如,通过支持向量机产生的n维平面)),具有最多的价值的自变量。在可选的实施方案中,未进行第一遗传算法,并使用来自原始数据的所有自变量。
应理解,变体的序列必然地设定另外的自变量-能量和结构约束变量的值。例如,存在于结合袋中的变体的组合将限定充当可用自变量的某些几何结构结合特征和相互作用能量值。然而,仅序列信息可能不足以有效训练序列活性模型以准确地预测活性。
在第二遗传算法中,每个自变量(除了序列之外)被精化(refined),以使得只有达到自变量的阈值的变体被选择用于数据子集。该精化可被应用于多个非序列自变量。换言之,第二遗传算法选择总的可用量级范围之内的子范围用于所选择的非序列自变量中的一个或更多个。作为一个方法的实例,给定的自变量可具有约
在该第二种类型的遗传算法的第一代中,自变量的每一个(除了序列变量之外)被划分到一部分。该划分是随机执行的。参见模块325。例如,自变量的每一个的量级的具体值是随机选择的。只考虑具有小于该划分点的值的变体。这有效地削减了用于序列活性模型的训练集的自变量。
在第一代中,个体数据子集对于每一个非序列自变量具有随机选择的截断点。模块325。第一代中的每个个体数据子集使用其自身的独特的序列活性模型进行训练。参见模块327。得到的模型被用于预测测试集的每个成员的活性。模块327。通过利用如以上描述的混淆矩阵,针对每个个体数据子集训练准确模型的能力对所述每个个体数据子集排名。参见模块331。这是适应度函数。替代性适应度函数是可能的。这些替代性适应度函数包含利用预测值和实际值之间的差异值的函数。适应度还可基于用于模型的自变量的类型和/或所用自变量值的完整范围的部分。
在某些实施方案中,数据子集包含原始数据集中的变体的子集的数据。这些变体的一部分的数据被用于训练序列活性模型。剩余变体的数据被用于测试产生的序列活性模型。换言之,每个数据子集被分为训练集和测试集。该划分可通过随机选择进行。在一些实施方案中,训练集包括子集中约20%和90%之间(或约50%和80%之间)的变体。不意图本发明限于子集和/或训练集中任何特定数目的变体。
在第一代中高得分的数据子集被选择用于第二代和/或被选择作为用于交配以产生第二代的后代的亲本。参见模块333。交配可使用任何合适的技术来进行。在一个实施方案中,应用成本加权(cost-weighting)方案,诸如差异的加权和,该方案对于每个给定的自变量,使用两个交配亲本的每一个的截断值(即,阈值)。在成本加权方案中,交配选择偏向于具有相对较高的适应度的个体(即,数据子集)。最适合的个体交配多于较不适合的个体。其他交配选择方案包括成比例的轮盘赌选择(proportionalroulettewheelselection)、基于排名的轮盘赌选择(rank-basedroulettewheelseletion)和锦标赛选择(tournamentseletion)。
实际交配过程可采取多种形式。一个实例是连续的参数交配。在该方法中,在子代数据子集中给定参数的截断值为两个亲本数据子集中同一参数的截断值之间的值。例如,一个亲本对于第一参数(距离x)可具有0.1埃截断值,而另一个亲本对于距离x可具有0.6埃的截断值。距离x的子代截断值将在0.1埃和0.6埃之间。可定义多种函数来确定子代的距离x的中间截断值。在连续的参数交配方案中,“β”值被随机选择并被应用来确定亲本的两个截断值之间的比例(fractional)距离。在以上实例中,如果β被选为0.7并产生两个子代,则可如下计算子代的截断值:
子代1的距离=0.1-(0.7)*0.1+(0.7)*0.6=0.45
子代2的距离=0.6+(0.7)*0.1-(0.7)*0.6=0.25
子代1=a+β*(b-a)
子代2=b+β*(a-b)
在第二代中,通过在第一轮中交配选择和/或产生的个体(定义的数据子集)通过对其每个应用适应度函数来评价。换言之,将模块327、331和333的过程应用于第二代。与第一代一样,可基于数据子集训练精确预测测试集的变体的活性的模型的能力来对数据子集排名。可使高排名的子集传递到下一代和/或如以上描述的交配。
其他代如同第二代一样继续,直到达到收敛。如在图3b中所示的,每一代经受收敛检查。参见模块329。在某些实施方案中,收敛准则确定当前代与前一代相比的改进是否小于针对一个或更多个连续代的阈值水平。测试收敛的其他方法包含测试最大/最小适应度值诸如100%的适应度、运行固定的代数、在固定的时间限制内运行或以上的组合。
在某些实施方案中,每一代产生并评价约5-100个数据子集。在某些实施方案中,每一代产生并评价约30-70个数据子集。在特定的实例中,在第二遗传算法的每一代中存在约45个个体数据子集。但是,不意图本发明被限制为每一代或任一代的特征为和/或使用任何特定数目的数据子集。
在一些方面,该数据集过滤过程可被描述为以下。首先,系统使用未经过滤的数据集以创建数据子集的群体。这些数据子集的每一个是遗传算法的一代的群体中的“个体”。使用表征配体与生物分子的结合位点的结合的几何参数的参数值阈值(截断)来鉴定每个数据子集。当系统应用参数值阈值时,其有效地将某些变体从未经过滤的数据集移出。换言之,每个数据子集包含未经过滤的数据集中所包含的仅一些变体的数据。
对于每个数据子集(即,个体),系统将组成变体分为属于训练集的那些变体和属于测试集的那些变体。属于训练集的变体被用于训练序列活性模型。可利用诸如支持向量机或偏最小二乘法的技术来完成训练。得到的经训练的序列活性模型被应用于测试集变体。模型对每个测试集变体预测活性,并且系统从而评价序列活性模型的准确性并由此评价其相关联的数据子集。以相同的方式评价遗传算法的世代的群体中的每个数据子集(即,个体)的准确性。
对于遗传算法的给定的一代,基于数据子集和相关联的序列活性模型准确预测相关联的测试集中的变体的活性的能力来对该数据子集和相关联的序列活性模型的每个排名。在该代中,过程选择排名靠前的子集用于升级至下一代。另外,该过程交配一些排名靠前的子集来产生子代子集,其也被提供至下一代。如以上描述的处理下一代数据子集(即,个体)。处理并评价多代,直到达到收敛。
c.第三遗传算法
在所描述的工作流中,通过过滤原始序列、活性和结构数据选择的数据子集训练高准确度的序列活性模型。支持向量机可被用来进行训练。得到的序列活性模型鉴定新的变体生物分子。在一些实施方案中,这些新的变体生物分子被用于至少一轮定向进化。在某些实施方案中,采用最后的遗传算法鉴定图1a的模块111中描述的新的生物分子变体。合适的遗传算法的实例被示于图3c中。如其所示的,该过程始于在结束第二遗传算法之后选择的序列活性模型。模块353。
如以上指出的,在该遗传算法和本文讨论的其他遗传算法之间存在差异。该算法将核酸、氨基酸或其他生物分子序列提供为群体中的个体。相对地,在本文讨论的其他遗传算法中,个体为模型或模型参数的集。在该ga的第一代中,遗传算法提供个体的随机群体,每一个表示不同的蛋白(或其他生物分子)序列。模块355。个体蛋白通过在给定的位置处突变而彼此不同。在一些实现中,至少在第一代中,突变是随即地产生的。可在一轮定向进化期间相对于单个蛋白骨架生成突变,所述单个蛋白骨架诸如野生型蛋白或鉴定的参考骨架。
利用适应度函数,即对在第二遗传算法结束处获得的数据子集训练的序列活性模型(即,在模块353中向前传递的模型)来排名或选择第一代中的个体。参见模块357和359。将每个个体生物分子的鉴定序列信息输入序列活性模型。该信息可以是一系列突变,任选地鉴定突变所处的位置的每一个处的起始残基和最终残基。模型通过对每个个体分配预测的活性来作用于该输入。模块357。选择具有排名靠前的活性值(如由模型预测的)的个体生物分子用于交配和/或用于转移至下一代。模块359和363。交配的个体提供突变的新组合,每个新组合为下一代的成员。在某些实施方案中,交配通过交叉操作完成。该遗传算法中的交叉操作的实例可理解如下。亲本1在位置12和25处有突变,且亲本2在位置15和30处有突变。第一后代可在来自亲本1的位置12和来自亲本2的位置30中有突变,并且第二后代将在来自亲本1的位置25和来自亲本2的位置12中有突变。
在一些情况中,使用任何合适的方法(包含但不限于点突变)使通过交配产生的后代中的一些(例如,其的20%)进一步突变。此类突变可被随机地进行。
如对第二代描述的,得到另外的世代的不同生物分子的群体。重复创建新的世代,直到由模型预测的活性持续限定的代数无显著改进。在该点处,生物分子的群体被视为已经收敛至由一组突变和预测的活性标识的被排名的个体的最终列表。在图3c的模块361示出了收敛状况。
在某些实施方案中,来自最终列表的个体生物分子是体外合成并筛选的。另外,可通过使用对接软件或其他工具来分析个体生物分子以提供几何约束或其他结构数据和/或相互作用能。然后组合得到的序列、活性和结构/能量数据来充当下一轮定向进化工作流的输入。换言之,在遗传算法之后筛选的蛋白提供了可充当用于第二轮分析的新训练集的数据。因此,数据过滤遗传算法被再次进行,但是使用完全新的训练集。在一些实施方案中,来自一轮定向进化的数据集和序列活性模型在下一轮中未被保留。即,下一轮重新开始,使用新的未经过滤的数据集寻找新的自变量组。
在一些实施方案中,使用能量和/或结构(几何)参数以及序列信息训练在第三遗传算法中采用的序列活性模型。然而,在某些实现中,最后的遗传算法只向模型输入序列信息,而不输入能量和/或结构信息。换言之,当使用序列和能量和/或结构自变量开发模型时,模型在评价第三遗传算法中的新序列时不接收能量和/或结构自变量。
在某些实施方案中,每一代中评价约10到10,000个生物分子。在某些实施方案中,每一代中评价约100到10,000个生物分子。在特定的实例中,在第三遗传算法的每一代中存在约500个个体生物分子。不意图本发明限于任何特定数目的被评价的生物分子。
在一些点处,以上描的过程被完成,并且当前代中的一个或更多个变体被选择用于另外的调查、合成、开发、产生等。在一个实例中,选择的生物分子变体被用来引发一轮或更多轮的体外定向进化。例如,一轮体外定向进化可包括(i)准备多个寡核苷酸,该寡核苷酸包含或编码选择的蛋白变体的至少一部分;以及(ii)使用该多个寡核苷酸进行一轮体外定向进化。寡核苷酸可通过基因合成、使编码一些或全部的选择的蛋白变体的核酸片段化等制备。在某些实施方案中,该轮体外定向进化包括片段化和重组该多个寡核苷酸。在某些实施方案中,该轮体外定向进化包括在该多个寡核苷酸上进行饱和诱变。
v.序列活性模型
本文公开的方法和系统提供了高预测力的序列活性模型。在一些实施方案中,序列活性模型是非线性模型。在其他的实施方案中,其为线性模型。线性和非线性序列活性模型的实例被描述于美国专利第7,747,391号、美国专利申请公布第2005/0084907号、美国临时专利申请第61/759,276号和美国临时专利申请第61/799,377号中,其每一个被通过引用以其全部并入本文。在本文描述的多种实施方案中,序列活性模型被实现为n维超平面,所述n维超平面可通过支持向量机产生。在以下说明中,当序列活性模型被示例为通过支持向量机产生的n维超平面时,意图该形式或模型可被其他类型的线性和非线性模型代替,诸如,最小二乘模型、偏最小二乘模型、多线性回归、主成分回归、偏最小二乘回归、支持向量机、神经网络、贝叶斯线性回归或靴襻法和这些的集成形式。
如以上所述,在一些实施方案中,用于本文的实施方案的序列活性模型将蛋白序列信息与蛋白活性联系起来。该模型使用的蛋白序列信息可采取很多种形式。在一些实施方案中,其是蛋白中的氨基酸残基的完整序列。但是,在一些实施方案中,完整的氨基酸序列是不必要的。例如,在一些实施方案中,只提供在特定的研究工作中待改变的那些残基已足够。在一些涉及后续研究阶段的实施方案中,很多残基是固定的,并且只有有限的序列空间的区域仍有待探索。在此类情况的一些中,提供这样的序列活性模型是方便的:作为输入,其只需要鉴定蛋白的需继续探索的区域中的那些残基。在一些另外的实施方式中,这些模型不要求知晓感兴趣的残基位置处的残基的准确身份。在一些此类实施方案中,表征特定残基位置处的氨基酸的一个或更多个物理或化学特性被鉴定。在一些实施方案中,描述结构信息(例如,部分之间的距离)的几何参数被包含在模型中。尽管结构信息可在结构模型中来实现,其还可被实现为序列活性模型的一部分。可选地,结构信息可被用来过滤掉数据,以选择序列活性数据的子集来训练序列活性模型。
此外,在一些模型中,采用了此类特性的组合。事实上,不意图本发明被限于任何特定方法,因为这些模型对于序列信息、活性信息、结构信息和/或其他物理特性(例如,疏水性等)的多种设置具备实用性。
在以上描述的一些实施方案中,氨基酸序列为序列活性模型提供自变量的信息。在其他实施方案中,与氨基酸序列相对,核酸序列提供自变量的信息。在较后的实施方案中,表示在特定位置处特定类型的核苷酸的存在或不存在的iv被用作模型的输入。源自核苷酸序列的蛋白提供活性数据,作为模型的输出。本领域技术人员认识到,由于密码子的简并性,不同的核苷酸序列可被翻译成相同的氨基酸序列,其中两个或更多个不同的密码子(即,三个一组的核苷酸)编码相同的氨基酸。因此,不同的核苷酸序列可潜在地与相同的蛋白和蛋白活性相关联。但是,将核苷酸序列信息当作输入并将蛋白活性当作输出的序列活性模型不需要关心此类简并性。实际上,在一些实施方案中,在输入和输出之间缺少一对一对应可将噪声引入模型,但此类噪声并不否定模型的实用性。在一些实施方案中,此类噪声甚至可提高模型的预测力,因为,例如,模型较不可能过度拟合数据。在一些实施方案中,模型通常将活性当作因变量,并将序列/残基值当作自变量。活性数据可利用本领域已知的任何合适的方法获得,包括但不限于被得当地设计以测量感兴趣的一种活性/多种活性的量级(magnitude)的测定和/或筛选方法。此类技术是本领域技术人员所熟知的并且对于本发明不是必需的。实际上,用于设计合适的测定或筛选方法的原则在本领域中是被广泛理解并已知的。用于获得蛋白序列的技术也被熟知,且对于当前发明不是关键的。如所提到的,可利用下一代测序技术。在一些实施方案中,感兴趣的活性可以是蛋白稳定性(例如,热稳定性)。但是,很多重要的实施方案考虑其他的活性诸如催化活性、对病原体和/或毒素的抗性、治疗活性、毒性等。事实上,不意图本发明被限制于任何特定的测定/筛选方法和/或测序方法,因为本领域已知的任何合适的方法在本发明中具备实用性。
在多种实施方案中,序列活性模型的形式可极大地变化,只要其如所期望的基于序列信息提供用于正确地接近蛋白的相对活性的工具。模型的数学/逻辑形式的实例包括但不限制于,相加、相乘、多阶的线性/非相互作用和非线性/相互作用数学表达式、神经网络、分类和回归树/图、聚类方法、递归分区、支持向量机等。
用于生成模型的多种技术是可获得的并且在本发明中具备实用性。在一些实施方案中,这些技术涉及优化模型或使模型误差最小化。具体实例包括但不限制于偏最小二乘法、集成回归、随机森林和多种其他的回归技术以及神经网络技术、递归分区、支持向量机技术、cart(分类和回归树)等。通常,所述技术应产生能将对活性具有显著影响的残基与那些对活性没有显著影响的残基区分开的模型。在一些实施方案中,这些模型还对个体残基或残基位置基于其对活性的影响排名。不意图本发明局限于用于产生模型的任何特定技术,因为本领域已知的任何合适的方法在本发明中具备实用性。
在涉及相加模型的一些实施方案中,这些模型通过回归技术产生,所述回归技术鉴定训练集中自变量和因变量的共变。多种回归技术是已知的并被广泛地使用。实例包括但不限制于多元线性回归(mlr)、主成分回归(pcr)和偏最小二乘回归(pls)。在一些实施方案中,模型利用涉及多个组分的技术,包括但不限于集成回归和随机森林产生。这些方法和任何其他合适的方法在本发明中具备实用性。不期望本发明局限于任何特定的技术。
mlr是这些技术中最基础的。其被用于简单地对训练集的成员的一组系数方程求解。每个方程涉及训练集成员的活性(即,因变量)与特定位置处特定残基的存在或不存在(即,自变量)。取决于训练集中残基选择的数目,这些方程的数目可以是相当大的。
像mlr一样,pls和pcr从将序列活性与残基值联系起来的方程产生模型。但是,这些技术以不同的方式产生模型。它们首先进行坐标转换来减少自变量的数目。然后它们对转换的变量进行回归。在mlr中,存在可能非常大量的自变量:在训练集内变异的每个残基位置有两个或三个自变量。假定感兴趣的蛋白和肽往往相当大并且训练集可提供很多不同的序列,那么自变量的数目可迅速地变得非常大。通过减少变量的数目以集中在数据集中提供最多变异的那些变量上,pls和pcr通常需要较少的样品并简化了参与产生模型的步骤。
实际的回归针对通过原始自变量(即,残基值)的坐标转换获得的相对少数目的潜在变量进行,在这方面pcr与pls回归相似。pls和pcr之间的不同在于,pcr中的潜在变量通过最大化自变量(即,残基值)之间的共变来构建。在pls回归中,潜在变量以最大化自变量和因变量(即,活性值)之间的共变的方式来构建。偏最小二乘回归被描述于hand,d.j.等人(2001)principlesofdatamining(adaptivecomputationandmachinelearning),boston,ma,mit出版社,以及geladi等人(1986)“partialleast-squaresregression:atutorial,”analyticachimicaacta,198:1-17。这些参考均为了所有的目被通过引用并入本文。
在pcr和pls中,回归分析的直接结果是:活性是加权的潜在变量的函数的表达式。通过进行将潜在变量转变回初始自变量的坐标转换,该表达式可转换成活性作为初始自变量的函数的表达式。
大体上,pcr和pls两者均首先减少训练集中包含的信息的维度,然后对已被转换以产生的新的自变量但保留了初始因变量值的经转换的数据集进行回归分析。转换形式的数据集可产生仅相对少的用于进行回归分析的表达式。在未进行维度减少的方案中,必须考虑可能存在变异的每个单独的残基。这会是很大的一组系数(例如,对于双向相互作用为2n个系数,其中n为在训练集中可能变异的残基位置的数目)。在典型的主成分分析中,只采用3、4、5、6个主成分。但是,不意图本发明限于任何特定数量的主成分。
机器学习技术拟合训练数据的能力往往被称为“模型拟合”,并且在诸如mlr、pcr和pls的回归技术中,模型拟合通常通过测量值和预测值之间的方差和来测量。对于给定的训练集,最优的模型拟合将利用mlr来完成,而pcr和pls往往具有较差的模型拟合(测量值和预测值之间较高的误差平方和)。但是,利用潜在变量回归技术诸如pcr和pls的主要优势在于此类模型的预测能力。获得具有很小的误差平方和的模型拟合绝对不能保证模型将能精确地预测训练集中未观察到的新样本-事实上,往往是相反的情况,特别是当存在很多变量并且只有少数观察值(即样本)时。因此,潜在变量回归技术(例如,pcr、pls)虽然往往具有对训练数据较差的模型拟合,但通常更稳健,并且能更精确地预测训练集之外的新样本。
支持向量机(svm)也可被用于产生在本发明中使用的模型。如以上解释的,svm将已基于活性分类成两个或更多个组的训练集序列作为输入。支持向量机通过根据训练集的不同成员如何接近超平面界面来不同地加权训练集的不同成员,所述超平面界面将训练集的“活性”和“非活性”成员分隔开。该技术需要科学家先决定将哪些训练集成员置于“活性”组以及将哪些训练集成员置于“非活性”组。在一些实施方案中,这通过选择针对活性水平的适当数值来完成,所述数值作为训练集的“活性”和“非活性”成员之间的分界线。支持向量机根据该分类生成向量w,其可为限定训练集中的活性和非活性组的成员的序列的个体自变量提供系数值。这些系数可被用于如本文其他处描述的对个体残基“排名”。该技术被用于鉴定超平面,所述超平面将该平面的相对侧上的最靠近的训练集成员之间的距离最大化。
vi.蛋白对接
在一些实施方案中,虚拟蛋白对接或筛选系统被配置成进行与计算上鉴定可能具有期望活性(诸如,有效地并选择性地在限定的温度催化反应)的生物分子相关的多种操作。虚拟蛋白对接系统可将意图与变体相互作用的至少一个配体的表示作为输入值。系统可将生物分子变体或这些变体的至少结合位点的表示作为其他输入值。这些表示可包括配体和/或变体的原子和/或部分的三维位置。同源模型是生物分子变体的表示的实例。在一些实施方案中,虚拟蛋白筛选系统可采用对接信息和活性约束来评价变体的功能。
在某些实施方案中,虚拟蛋白对接和筛选系统确定与两个不同分子上的部分之间的关系有关的一个或更多个能量值和一个或更多个几何值。在一些实施方案中,能量值可包括底物和酶之间的相互作用能,其中底物处于与酶对接的一个或更多个位姿。在一些实施方案中,能量值可包括总对接能,所述总对接能包括相互作用能和结合相互作用的参与者的内能。在一些实施方案中,几何值可包括两个分子的部分之间的距离、角度或扭力值。在一些实施方案中,几何值包括均被对接到同一个酶的原始底物和期望的底物上的对应部分之间的距离。在其他实施方案中,几何值包括彼此对接的底物和酶之间的距离。
当将底物的催化转化当做活性时,虚拟蛋白筛选系统可被配置成鉴定已知与特定反应相关的位姿。在一些实施方案中,这涉及到考虑反应中间体或过渡态,而不考虑底物自身。除了转化之外,可针对其他类型的活性评价位姿,所述其他类型的活性诸如,对映异构体的立体选择性合成、与被鉴定对药物发现重要的靶生物分子的受体结合等。在一些情况中,活性是不可逆或可逆的共价结合,诸如,靶向共价抑制(tci)。
在某些实施方案中,计算结合能的协议被执行以评价变体的每个活性位姿的能量学。在一些实现中,该协议可考虑范德瓦尔斯力、静电相互作用和溶剂化能。在通过对接器进行的计算中通常不考虑溶剂化。多种溶剂化模型对于计算结合能是可用的。这些溶剂化模型包括但不限于距离依赖性电介质、具有配对求和的广义博恩(generalizedborn,genborn)、具有隐含膜的广义博恩(generalizedbornwithimplicitmembrane,gbim)、具有分子体积集成的广义博恩(gbmv)、具有简单转换的广义博恩(gbsw)以及具有非极性表面区域的poisson-boltzmann方程(pbsa)。用于计算结合能的协议不同于或独立于对接程序。它们通常产生比对接得分更准确的结果,部分地因为在它们的计算中包含溶剂化效应。在多种实现中,只对被视为是活性的位姿计算结合能。
a.生物分子及其结合位点的结构模型
在某些实施方案中,计算机系统为蛋白变体(或其他生物分子)提供了三维模型。三维模型是蛋白变体的全长序列中的一些或全部的计算表示。通常,在最低限度上,计算表示覆盖至少蛋白变体的结合位点。
如在本文中描述的,三维模型可以是使用适当设计的计算机系统准作的同源模型。三维模型采用结构模板,在所述结构模板中蛋白变体在其氨基酸序列上彼此不同。通常,结构模版是与模型序列同源的序列的先前通过x射线晶体学或nmr解决的结构。同源模型的质量依赖于结构模板的序列身份和分辨率。在某些实施方案中,三维模型可被存储在数据库中,以在需要时被用于当前项目或未来项目。
蛋白变体的三维模型可通过除了同源建模之外的技术产生。一个实例是蛋白线程,其也需要结构模版。另一个实例是从头开始或重新蛋白建模,其不需要结构模板且基于基础物理原则。从头开始技术的实例包括分子动态模拟和使用rosetta软件套件的模拟。
在一些实施方案中,蛋白变体在其结合位点方面彼此不用。在一些情况中,结合位点通过结合位点的氨基酸序列中的至少一个突变彼此不同。可在野生型蛋白序列或一些其他参考蛋白序列中进行突变。在一些情况中,两个或更多个蛋白变体共有结合位点的相同氨基酸序列,但在蛋白的另一个区域的氨基酸序列中不同。在一些情况中,两个蛋白变体通过至少约2个氨基酸或至少约3个氨基酸或至少约4个氨基酸彼此不同。然而,不期望本发明限于蛋白变体之间任何特定数目的氨基酸差异。
在某些实施方案中,多个变体包括通过一轮或更多轮定向进化产生的文库的成员。用于定向进化的多样性生成技术包括基因重排、定点诱变等。定向进化技术的实例被描述于美国专利第7,024,312号、美国专利申请公布第2012/0040871号、美国专利第7,981,614号、wo2013/003290、pct申请第pct/us2013/030526号中,其每一个被通过引用以其全部并入本文。
b.将配体对接到蛋白变体
如在本文中解释的,对接可被用于鉴定用于在训练序列活性模型中使用的相互作用能和/或几何参数。通常,对接由使用配体的计算表示和产生的多个变体的结合位点的计算表示的适当编程的计算机系统来执行。
例如,对接器可被配置成进行以下操作的一些或所有:
1.利用高温分子动力学与随机种子产生一组配体构象。对接器可产生此类构象而在不考虑配体的环境。因此,对接器可通过只考虑特定于配体自身的内部张力或其他考虑来鉴定有利构象。要产生的构象数目可被任意地设置。在一个实施方案中,产生至少约10个构象。在另一个实施方案中,产生至少约20个构象、或者至少约50个构象、或者至少约100个构象。然而,不期望本发明限制于特定数目的构象。
2.通过将配体的中心转移到受体活性位点内的特定位置并进行一系列的随机旋转来产生构象的随机方位。要精化的方位数目可被任意地设置。在一个实施方案中,产生至少大约10个方位。在另一个实施方案中,产生至少大约20个方位、或者至少大约50个方位、或者至少大约100个方位。然而,不期望本发明限制于特定数目的方位。在某些实施方案中,对接器计算“软化(softened)”能以产生方位和构象的另外的组合。对接器利用关于结合位点中的某些方位的容许性的物理上不现实的假设来计算软化能。例如,对接器可假设配体原子和结合位点原子能占据基本上相同的空间,基于pauli排斥和空间考虑,这是不可能的。当探索构象空间时,该软化假设可通过例如采用伦纳德—琼斯势的松弛形式来实现。与使用物理上现实的能量考虑可获取的相比,通过使用软化能计算,对接器允许更完全的构象探索。如果特定方位中的构象退火能小于特定阈值,则保留该构象-方位。这些低能构象被保留为“位姿”。在某些实现中,该过程继续,直到找到期望的数目的低能位姿或找到最大数目的差位姿。
3.使来自于步骤2的每个保留的位姿经受模拟退火分子动力学以精化位姿。温度被提高至高的值,然后被冷却至目标温度。对接器可完成这以提供比由软化能计算结果提供的在物理上更现实的方位和/或构象。
4.使用非软化势(non-softenedpotential)来进行配体在刚性受体中的最终最小化。这为保留的位姿提供了更准确的能量值。然而,计算可只提供关于位姿能的部分信息。
5.对于每个最终位姿,计算总能量(受体-配体相互作用能加上配体内部张力)和单独的相互作用能。可使用charmm进行计算。通过charmm能对位姿分选,并且得分靠前(最负的,因此对结合有利)位姿被保留。在一些实施方案中,该步骤(和/或步骤4)移出在能量上不利的位姿。
以下参考提供对接器的运作的实例:wu等人,detailedanalysisofgrid-basedmoleculardocking:acasestudyofcdocker–acharmm-basedmddockingalgorithm、j.computationalchem.,24卷,13号,1549-62页(2003),其被通过引用以其全部并入本文。
对接器诸如此处描述的对接器可提供诸如以下的信息:与期望的底物对接不太可能的变体的身份;可被考虑活性的多组位姿(每个变体一组);以及所述多组中的位姿的相互作用能。
c.确定对接的配体的几何参数
对于成功与配体对接的蛋白变体,几何结合参数可鉴定一个或更多个活性位姿。活性位姿是满足对配体的一个或更多个约束以在限定的条件(而不是任意的结合条件)下结合的位姿。如果配体是底物且蛋白是酶,则活性结合可以是允许底物经历催化的化学转化、特别地立体专一性转化的结合。在一些实现中,几何结合特征限定配体中的一个或更多个原子和蛋白和/或与蛋白相关的辅因子中的一个或更多个原子的相对位置。
在一些情况中,当原始底物通过野生型酶经历催化的化学转化时,从原始底物和/或随后的中间体鉴定几何参数。在某些实施方案中,几何参数包括:(i)底物和/或随后的中间体上的特定部分和催化位点中的特定残基或残基部分之间的距离;(ii)底物和/或随后的中间体上的特定部分和催化位点中的特定辅因子之间的距离;和/或(iii)底物和/或随后的中间体上的特定部分和在催化位点中理想地放置的原始底物和/或随后的中间体上的特定部分之间的距离。距离的替代项包括键与键之间的角度或化合物内原子排列、围绕共同轴的扭转位置等。这些几何参数的实例被描述在与本发明同日提交的美国专利申请第61/883,838号中,并且其被通过引用以其全部并入本文。
底物和/或随后的中间体的计算表示的多个位姿可相对于在考虑中的蛋白变体的计算表示产生。可通过多种技术产生该多个位姿。此类技术的普通实例包括但不限制于关于可旋转键的系统的或随机的扭转搜索、分子动力学模拟和被设计以查找低能构象的遗传算法。在一个实例中,利用高温分子动力学来产生位姿,然后随机旋转、通过基于网格的模拟退火的精化和/或最后的基于网格的或力场最小化,以产生底物和/或随后的中间体在计算表示的催化位点中的构象和/或方位。这些操作中的一些是任选的,例如,通过基于网格的模拟退火精化和基于网格的或力场最小化。
在某些实施方案中,所考虑的位姿数目为至少大约10个、或至少大约20个、或至少大约50个、或至少大约100个、或至少大约200个、或至少大约500个。然而,不期望本发明限制于考虑的特定位姿数。
vii.通过修改模型预测的序列产生具有期望的活性的蛋白
本发明的目标之一是,通过定向进化产生优化的蛋白变体文库。本发明的一些实施方案提供了利用所产生的序列-活性模型指导蛋白变体的定向进化的方法。根据以上描述的方法制作并精化的多种序列-活性模型适于指导蛋白或生物分子的定向进化。作为过程的一部分,该方法可鉴定将用于产生下一轮的定向进化所用的新的蛋白变体的序列,如图1a的模块111所示。此类序列包括对以上鉴定的特定残基的变异,或者是用来随后引入此类变异的前体。可通过进行诱变和/或基于重组的多样性生成机制来修改这些序列,以生成新的蛋白变体文库。在一些实施方案中,可对新的变体测定感兴趣的活性。参见图1a的模块113。在一些应用中,可为新的变体产生结构模型,该结构模型可为变体提供能量值和几何值。参见图1a的模块113。在一些实施方案中,这些数据可以随后被用于在新一轮的定向进化中开发新的序列活性模型。参见图1a的模块115。
在一些实施方案中,寡核苷酸或核酸序列的制备通过利用核酸合成仪合成所述寡核苷酸或核酸序列来完成。本发明的一些实施方案包括利用所制备的寡核苷酸或蛋白序列作为用于定向进化的组成模块(buildingblock)进行一轮定向进化。本发明的多个实施方案可将重组和/或诱变应用于这些组成模块以产生多样性。
在一些实施方案中,该过程鉴定具有有利特性的一个或更多个序列。然后在新一轮的定向进化中,从作为序列活性模型的训练集的经鉴定的序列产生变体。参见图3c的框355和357。
作为一个具体的实例,为了产生变体,一些实施方案对寡核苷酸应用重组技术。在这些实施方案中,所述方法包括通过评价序列-活性模型的项的系数来选择用于一轮定向进化的一个或更多个突变。突变从特定位置处的特定氨基酸或特定残基类型的核苷酸的组合中基于通过所述模型预测的它们对蛋白活性的贡献选择。在一些实施方案中,突变的选择包括鉴定被确定比其他系数大的一个或更多个系数。每个系数与残基对蛋白活性的贡献相关,且该残基被限定为特定位置处的特定类型。突变的选择包括选择与被如此鉴定的一个或更多个系数相关的残基。在一些实施方案中,在根据序列活性模型选择突变之后,该方法包括制备含有或编码至少一个突变的多个寡核苷酸,并进行一轮定向进化。在一些实施方案中,定向进化技术包括组合和/或重组所述寡核苷酸。
其他实施方案对蛋白序列应用重组技术。在一些实施方案中,所述方法包括鉴定新的蛋白序列或新的核酸序列,以及制备并测定所述新的蛋白或由所述新的核酸序列编码的蛋白。在一些实施方案中,所述方法还包括使用所述新的蛋白或由所述新的核酸序列编码的蛋白作为用于进一步的定向进化的起始点。在一些实施方案中,定向进化过程包括片段化并重组由模型预测的具有期望的活性水平蛋白序列。
在一些实施方案中,所述方法基于被所述模型预测为重要的个体突变来鉴定和/或制备新的蛋白或新的核酸序列。这些方法包括:通过评价序列-活性模型的项的系数选择一个或更多个突变,以鉴定对活性有贡献的特定位置处的一个或更多个特定的氨基酸或核苷酸;鉴定包含以上选择的一个或更多个突变的新蛋白序列或新核酸序列,并制备和测定所述新蛋白或由所述新核酸序列编码的蛋白。
在其他的实施方案中,所述方法基于整个序列(而不是个体突变)的预测活性来鉴定和/或制备新蛋白或新核酸序列。在这些实施方案的一些中,所述方法包括将多个蛋白序列或多个氨基酸序列应用于序列-活性模型,以及确定所述序列-活性模型预测的所述多个蛋白序列或核酸序列中的每一个的活性值。所述方法还包括通过评价由所述序列-活性模型针对多个序列预测的活性值,从以上应用的多个蛋白序列或多个氨基酸序列中选择新蛋白序列或新核酸序列。所述方法还包括制备并测定具有所述新蛋白序列的蛋白或由所述新核酸序列编码的蛋白。
在一些实施方案中,不是简单地合成单个最佳预测蛋白,而是基于对蛋白中每个位置处的残基选择的最佳改变的敏感性分析来产生组合蛋白文库。在该实施方案中,对于所预测的蛋白,给定的残基选择越敏感,预测到的适应度改变将越大。在一些实施方案中,这些敏感性从最高至最低并且敏感性得分被用于在随后的轮次中创建组合蛋白文库(即,通过基于敏感性并入那些残基)。在一些实施方案中,其中使用线性/非相互作用模型,通过简单地考虑与该模型中给定的残基项相关的系数的大小来确定敏感性。但是,对于非线性/相互作用模型这是不可能的。相反,在利用非线性/相互作用模型的实施方案中,残基敏感性通过使用模型计算当“最佳”预测的序列中单个残基被改变时的活性的改变来确定。
本发明的一些实施方案包括选择蛋白序列或核酸序列中的一个或更多个位置,以及在如此鉴定的一个或更多个位置处进行饱和诱变。在一些实施方案中,这些位置通过评价序列-活性模型的项的系数来选择,以鉴定对活性有贡献的特定位置处的一个或更多个特定的氨基酸或核苷酸。相应地,在一些实施方案中,一轮定向进化包括在使用所述序列-活性模型选择的位置处对蛋白序列进行饱和诱变。在一些涉及包括一个或更多个相互作用项的模型的实施方案中,每个相互作用项与两个或更多个残基相关。所述方法包括在两个或更多个相互作用残基处同时应用诱变。
在一些实施方案中,残基以其排列顺序被考虑。在一些实施方案中,对于所考虑的每个残基,所述程序确定是否“切换”那个残基。术语“切换”指在优化的文库中的蛋白变体的序列中的特定位置处包含或排除特定的氨基酸残基。例如,丝氨酸可出现在一个蛋白变体的位置166,然而苯丙氨酸可出现在相同文库的另一个蛋白变体的位置166。在训练集中的蛋白变体序列之间不发生变化的氨基酸残基在优化的文库中通常保持固定。但是,情况并非总是这样,因为在优化的文库中可能存在变异。
在一些实施方案中,优化的蛋白变体文库被设计成使得所有被鉴定的回归系数排名“高”的残基被固定,而剩余的回归系数排名较低的残基被切换。该实施方案的基本原理是,‘最佳’预测的蛋白周围的局部空间应被搜索。应注意,其中切换被引入的起点“骨架”可能是模型预测的最佳蛋白和/或已经被验证为来自被筛选的文库的‘最佳’蛋白。事实上,不期望起点骨架被限制于任何特定蛋白。
在可选的实施方案中,至少一个或更多个(但并非全部的)被鉴定的回归系数排名高的残基在优选的文库中被固定,而其他的残基被切换。在一些实施方案中,如果不期望通过一次性并入太多改变而显著地改变其他氨基酸残基的背景,则推荐该方法。再次地,用于切换的起点可以是所述模型预测的最佳的残基组、来自现有文库的最佳验证的蛋白或模拟得很好的“平均”克隆。在后一种情况中,切换被预测为较高重要性的残基可能是期望的,因为在对之前被从抽样中遗漏的活性峰的搜索中应探索更大的空间。该类型的文库通常在早期的多轮文库制备中更为重要,因为其产生了对随后的轮次的更精确的描绘(picture)。不期望起始点骨架被限于任何特定蛋白。
以上实施方案的一些备选方案涉及在确定切换哪些残基时使用残基重要性(排名)的不同程序。在一个这样的备选实施方案中,排名较高的残基位置是切换更强烈地偏爱的。该方法中所需的信息包括来自训练集的最佳蛋白的序列、pls或pcr预测的最佳序列和来自pls或pcr模型的残基排名。在一些实施方案中,“最佳”蛋白为数据集中经湿实验室验证的“最佳”克隆(即,具有最高的测量功能但是由于其相对接近交叉验证的预测值仍模拟得良好的克隆)。所述方法将来自该蛋白的每个残基与来自具有期望活性的最高值的“最佳预测”序列的相应残基比较。如果具有最高载荷或回归系数的残基不存在于‘最佳’克隆中,则所述方法引入该位置作为切换位置用于随后的文库。如果所述残基存在于最佳克隆中,则所述方法不将该位置视作切换位置,并且将按顺序移至下一个位置。对多个残基重复该过程,连续地穿过较低的载荷值,直到生成具有足够容量的文库。
在一些另外的实施方案中,湿实验室验证的当前优化的文库中的‘最佳’(或最佳之一)蛋白(即,具有最高的测量功能或最高之一的测量功能、仍很好地模拟即相对地接近交叉验证中的预测值的蛋白)被用作其中多个改变被并入的骨架。在另一个方法中,湿实验室验证的当前文库中的‘最佳’(或最佳之一)的、可能模拟不好的蛋白被用作其中多个改变被并入的骨架。在一些其他的方法中,序列-活性模型预测为具有最高的期望活性值(或最高值之一)的序列被用作为骨架。在这些方法中,用于“下一代”文库(以及可能地相应的模型)的数据集通过改变最佳蛋白的至少一个中的残基来获得。在一个实施方案中,这些改变构成骨架中的残基的系统变异。在一些情形中,这些改变包括多种诱变、重组和/或亚序列选择技术。这些改变中的每个可体外、体内和/或经由计算机模拟进行。事实上,不期望本发明被限于任何特定的形式,因为任何合适的形式具备实用性。
在一些实施方案中,优化的蛋白变体文库利用本文描述的重组方法或可选地通过基因合成方法随后通过体内或体外表达来产生。在一些实施方案中,在优化的蛋白变体文库被筛选期望的活性之后,对其测序。如以上指出的,来自优化的蛋白变体文库的活性和序列信息可被采用以生成另一个序列-活性模型,可利用本文描述的方法由所述另一个序列-活性模型设计进一步优化的文库。在一个实施方案中,所有来自该新文库的蛋白被用作数据集的一部分。
viii.对多核苷酸和多肽测序
在一些实施方案中,多核苷酸和多肽序列信息被用于产生序列活性模型或蛋白变体的活性位点的计算表示。在一些实施方案中,多核苷酸和多肽序列信息被用于定向进化过程以获得具有期望的特性的蛋白变体。
在多个实施方案中,通过蛋白测序方法从物理生物分子确定蛋白变体的序列,这些方法中的一些在下文中被详细描述。蛋白测序包括确定蛋白的氨基酸序列。一些蛋白测序技术还确定蛋白采用的构象和其与任何非肽分子复核的程度。质谱分析法和埃德曼降解反应可被用于直接确定蛋白的氨基酸的序列。
埃德曼降解反应允许发现蛋白的有序的氨基酸组成。在一些实施方案中,自动埃德曼序列可被用于确定蛋白变体的序列。自动埃德曼序列能够对逐渐增长,例如,多达大约50个氨基酸长的肽进行测序。在一些实施方案中,实现埃德曼降解的蛋白测序方法包括以下中的一个或更多个:
--使用例如2-巯基乙醇的还原剂来打破蛋白中的二硫键。诸如碘乙酸的保护基团可被用于防止键重新形成。
--如果有多于一条链,分离并纯化蛋白复合物的个体链。
--确定每条链的氨基酸组成。
--确定每条链的末端氨基酸。
--将每条链打断为片段,例如,少于50个氨基酸长的片段。
--分离并纯化片段。
--利用埃德曼降解反应确定每个片段的序列。
--应用不同的裂解模式来重复以上步骤,以提供氨基酸序列的另外的读段。
--从氨基酸序列读段构建整个蛋白的序列。
在多种实现中,长于大约50-70个氨基酸的肽被打断为小的片段,以利于通过埃德曼反应测序。较长序列的消化可通过诸如胰蛋白酶或胃蛋白酶的内切肽酶或通过诸如溴化氰的化学试剂来进行。不同的酶给出不同的裂解模式,且在片段之间的重叠可被用来构建整个序列。
在埃德曼降解反应期间,待测序的肽被吸附到底物的固体表面上。在一些实施方案中,一个合适的底物是涂覆阳离子聚合物聚凝胺的玻璃纤维。埃德曼试剂、异硫氰酸苯酯(pitc)与三甲胺的弱碱性缓冲溶液一起被添加至被吸附的肽。该反应溶液与n-末端氨基酸的氨基反应。该末端氨基酸然后可通过添加无水酸被选择性地分离。随后衍生物异构化,以给出取代的乙内酰苯硫脲,其可被洗涤掉并通过色谱分析法鉴定。然后可重复该循环。
在一些实施方案中,质谱分析法可被用于通过确定氨基酸序列的片段的质荷比来确定氨基酸序列。包含对应于多电荷片段的峰的质谱可被确定,其中在对应于不同的同位素的峰之间的距离与片段上的电荷成反比。质谱例如通过与先前测序的蛋白的数据库比较来分析,以确定片段的序列。然后用不同的消化酶重复该过程,且序列中的重叠被用来构建完整的氨基酸序列。
肽通常比整个蛋白更容易制备并且对于质谱分析法更容易分析。在一些实施方案中,电喷射离子化被用来将肽传送到分光仪。蛋白被内切蛋白酶消化,且得到的溶液被传送到高压液相色谱柱。在该柱的末端,将所述溶液喷射到质谱仪中,所述溶液带有正电势。溶液液滴上带的电荷导致它们碎成单个离子。然后使肽片段化,并测量片段的质荷比。
直接从编码蛋白的dna或mrna序列确定氨基酸序列也是可能的。核酸测序方法例如多种下一代测序方法可被用于确定dna或rna序列。在一些实现中,蛋白序列被新分离出,而不知道编码蛋白的核苷酸。在这种实现中,技术人员可利用直接的蛋白测序方法先确定短的多肽序列。可从该短序列确定蛋白的rna的互补标记物。这然后可被用于分离编码蛋白的mrna,其然后可在聚合酶链式反应中被复制以产生大量的dna,然后可使用dna测序方法来对其测序。然后可从dna序列推断出蛋白的氨基酸序列。在推断中,考虑在mrna已被翻译之后被移出的氨基酸是必要的。
在多种实施方案中,多核苷酸的序列信息被用于产生序列活性模型或蛋白活性位点的计算表示。可通过核酸测序方法从物理生物分子确定核酸序列信息,这些方法中的一些在以下被进一步描述。
在一个或更多个实施方案中,序列数据可利用被认为是第一代测序方法的大量(bulk)测序方法来获得,包括例如桑格测序或maxam-gilbert测序。涉及使用带标记的双脱氧链终止剂的桑格测序是本领域熟知的;参见例如,sanger等人,proceedingsofthenationalacademyofsciencesoftheunitedstatesofamerica74,5463-5467(1997)。涉及在核酸样品的片段上进行多部分化学降解反应,接着检测并分析所述片段来推断序列的maxam-gilbert测序也是本领域熟知的;参见例如maxam等人,proceedingsofthenationalacademyofsciencesoftheunitedstatesofamerica74,560-564(1977)。另一种大量测量方法是通过杂交测序,其中样品的序列基于其与例如微阵列或基因芯片上的多个序列的杂交特性来推导;参见,例如,drmanac等人,naturebiotechnology16,54-58(1998)。
在一个或更多个实施方案中,序列数据利用下一代测序方法来获得。下一代测序也被称为高通量测序。这些技术使测序过程平行化,一次产生数以千计或数百万计的序列。合适的下一代测序方法的实例包括但不限于,单分子实时测序(例如,pacificbiosciences,menlopark,california)、离子半导体测序(例如,iontorrent,southsanfrancisco,california)、焦磷酸测序(例如,454,branford,connecticut)、连接测序(例如,lifetechnologies,carlsbad,california所有的solid测序)、合成测序和可逆性终止物测序(例如,illumina,sandiego,california)、核酸成像技术诸如透射电子显微术等。
一般地,下一代测序方法通常利用体外克隆步骤以扩增个体dna分子。乳液pcr(empcr)分离伴随在油相内的水滴中的引物所包被的珠中的个体dna分子。pcr产生与珠上的引物结合的dna分子的拷贝,然后固定用于后续测序。empcr被marguilis等人(被454lifesciences,branford,ct商业化)、shendure和porreca等人(也称为“聚合酶克隆测序”)和solid测序(appliedbiosystemsinc.,fostercity,ca)用于这些方法中。参见m.margulies等人(2005)“genomesequencinginmicrofabricatedhigh-densitypicolitrereactors”nature437:376–380;j.shendure等人(2005)“accuratemultiplexpolonysequencingofanevolvedbacterialgenome”science309(5741):1728–1732。体外克隆扩增还可通过“桥式pcr”进行,在所述“桥式pcr”中,片段在引物附接至固体表面后被扩增。braslavsky等人开发了省去该扩增步骤、直接将dna分子固定至表面的单分子方法(由helicosbiosciencescorp.,cambridge,ma商业化)。i.braslavsky等人(2003)“sequenceinformationcanbeobtainedfromsinglednamolecules”proceedingsofthenationalacademyofsciencesoftheunitedstatesofamerica100:3960-3964。
与表面自然结合的dna分子可被平行地测序。在“合成测序”中,基于模板链的序列使用dna聚合酶形成互补链,如染料-终止电泳测序,“可逆”终止剂法(由illumina,inc.,sandiego,ca和helicosbiosciencescorp.,cambridge,ma商业化)利用可逆形式的染料-终止剂,通过重复移除封闭基团以允许另一个核苷酸的聚合而一次添加一个核苷酸,并实时检测每个位置处的荧光。“焦磷酸测序”也利用dna聚合,一次添加一个核苷酸并通过由所附接的焦磷酸的释放发射的光来检测和定量被添加到给定位置的核苷酸的数目(由454lifesciences,branford,ct商业化)。参见m.ronaghi,等人(1996).“real-timednasequencingusingdetectionofpyrophosphaterelease”analyticalbiochemistry242:84-89。
下文更详细地描述了下一代测序方法的具体实例。本发明的一个或更多个实施方案可利用以下测序方法的一种或更多种而不背离本发明的原理。
单分子实时测序(也被称为smrt)是pacificbiosciences开发的并行化单分子dna合成测序技术。单分子实时测序利用零模式波导(zmw)。单个dna聚合酶被附着在zmw的底部,单分子的dna作为模板。zmw是产生足够小以观察到被dna聚合酶并入的dna的仅单个核苷酸(也被称为碱基)的被照射的观察体积的结构。四种dna碱基的每个被附接至四种不同的荧光染料之一。当核苷酸被dna聚合酶并入时,荧光标签被裂解掉并从zmw的观察区域扩散出来,在所述观察区域处其荧光不再可观察。检测器检测核苷酸并入的荧光信号,并根据相应的染料荧光进行碱基响应(basecall)。
另一种可应用的单分子测序技术是helicostruesinglemoleculesequencing(tsms)技术(例如,如harrist.d.等人,science320:106-109[2008]中描述的)。在tsms技术中,dna样品被裂解成约100至200个核苷酸的链,并将多聚a序列添加至每条dna链的3’末端。通过添加荧光标记的腺苷核苷酸来标记每条链。然后使dna链与流动池杂交,所述流动池包含数以百万计的被固定至所述流动池表面的寡聚t捕获位点。在某些实施方案中,模板可以是约1亿模板/cm2的密度。然后将流动池装载至设备例如heliscopetm测序仪,并且激光照亮流动池的表面,显示出每个模板的位置。ccd摄像机可绘制模板在流动池表面上的位置。然后模板荧光标签被裂解并被冲走。测序反应通过引入dna聚合酶和荧光标记的核苷酸开始。寡聚t核酸作为引物。聚合酶以模板指导的方式将带标记的核苷酸并入至引物。聚合酶和未并入的核苷酸被移出。具有荧光标记的核苷酸的指导性并入的模板通过对流动池表面成像来识别。成像后,裂解步骤移出荧光标签,并且用其他的荧光标记的核苷酸重复该程序直到达到期望的读段长度。随着每个核苷酸添加步骤收集序列信息。在测序文库的制备中,借助于单分子测序技术的整个基因组测序不包括或通常排除基于pcr的扩增,并且所述方法允许直接测量样品,而不是测量该样品的拷贝。
离子半导体测序是基于检测dna的聚合期间释放的氢离子的dna测序方法。这是“合成测序”方法,在该方法期间,互补链基于模板链的序列形成。将包含待测序的模板dna链的微孔用单一种类的脱氧核糖核苷三磷酸(dntp)充满。如果所引入的dntp与引导性模板核苷酸互补,则其被并入正在增长的互补链。这导致氢离子的释放,氢离子的释放引发isfet离子传感器,其指示反应已经发生。如果均聚物的重复片段存在于模板序列中,则多个dntp分子将在单个循环中被并入。这导致相应数目的释放的氢和成比例地更高的电子信号。该技术不同于其他测序技术,因为没有使用被修饰的核苷酸或光学。离子半导体测序也可被称作离子激流测序(iontorrentsequencing)、ph介导的测序、硅测序或半导体测序。
在焦磷酸测序中,聚合反应释放的焦磷酸根离子通过atp硫酸化酶与腺苷5'磷酰硫酸反应以产生atp;然后atp驱动荧光素通过荧光素酶转化为氧化荧光素加光。由于荧光是瞬时的,所以在该方法中不需要单独的消除荧光的步骤。一次添加一种类型的脱氧核糖核苷三磷酸(dntp),并根据哪种dntp在反应位点产生明显信号来鉴定序列信息。市购可得的rochegsflx仪器利用这种方法获得序列。例如在ronaghi等人,analyticalbiochemistry242,84-89(1996)和margulies等人,nature437,376-380(2005)(勘误表在nature441,120(2006))中详细地讨论了该技术及其应用。市购可得的焦磷酸测序技术为454测序(roche)(例如,如在margulies,m.等人nature437:376-380[2005]中描述的)。
在连接测序中,连接酶被用来将具有突出端的部分双链的寡核苷酸连接至具有突出端的正被测序的核酸;为了使连接发生,这些突出端必须是互补的。部分双链的寡核苷酸的突出端中的碱基可根据偶联至该部分双链的寡核苷酸和/或偶联至与该部分双链的寡核苷酸的另一部分杂交的第二寡核苷酸的荧光团而被鉴定。在获得荧光数据之后,连接的复合体在连接位点上游被裂解,诸如通过在离其识别位点(其被包含于该部分双链的寡核苷酸中)固定距离的位点处切割的ii型限制酶例如bbvl。该裂解反应使刚好在之前的突出端上游的新突出端暴露,并重复所述程序。例如,在brenner等人,naturebiotechnology18,630-634(2000)中详细讨论了该技术及其应用。在一些实施方案中,连接测序通过获得环状核酸分子的滚环扩增产物并将该滚环扩增产物用作连接测序的模板而适合于本发明的方法。
连接测序技术的市购可得的实例是solidtm技术(appliedbiosystems)。在solidtm连接测序中,基因组dna被剪切成片段,并且衔接子被附接至片段的5’和3’末端以产生片段文库。可选地,内部衔接子可通过以下步骤而被引入:将衔接子连接至片段的5’和3’末端、使片段环化、消化环化的片段以产生内部衔接子,并将衔接子连接至所得到的片段的5’和3’末端以产生配对文库。接下来,在包含珠、引物、模板和pcr组分的微反应器中制备克隆珠群体。伴随pcr,模板被变性并且珠被富集以分离带有延伸的模板的珠。对所选择的珠上的模板进行3’修饰,所述3'修饰允许与载玻片结合。序列可通过特定的荧光团被鉴定的部分随机的寡核苷酸与中心的已确定的碱基(或碱基对)顺序性杂交和连接来确定。记录颜色之后,连接的寡核苷酸被裂解和移出,然后重复该程序。
在可逆终止物测序中,荧光染料标记的核苷酸类似物即可逆的链种终止物由于封闭基团的存在而被掺入单碱基延伸反应。碱基的身份根据荧光团来确定;换言之,每个碱基与不同的荧光团配对。获得荧光/序列数据之后,荧光团和封闭基团通过化学方法移出,并重复该循环以获得序列信息的下一个碱基。illuminaga仪器通过该方法运行。例如,在ruparel等人,proceedingsofthenationalacademyofsciencesoftheunitedstatesofamerica102,5932-5937(2005)和harris等人,science320,106-109(2008)中详细讨论了该技术及其应用。
可逆终止剂测序方法的商业上可获得的实例是illumina的合成测序和基于可逆终止剂的测序(例如,在bentley等人,nature6:53-59[2009]中描述的)。illumina的测序技术依赖于片段化的基因组dna与平面、任选地其上结合寡核苷酸锚的透明表面的附接。模板dna的末端被修复以生成5'磷酸化的平末端,并且klenow片段的聚合酶活性被用来将单个a碱基添加至平端的磷酸化dna片段的3'末端。该添加制备了连接至寡核苷酸衔接子的dna片段,所述寡核苷酸衔接子在其3'末端具有单个t碱基的突出端以增加连接效率。该衔接子寡核苷酸与所述流动池锚互补。在有限稀释条件下,衔接子修饰的单链模板dna被添加至流动池并通过与锚的杂交而被固定。附接的dna片段被延伸并被桥式扩增以生成具有数以亿计的簇的超高密度测序流动池,每个簇包含相同模板的~1,000个拷贝。这些模板利用稳健的四色dna合成测序技术来测序,所述四色dna合成测序技术采用具有可移除的荧光染料的可逆终止剂。高密度荧光检测利用激光激发和全内反射光学来完成。将约20-40bp例如36bp的短序列读段与重复片段-掩盖的(repeat-masked)参考基因组比对,并利用专门开发的数据分析流水线软件鉴定短序列读段在参考基因组上的独特位置。也可使用非重复片段-掩盖的参考基因组。不论使用重复片段-掩盖的参考基因组或非重复片段-掩盖的参考基因组,只计算独特地匹配至参考基因组的读段。第一次读取完成以后,可通过计算机模拟重新生成模板使得能够进行来自片段的相反末端的第二次读取。因此,可使用dna片段的单末端或成对末端测序。进行对存在于样品中的dna片段的部分测序,并对被绘制至已知参考基因组的包含预定长度例如36bp的读段的序列标签计数。
在纳米孔测序中,例如利用电泳驱动力使单链核酸分子穿过孔,并通过分析随着单链核酸分子通过孔获得的数据来推导序列。数据可以是离子电流数据,其中每个碱基,例如通过部分地阻断通过孔的电流而将电流改变至不同的可辨别的程度。
在另一个示例性但非限制性实施方案中,本文描述的方法包括利用透射电子显微术(tem)获得序列信息。所述方法包括,利用选择性标记有重原子标记物的高分子量(150kb或更大)dna的单原子分辨率透射电子显微成像,并将这些分子以具有一致的碱基间间距的超密(链和链之间3nm)平行阵列排列在超薄的膜上。电镜被用来对薄膜上的分子成像,以确定重原子标记物的位置并提取dna中的碱基序列信息。所述方法被进一步描述于pct专利公布wo2009/046445中。
在另一个示例性但非限制性实施方案中,本文描述的方法包括利用第三代测序获得序列信息。在第三代测序中,具有带有很多小的(~50nm)孔的铝涂层的玻片被用作零模式波导(参见,例如,levene等人,science299,682-686(2003))。铝表面通过聚磷酸酯化学例如聚乙烯膦酸酯化学避免附连dna聚合酶(参见例如korlach等人,proceedingsofthenationalacademyofsciencesoftheunitedstatesofamerica105,1176-1181(2008))。这导致dna聚合酶分子优先附连至铝涂层的孔中暴露的硅。该设置允许渐失波现象(evanescentwavephenomena)被用于减少荧光背景,允许使用较高浓度的荧光标记的dntp。荧光团被附接至dntp的末端磷酸根,以使得荧光在并入dntp后释放,但荧光团不保持与新并入的核苷酸附接,意味着复合物立即准备好另一轮并入。通过该方法,dntp至铝涂层的孔中存在的个体引物-模板复合物的并入可被检测到。参见,例如eid等人,science323,133-138(2009)。
ix.测定基因和蛋白变体
在一些实施方案中,结合本发明的方法产生的多核苷酸任选地被克隆到细胞中,以表达用于活性筛选的蛋白变体(或者被用于体外转录反应以制备被筛选的产物)。此外,编码蛋白变体的核酸可被富集、测序、表达、体外扩增或以任何其他常见的重组方法处理。
描述在本文中有用的分子生物技术(包含克隆、诱变、文库构建、筛选测定、细胞结构等)的常规文本包括:berger和kimmel,guidetomolecularcloningtechniques,methodsinenzymology152卷academicpress,inc.,sandiego,ca(berger);sambrook等人,molecularcloning-alaboratorymanual(第二版),1-3卷,coldspringharborlaboratory,coldspringharbor,newyork,1989(sambrook)以及currentprotocolsinmolecularbiology,f.m.ausubel等人编辑,currentprotocols,ajointventurebetweengreenepublishingassociates,inc.andjohnwiley&sons,inc.,newyork(2000年补编(ausubel))。用核酸转化细胞包括植物细胞和动物细胞的方法通常是可得的,表达由此类核酸编码的蛋白的方法同样是可得的。除了berger、ausubel和sambrook之外,对培养动物细胞有用的一般参考包括freshney(cultureofanimalcells,amanualofbasictechnique,thirdeditionwiley-liss,newyork(1994))以及本文引用的参考humason(animaltissuetechniques第四版w.h.freemanandcompany(1979))和ricciardelli等人,invitrocelldev.biol.25:10161024(1989)。植物细胞克隆、培养和再生的参考包括payne等人(1992)plantcellandtissuecultureinliquidsystemsjohnwiley&sons,inc.newyork,ny(payne);以及gamborg和phillips(编辑)(1995)plantcell,tissueandorganculture;fundamentalmethodsspringerlabmanual,springer-verlag(berlinheidelbergnewyork)(gamborg)。多种细胞培养培养基被描述于atlas和parks(编辑)thehandbookofmicrobiologicalmedia(1993)crcpress,bocaraton,fl(atlas)。用于植物细胞培养的另外的信息发现于可商业获得的文献中,诸如来自sigma-aldrich,inc(stlouis,mo)的lifescienceresearchcellculturecatalogue(1998)(“sigma-lsrccc”)和,例如,同样来自sigma-aldrich,inc(stlouis,mo)的theplantculturecatalogueandsupplement(1997)(“sigma-pccs”)。
足以指导技术人员通过体外扩增方法用来例如扩增寡核苷酸重组核酸的技术的实例包括聚合酶链式反应(pcr)、连接酶链式反应(lcr)、qβ-复制酶扩增和其他rna聚合酶介导的技术(例如,nasba)。这些技术被发现于berger、sambrook和ausubel,同上,以及mullis等人,(1987)美国专利第4,683,202号;pcrprotocolsaguidetomethodsandapplications(innis等人编辑)academicpressinc.sandiego,ca(1990)(innis);arnheim&levinson(1990年10月1日)c&en36-47;thejournalofnihresearch(1991)3,81-94;kwoh等人(1989)proc.natl.acad.sci.usa86,1173;guatelli等人(1990)proc.natl.acad.sci.usa87,1874;lomell等人(1989)j.clin.chem35,1826;landegren等人,(1988)science241,1077-1080;vanbrunt(1990)biotechnology8,291-294;wu和wallace,(1989)gene4,560;barringer等人(1990)gene89,117,以及sooknanan和malek(1995)biotechnology13:563-564。克隆体外扩增的核酸的改进方法被描述在wallace等人,美国专利第5,426,039中。通过pcr扩增大的核酸的改进方法被总结在cheng等人(1994)nature369:684-685以及本文的参考文献中,其中产生了多达40kb的pcr扩增子。技术人员将领会到使用反转录酶和聚合酶基本上可将任何rna转化成适于限制性消化、pcr扩增和测序的双链dna。参见,ausubel、sambrook和berger,均同上。
在一个优选的方法中,对重装序列(reassembledsequences)检查基于家族的重组寡核苷酸的并入。这可通过对核酸进行克隆并测序和/或通过限制性消化来完成,例如,如在sambrook、berger和ausubel,同上中主要教导的。此外,可对序列pcr扩增并直接测序。因此,除了例如sambrook、berger、ausubel和innis(同上)之外,另外的pcr测序方法也是特别有用的。例如,通过在pcr期间选择性地将含硼核酸酶抗性核苷酸并入到扩增子中并用核酸酶消化扩增子以产生一定大小的模板片段来直接对pcr产生的扩增子测序已被进行(porter等人(1997)nucleicacidsresearch25(8):1611-1617)。在这些方法中,对模型进行四次pcr反应,在每次中pcr反应中,pcr反应混合物中的核苷三磷酸中的一种被2’脱氧核苷5’-[p-硼烷]-三磷酸部分地取代。在模板的一组嵌套pcr片段中,含硼核苷酸被沿着pcr扩增子在不同的位置处随机地并入到pcr产物。使用被并入的含硼核苷酸阻塞(blocked)的核酸外切酶来裂解pcr扩增子。然后利用聚丙烯酰胺凝胶电泳将被裂解的扩增子按大小分离,提供扩增子的序列。该方法的优势是它与进行prc扩增子的标准桑格类测序相比使用更少的生物化学操作。
合成基因服从传统的克隆和表达方法;因此,它们编码的基因和蛋白的特性在它们在宿主细胞中表达之后可被容易地检测。合成基因也可被用来通过体外(无细胞)转录和翻译生成多肽产物。多核苷酸和多肽可因此被检测其与多种预先确定的配体、小分子和离子或聚合和杂聚物质(包含其他蛋白和多肽表位)以及微生物细胞壁、病毒颗粒、表面和膜结合的能力。
例如,许多物理方法可被用来检测编码与化学反应的催化相关的表型(phenotypes)的多核苷酸,所述检测通过多核苷酸直接或由编码的多肽进行。单纯为了说明的目的,并根据特定的预先确定的感兴趣的化学反应的特殊性,这些方法可包括本领域已知的说明底物和产物之间的物理差距、或者说明与化学反应相关的反应介质的变化(例如,电磁辐射、吸收、消耗和荧光的变化,不管是uv、可见的或红外的(热))的众多技术。这些方法还可选自以下的任何组合:质谱分析法;核磁共振;说明同位素分布或带标记的产物形成的同位素标记物、划分法和谱方法;检测反应产物的离子或元素组成的伴随变化(包含ph、无机离子和有机离子等的变化)的谱方法和化学方法。适用于本文的方法的其他物理测定方法可基于反应产物特异性生物传感器的使用,所述反应产物特异性生物传感器包括:包含具有报告物特性的抗体的那些;或者基于与报告基因的表达和活性偶联的体内亲和力识别的那些。用于反应产物检测的酶偶联测定和体内细胞生活-死亡-生长选择当适合时也可被使用。不管物理测定的特定特征,他们均被用于选择由感兴趣的生物分子提供或编码的期望的活性或者期望的活性的组合。
用于选择的特定测定将取决于应用。用于蛋白、受体、配体、酶、底物等的许多测定是已知的。形式包括与固定的组分结合、细胞或有机体的生活力、报告物组分的产生等。
高通量测定尤其适合用于筛选本发明中采用的文库。在高通量测定中,在单日内筛选出几千个不同的变体是可能的。例如,微量滴定板的每个孔可被用于运行独立的测定,或者,如果要观察浓度或孵育时间效应,每5-10个孔可测试单个变体(例如,以不同的浓度)。因此,单标准微量滴定板可测定大约100(例如,96)个反应。如果使用1536孔板,那么单个板可轻易地测定从大约100至大约1500个不同的反应。每天测定若干个不同的板是可能的;使用本发明的集成系统,测定筛选多达约6,000-20,000个不同的测定(即,涉及到不同的核酸、编码的蛋白、浓度等)是可能的。最近,例如calipertechnologies(mountainview,ca)已研发出了试剂操作的微流体方法,其可提供非常高通量的微流体测定方法。
高通量筛选系统是市售可得的(参见,例如,zymarkcorp.,hopkinton,ma;airtechnicalindustries,mentor,oh;beckmaninstruments,inc.fullerton,ca;precisionsystems,inc.,natick,ma等)。这些系统通常自动化整个程序,包含所有样本和试剂的移液、液体分配、定时孵育以及在适合于测定的检测器中最终读取微板。这些可配置的系统提供高通量和快速启动以及高度的灵活性和用户化。
此类系统的制造商提供了针对多种高通量筛选测定的详细方案。因此,例如,zymarkcorp.提供了描述用于检测基因转录、配体结合等的调整的筛选系统的技术通报。
多种市售可得的外围设备和软件可用于例如使用pc(intelx86或者pentium芯片兼容的macos、windowstm系列或者基于unix的(例如,suntm工作站)计算机)来数字化、存储和分析数字化视频或数字化光图像或其他测定图像。
用于分析的系统通常包含经特别编程以使用用于指导本文的一个或更多个方法的一个或更多个步骤的软件来执行专用算法的数字计算机,并且任选地还包含:例如,下一代测序平台控制软件、高通量液体控制软件、图像分析软件、数字解释软件、用于将溶液从源传输到可操作地连接到数字计算机的目的地的机械液体控制电枢、用于将数据输入到数字计算机以控制机械液体控制电枢的操作或高通量液体传输的输入设备(例如,计算机键盘)、以及任选地用于将来自带标记的测定组分的标记信号数字化的图像扫描仪。图像扫描仪可与图像分析软件交互以提供探针标记强度的测量值。通常,探针标记强度测量值通过数据解释软件解释,以示出带标记的探针是否与固体支持物上的dna杂交。
在一些实施方案中,包含体外寡核苷酸介导的重组产物或计算机模拟的重组核酸的物理实施物的细胞、病毒空斑、孢子等可在固体介质上被分离,以产生个体集落(或空斑)。使用自动集落挑选仪(例如,q-bot,genetix,u.k)鉴定、挑选集落或空斑,并将多达10,000个不同的突变体接种到包含两个3mm玻璃球/孔的96孔微量滴定盘中。q-bot不挑选整个集落,而是通过集落的中心插入针,并带出细胞(或菌丝)和孢子(或空斑应用中的病毒)的小样本。针在集落中的时间、用于接种培养基的带出物的数目和针在该培养基中的时间,每一个均影响接种量,并且每一个参数可被控制并优化。
诸如q-bot的自动集落挑选仪的一致的过程降低了人类操作失误并增加了建立培养物的速率(大约10,000/4小时)。任选地在控制温度和湿度的培养箱中摇动这些培养物。微量滴定板中的任选的玻璃球与发酵罐的叶片相似发挥促进细胞的均匀通气以及细胞(例如,菌丝)碎片的分散的作用。可通过有限稀释来分离来自培养物的感兴趣的克隆。也如上文描述的,还可通过检测杂交、蛋白活性、与抗体结合的蛋白等对组成文库的空斑或细胞直接筛选蛋白的产生。为了增加鉴定足够大小的池的机会,可使用使处理的突变体的数目增加10-倍的预筛选。初筛选的目的是快速鉴定具有与亲株相等或比亲株更好的产物滴度的突变体,并只将这些突变体转向液体细胞培养基用于后续分析。
筛选多样的文库的一种方法是使用大规模并行固相程序,以筛选表达多核苷酸变体,例如编码酶变体的多核苷酸的细胞。利用吸收、荧光或fret的大规模并行固相筛选装置是可得的。参见,例如,美国专利第5,914,245号到bylina等人(1999);还参见,http://www|.|kairos-scientific.com/;youvan等人(1999)“fluorescenceimagingmicro-spectrophotometer(fims)”biotechnologyetalia,<www|.|et-al.com>1:1-16;yang等人(1998)“highresolutionimagingmicroscope(hirim)”biotechnologyetalia,<www|.|et-al.com>4:1-20;以及在www|.|kairos-scientific.com发布的youvan等人(1999)“calibrationoffluorescenceresonanceenergytransferinmicroscopyusinggeneticallyengineeredgfpderivativesonnickelchelatingbeads”。在通过这些技术筛选之后,利用本领域已知的技术,感兴趣的分子通常被分离,并任选地被测序。然后如本文列出的使用序列信息来设计新的蛋白变体文库。
相似地,还已开发出许多已知的机械系统,用于在测定系统中使用的溶液相化学。这些系统包括:自动工作站,像由akedachemicalindustries,ltd(osaka,janpan)研发的自动合成装置,和使用机械臂的很多机械系统(zymateii,zymarkcorporation,hopkinton,mass.;orca,beckmancoulter,inc.(fullerton,ca)),其模拟由科学家执行的手动合成操作。以上设备的任一个适合于与本发明一起使用,例如,用于高通量筛选由如本文描述地进化的核酸编码的分子。对于相关领域的技术人员,对这些设备的修改(如果有任何修改)以使得他们能如本文讨论的操作的本质和实施方式将是明显的。
x.数字装置和系统
明显的是,本文描述的实施方案采用在指令的控制下行动的程序和/或储存在一个或更多个计算机系统中或经其转换的数据。本文公开的实施方案还涉及用于进行这些操作的装置。在一些实施方案中,所述装置针对所需的目的而被专门设计和/或构建,或其可以是通过计算机程序和/或储存于计算机中的数据结构选择性地激活或重新配置的通用型计算机。本公开内容提供的程序并不固有地与任何特定的计算机或其他特定装置有关。具体地,多种通用型机器在根据本文的教导书写的程序中具备实用性。但是,在一些实施方案中,构建专门的装置以进行所需方法的操作。下文描述了用于各种各样的这些机器的特定结构的一个实施方案。
另外,本公开内容的某些实施方案涉及包括用于进行多种计算机实施的操作的程序指令和/或数据(包括数据结构)的计算机可读介质或计算机程序产品。计算机可读介质的实例包括但不限制于:磁性介质,诸如硬盘;光学介质,诸如cd-rom设备和全息设备;磁-光介质;和半导体存储设备,诸如闪存存储器。诸如只读存储器设备(rom)和随机访问存储器设备(ram)的硬件设备可被配置成存储程序指令。诸如专用集成电路(asic)和可编程逻辑设备(pld)的硬件设备可被配置成存储程序指令并执行。不期望本公开内容被限制于包含用于执行计算机实施的操作的指令和/或数据的任何特定的计算机可读介质或任何其他计算机程序产品。
程序指令的实例包括但不限于诸如由编译器产生的那些的低阶码和可由计算机利用解释器执行的包含较高阶代码的文件。另外,程序指令包括但不限于直接或间接地控制根据本公开内容的计算机的操作的机器代码、源代码和任何其他代码。代码可规定输入、输出、计算、条件式、分支、迭代循环等。
在一个示例性实例中,本文公开的代码体现方法在含有逻辑指令和/或数据的固定的介质或可传输程序部件中体现,所述逻辑指令和/或数据当被加载到合适地配置的计算装置时导致所述装置对一个或更多个字符串进行模拟的遗传操作(go)。图4显示了示例性数字设备800,其是一种逻辑装置,能够从介质817、网络端口819、用户输入键盘809、用户输入811或其他输入工具读取指令。之后装置800能利用那些指令在数据空间中指导统计操作,例如,以构建一个或更多个数据集(例如,以确定数据空间的多个代表性成员)。可体现所公开的实施方案的一种类型的逻辑装置是如包括cpu807、光学用户键盘输入设备809和gui定点设备811,以及周围部件诸如磁盘驱动器815和监视器805(其显示go修饰的字符串并提供用户对此类字符串的子集的简化的选择)的计算机系统800中的计算机系统。固定介质817被任选地用来为整个系统提供程序并可包括例如,盘式光学或磁性介质(disk-typeopticalormagneticmedia)或其他电子存储元件。通信端口819可被用来为系统提供程序并可代表任何类型的通信连接。
某些实施方案还可体现于专用集成电路(asic)或可编程的逻辑设备(pld)的电路中。在该情况中,所述实施方案以可被用来生成asic或pld的计算机可读描述符实施。本公开内容的一些实施方案在各种各样的其他数字化装置的电路或逻辑处理器,例如pda、笔记本电脑系统、显示器、图像编辑设备等内实施。
在一些实施方案中,本公开内容涉及包括计算机可执行指令存储于其上的一个或更多个计算机可读存储介质的计算机程序产品,所述计算机可执行指令当被计算机系统的一个或更多个处理器执行时,导致计算机系统实施用于虚拟筛选蛋白变体和/或具有期望的活性的蛋白的计算机模拟定向进化的方法。该方法可以是本文描述的任何方法,诸如由附图和伪代码涵盖的那些方法。在一些实施方案中,例如,该方法接收多个酶的序列数据,生成生物分子的三维同源模型,将酶的同源模型与底物的一个或更多个计算表示对接,以及参考酶和底物导出关于几何参数的结构数据。在一些实施方案中,该方法可通过参考建模的结构数据过滤数据来进一步开发序列活性模型。变体文库可被用于重复迭代的定向进化,其可导致具有期望的有益特性的酶。
在一些实施方案中,酶的同源模型与底物的一个或更多个计算表示的对接以本文描述的方式通过计算系统上的对接程序来进行,所述对接程序使用配体的计算表示和多个变体的结合位点的计算表示。在多种实施方案中,对接程序评价底物的位姿和酶之间的结合能。对于成功地与配体对接的蛋白变体,系统确定关于参与的配体和蛋白的几何值。在多种实施方案中,计算机系统通过训练支持向量机来构建序列活性模型。在多种实施方案中,计算机系统利用几何算法来过滤掉不提供信息的数据,从而提供用于训练支持向量机的数据的子集。
xi.网站和云计算的实施方案
互联网包括通过通信链路互相连接的计算机、信息用品和计算机网络。互相连接的计算机使用多种服务来交换信息,所述多种服务诸如电子邮件、ftp、万维网(“www”)和其他服务,包括安全服务。www服务可被理解为允许服务器计算机系统(例如,web服务器或web站点)向远程客户端信息设备或计算机系统发送信息的网页。远程客户端计算机系统然后可显示网页。通常,www的每一个资源(例如,计算机或网页)可被统一资源定位符(“url”)唯一地识别。为了查看特定网页或与其交互,客户端计算机系统对所请求的该网页指定url。该请求被转送到支持该网页的服务器。当服务器接收请求时,它将该网页发送给客户端信息系统。当客户端计算机系统接收该网页时,它可使用浏览器显示该网页或者可与该网页或以其他方式提供的界面交互。浏览器是影响网页的请求并显示网页或与网页交互的逻辑模块。
当前,通常使用超文本标记语言(“html”)来定义可显示的网页。html提供定义如何显示网页的标准的标签组。html文档包含控制文本、图像、控件和其他特征的显示的多种标签。html文档可包含在该服务器计算机系统或其他服务器计算机系统上可获得的其他网页的url。url还可指示其他类型的界面,包含信息设备使用以与远程信息设备或服务器进行通信而不必然地向用户显示信息的如cgi脚本或可执行界面的事物。
互联网尤其有利于向一个或更多个远程客户提供信息服务。服务可包括在互联网上向买家电子传递的项(items)(例如,音乐或股票报价)。服务还可包括处理可通过传统的分配渠道(例如,普通的载体)传送的项(例如,杂货、书或化学或生物化合物等)的订单。服务还可包括处理买家稍后访问的诸如航线或电影院预定的项的订单。服务器计算机系统可提供列出可得的项或服务的电子版界面。用户或潜在买家可使用浏览器访问界面并选择感兴趣的多个项。当用户已经完成选择期望的项时,服务器计算机系统然后可向用户提示完成服务所需要的信息。该交易特定的订单信息可包括买家的姓名或其他身份、用于支付的身份(诸如企业购买订单号或账号)或者完成服务所需的另外的信息,诸如,航班信息。
在可在互联网上和在其他网络上提供的服务中特定感兴趣的是生物数据和生物数据库。此类服务包括由nationalinstitutesofhealth(nih)的nationalcenterforbiotechnologyinformation(ncbi)提供的多种服务。ncbi承担创建存储并分析关于分子生物学、生物化学和遗传学的知识的自动系统;促进此类数据库和软件为研究和医疗界所使用;国家性地和国际性地协调为汇总生物技术信息做出努力;并研究用于分析生物学上重要分子的结构和功能的、基于计算机的信息处理的先进方法。
ncbi负责
在互联网上可得的一些生物数据是通常用特殊浏览器“插件(plug-in)”或其他可执行代码查看的数据。此类系统的一个实例是chime,允许分子结构包括生物分子结构的交互式虚拟3维显示的浏览器插件。关于chime的另外的信息在www|.|mdlchime.com/chime/可得。
多个公司和研究所提供用于订购生物化合物的在线系统。在www|.|genosys.com/oligo_custinfo.cfm或www|.|genomictechnologies.com/qbrowser2_fp.html可找到此类系统的实例。通常,这些系统接收期望的生物化合物(诸如,寡核苷酸、dna链、rna链、氨基酸序列等)的一些描述符,并且然后制备所请求的化合物并以液体溶液或其他合适的形式运送至客户。
由于本文提供的方法可如以下进一步描述的被实现在网站上,可以通过互联网以与上文描述的生物信息和化合物相似的方式提供涉及通过本公开内容的一些实施方案产生的多肽或多核苷酸的计算结果或物理结果。
为了进一步说明,本发明的方法可在本地的或分布式计算环境中被实现。在分布式环境中,该方法可在包括多个处理器的单个计算机上或在多个计算机上被实现。计算机可例如通过公共总线连接,但更优选地,计算机为网络上的节点。网络可以是通用网络或本地专用网络或广域网络,并在某些优选的实施方案中,计算机可以是内联网或互联网的组件。
在一个互联网实施方案中,客户端系统通常执行web浏览器并被耦合到执行web服务器的服务器计算机上。web浏览器通常为诸如ibm的webexplorer、microsoft的internetexplorer、netscape、opera或mosaic的程序。web服务器通常是但并必然是诸如ibm的httpdaemon或其他www守护进程(例如,基于linux的形式的程序)的程序。客户端计算机在线路上或经由无线系统与服务器计算机双向耦合。反过来,服务器计算机与提供对实现本发明的方法的软件的访问权的网站(托管网站的服务器)双向耦合。
如所提到的,连接到内联网或互联网的客户端的用户可使得客户端请求为网站的一部分的资源,所述网站托管提供本发明的方法的实现的应用。然后服务器程序处理该要求以返回指定的资源(假设它们当前是可得的)。标准命名约定(即,统一资源定位符(“url”))包括若干类型的位置名,当前包括诸如超文本传输协议(“http”)、文件传输协议(“ftp”)、信息鼠和广域信息服务(“wais”)的子分类。当资源被下载时,其可包含另外的资源的url。因此,客户端的用户可轻易地获悉他或她未特别地请求的新资源的存在。
实现本发明的方法的软件可本地运行于真实的客户端-服务器架构中的托管网站的服务器上。因此,客户端计算机向主机服务器发布请要,主机服务器在本地运行所请要的过程并然后下载返回到客户端的结果。可选地,本发明的方法可以“多层”形式被实现,其中所述方法的组成部分可由客户端本地执行。客户端请求后,这可由从服务器下载的软件来实现(例如,java应用),或者它可由在客户端上“永久性”安装的软件来实现。
在一个实施方案中,实现本发明的方法的应用可被分为框架。在该范例中,甚至不将应用视为特征或功能的集合而是视为离散框架或视图的集合是有用的。例如,典型的应用通常包含一组菜单项,其每一个调用特定的框架-即表明应用的特定功能的形式。以该视角,应用不被视为代码的整体性主体,而是被视为小应用程序的集合或功能集。以此方式,用户可从浏览器之内选择网页链接,其反过来会调用应用的特定框架(即,子应用)。因此,例如,一个或更多个框架可提供用于将生物分子输入和/或编程到一个或更多个数据空间的功能,而另一个框架提供用于精化数据空间的模型的工具。
在某些实施方案中,本发明的方法被实现为提供例如以下功能的一个或更多个框架:将两个或更多个生物分子编程为字符串以提供两个或更多个不同初始字符串的集合的功能,其中,所述生物分子的每一个包括一组选择的亚单位;从字符串选择至少两个子串的功能;连接子串以形成与一个或更多个初始字符串具有大约相同的长度的一个或更多个产物字符串的功能;将产物字符串添加(放置)到字符串的集合的功能;产生并操作酶和底物的计算表示/模型的功能;将底物(例如,配体)的计算表示与酶(例如,蛋白)的计算表示对接的功能;将分子动力学应用到分子模型的功能;计算影响涉及分子的化学反应的所述分子之间的多种约束(例如,在底物部分和酶活性位点之间的距离或角度)的功能;以及实现本文列出的任何特征的功能。
这些功能中的一个或更多个还可在服务器上或在客户端计算机上被单独地实现。这些功能,例如用于产生并操作生物分子的计算模型的功能,可提供其中用户可插入或操作生物分子的表示的一个或更多个窗口。另外,这些功能还任选地提供通过局域网和/或内联网可访问的私人和/或公共数据库的访问权,数据库中包含的一个或更多个序列可藉以被输入到本发明的方法中。因此,例如,在一个实施方案中,用户可任选地具有请求搜索
实现计算和/或数据访问过程的内联网和/或内联网实施方案的方法对于本领域技术人员是熟知的,并被很详细地记录(参见,例如,cluer等人(1992)“ageneralframeworkfortheoptimizationofobject-orientedqueries,”procsigmodinternationalconferenceonmanagementofdata,sandiego,california,1992年6月2-5日,sigmodrecord,21卷,2期,1992年6月;stonebraker,m.,编辑;acmpress,383-392页;iso-ansi,workingdraft,“informationtechnology-databaselanguagesql,”jimmelton,编辑,internationalorganizationforstandardizationandamericannationalstandardsinstitute,1992年7月;microsoftcorporation,“odbc2.0programmer'sreferenceandsdkguidethemicrosoftopendatabasestandardformicrosoftwindows.tmandwindowsnttm,microsoftopendatabaseconnectivity.tm.softwaredevelopmentkit,”1992,1993,1994microsoftpress,3-30页和41-56页;isoworkingdraft,“databaselanguagesql-part2:foundation(sql/foundation),”cd9075-2:199.chi.sql,1997年9月11日,等)。关于基于网络的应用的另外的相关细节可发现于selifonov和stemmer的标题为“methodsofpopulatingdatastructuresforuseinevolutionarysimulations,”的wo00/42559中。
在一些实施方案中,用于探索、筛选和/或开发多核苷酸或多肽序列的方法可被实现为具有多个处理单元和分布在计算机网络上的存储器的计算机系统上的多用户系统,其中网络可包括lan上的内联网和/或互联网。在一些实施方案中,分布式计算架构涉及到“云”,其为在计算机网络上可得的计算机系统的集合,用于计算和数据存储。涉及云的计算环境被称为云计算环境。在一些实施方案中,一个或更多个用户可访问分布在内联网和/或互联网上的云的计算机。在一些实施方案中,用户可通过网络客户端来远程访问实现用于筛选和/或开发以上描述的蛋白变体的服务器计算机。
在一些涉及到云计算环境的实施方案中,在服务器计算机上提供虚拟机(vm),且虚拟机的结果可被发回到用户。虚拟机(vm)是计算机的基于软件的仿真。虚拟机可基于假设的计算机的规格,或者模仿现实世界计算机的计算架构和功能。vm的结构和功能在本领域是熟知的。通常,vm被安装在包含系统硬件的主机平台上,且vm自身包含虚拟系统硬件和访客软件。
用于vm的主机系统硬件包括一个或更多个中央处理器(cpu)、存储器、一个或更多个硬盘和多个其他设备。vm的虚拟系统硬件包括一个或更多个虚拟cpu、虚拟存储器、一个或更多个虚拟硬盘和一个或更多个虚拟设备。vm的访客软件包含访客系统软件和访客应用程序。在一些实现中,访客系统软件包含具有用于虚拟设备的驱动器的访客操作系统。在一些实现中,vm的访客应用程序包含以上描述的虚拟蛋白筛选系统的至少一个实例。
在一些实施方案中,提供的vm的数量可与待解决的问题的计算负荷成比例。在一些实施方案中,用户可从云请求虚拟机,vm包含虚拟筛选系统。在一些实施方案中,云计算环境可基于用户请求来提供vm。在一些实施方案中,vm可存在于先前存储的vm图像中,vm图像可被存储在图像库中。云计算环境可搜索图像并将图像传送到服务器或用户系统。然后云计算环境可将图像引导到服务器上或用户系统上。
虽然前面已经为了清楚和理解的目的以一定的细节进行了描述,但对于本领域技术人员通过阅读该公开内容将清楚的是,可进行多种形式和细节上的多种改变而不偏离本公开内容的真实范围。例如,以上描述的所有技术和装置可以多种组合使用。本申请中引用的所有出版物、专利、专利申请或其他文件为了所有的目的被通过引用以其全部并入,如同每个单独的出版物、专利、专利申请和其他文件被单独地指明为了所有目的被通过引用并入一样。
- 还没有人留言评论。精彩留言会获得点赞!