一种药物-DBPs结合位点的预测方法与流程
本发明涉及一种药物-dbps结合位点的预测方法,属于药物-蛋白质结合位点预测技术领域。
背景技术:
药物-dna结合蛋白(dnabindingproteins,dbps)相互作用的研究为治疗遗传病和癌症开辟了新途径。药物和dbps结合位点的结合机制对dbps的药物开发和相关疾病研究具有重要意义。因此,研究人员希望开发出预测药物-dbps结合位点相互作用的新方法。
目前机器学习的方法被广泛应用在药物-蛋白质相关预测,如基于自动编码器(ae)和支持向量机(svm)。虽然这些方法表现优异,但它们需要蛋白质和药物的相关信息。例如,深度学习中的自动编码器被用来预测dtis,尽管它只需要蛋白质的序列,但是药物的结构信息却不容易被描述。并且当药物和蛋白质的相互作用模式未知时,该模型不能产生令人满意的结果。这是有缺陷的,因为包含dti的数据库可能只是部分注释或者是未注释的。例如在drugbank中仅提供对蛋白质的注释,但不提供药物-蛋白质相互作用。此外,基于化学结构或蛋白质序列之间相似性的dti预测具有局限性,因为相似的药物共享相似蛋白质的假设不一定是正确的。
在传统的药物-蛋白质网络中,许多方法都是将蛋白质作为整体考虑,很少有人将蛋白质的药物结合位点作为研究的对象,忽略了药物和蛋白质结合位点的结合机制。
技术实现要素:
本发明的目的是提供药物-dbps结合位点的预测方法,该方法以三个氨基酸组成的结合位点片段为研究对象,在缺乏特征信息的情况下,该方法能够被用来预测新的药物-dbps结合位点片段相互作用。
为了实现上述目的,本发明所采用的技术方案是:
一种药物-dbps结合位点的预测方法,包括如下步骤:
1)建立由药物和可与药物结合的dna结合蛋白构成的数据集;提取dna结合蛋白序列上可与药物结合的结合位点,每三个结合位点作为一个结合位点片段;
2)根据结合位点片段的氨基酸理化性质通过层次聚类法分为不同的簇,根据药物与dna结合蛋白结合位点片段的结合关系构建药物与所述簇相互作用的二分网络;
3)根据二分网络计算药物与簇之间的相互作用关系程度,挑选出相互作用关系强的药物-簇组合作为预测组,在该预测组中,簇所包含未与药物结合的结合位点片段作为预测的药物-dbps结合位点。
本发明中提出一种基于二分网络的药物-dbps结合位点的预测方法,以三个氨基酸组成的结合位点片段为研究对象,根据结合位点片段理化性质进行聚类,建立药物-簇网络关系,然后使用cn方法计算药物-簇相互作用分数,挑选出得分≥平均标准误差的药物-簇;簇中的结合位点片段即与该药物具有显著的相互作用,簇之前未记载与该药物有相互作用的结合位点片段为预测的药物-dbps结合位点。
本发明中通过网络分析显示药物倾向与带正电荷的结合位点片段结合,并且结合过程更可能发生在dbp内部。网络分析还发现了药物-dbps结合位点片段的一些结合特点,例如药物倾向结合疏水性的片段。在缺乏特征信息的情况下,基于结构的链路预测算法被用来预测新的药物-dbps结合位点片段相互作用。基于此,还可以进一步分析预测结果中药物和结合位点片段的结合机制。
在结合位点片段的划分时,存在多种划分结合位点片段的方式。优选的,步骤1)中dna结合蛋白序列上临近的三个结合位点作为一个结合位点片段。更为优选的,自dna结合蛋白序列的n端开始依次临近的三个结合位点作为一个结合位点片段。
优选的,步骤2)中所述结合位点片段的氨基酸理化性质采用如下方法量化:
ɑtri=x(α00)+(x(α01)+x(α10))/k;
其中ɑtri表示结合位点片段的氨基酸理化性质量化值;x(*)为结合位点片段的结合位点的氨基酸理化性质量化值,*为ɑ00、ɑ01或ɑ10,ɑ00是中心氨基酸,ɑ01和ɑ10分别是边侧氨基酸;k为边侧氨基酸理化性质量化值修正系数,2≤k≤6;
所述x(*)的计算方法为:
其中e1~en代表结合位点的不同氨基酸理化性质的特征值,λ1~λn代表结合位点的不同氨基酸理化性质的权重;其中n为氨基酸理化性质经主成分分析法所得到的主成分的第n位,n≥5。
pca(principalcomponentanalysis),即主成分分析方法,是一种使用最广泛的数据降维算法;目前氨基酸被237种特征表示成向量的形式,这些特征来源于从公共数据库swissprot和dbget。为了降低维数并简化后续分析,对237种特征进行主成分分析,并保留前n个主成分。氨基酸可以表示为n维载体,计算出结合位点片段的n维向量。利用上述的计算公式能够计算出结合位点片段中各个氨基酸的n维向量。上述公式中,优选的k=4。
优选的,所述n=5,所述氨基酸理化性质的前5个主成分是疏水性、氨基酸大小、氨基酸在α-螺旋中的偏好、简并三联体密码子的数量和β-链中氨基酸残基的出现频率。具体的,在氨基酸的5维向量的计算中,e1、e2、e3、e4、e5的数值分别为:1961.504、788.2、539.776、276.624、244.1。
优选的,步骤2)中所述簇的数目为数据集中药物种类数量的80%-120%。更为优选的,步骤2)中所述簇的数目与数据集中药物种类的数目相同。在本发明中希望每种药物聚集对应一类理化基团,因此将所述簇的数目与数据集中药物种类的数目相同。
具体的,步骤2)中在构建药物与所述簇相互作用的二分网络时,首先找到某dna结合蛋白的各个结合位点片段所在的簇,然后将与该dna结合蛋白结合的药物和这些簇建立相互作用关系,并根据这些相互作用关系构建二分网络。
将药物集合表示为d={d1,d2,...,dn},簇设为c={c1,c2,...,cn},dci描述为二分dc图g(d,c,e),其中e(eij:di∈d,cj∈c);当药物di与簇cj结合时,在di和cj之间存在连接;dc二分网络可以由相邻矩阵{aij}表示,其中如果di和cj被链接,则aij=1,否则aij=0。例如,药物mrc中的结合位点片段为:tlg、ppy、hmg,而tlg、ppy、hmg分别位于簇2、簇45、簇74,这样药物mrc就和簇2、簇45、簇74建立连接。
优选的,步骤3)中通过commonneighbor方法的计算药物与簇之间的相互作用关系得分,计算公式为:
在实际的计算中,例如簇96连接药物5jz,5jz连接的簇集合是{95,91,7,5},则
在显著的相互作用分析中,只有那些值大于平均标准误差的元素才被认为是显著作用,而大于前20%的相互作用被视为重要的相互作用。因此具体的,步骤3)中挑选出得分大于平均标准误差的药物-簇作为预测组。优选的,步骤3)中挑选出得分为前20%的药物-簇作为预测组。
附图说明
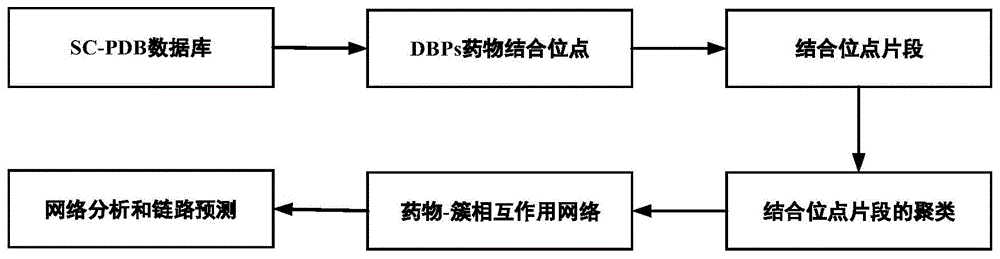
图1为本发明中药物-dbps结合位点的预测方法的整个工作流程图;
图2为本发明中氨基酸三聚体结合位点片段生成过程图;
图3为本发明中结合位点片段层次聚类法聚类示意图;
图4为本发明中所构建的药物-簇相互作用网络示意图;
图5为本发明中药物i和蛋白质j经过两个节点连通示意图;
图6为本发明数据集中不同蛋白质中结合位点片段的重叠程度调查图;
图7为本发明簇的疏水性和电荷强度分析图;
图8为本发明药物-簇相互作用网络中药物的度分布图;
图9为本发明药物-簇相互作用网络中簇的度分布图;
图10为本发明三种链路预测方法中相似性指数的roc曲线和基线展示图;
图11为本发明cn方法获得的预测得分矩阵图;
图12为本发明预测得到的药物-簇相互作用网络图;
图13为本发明中预测结果中药物-结合位点片段的结合机制展示图。
具体实施方式
下面结合具体实施例对本发明做进一步的详细说明。
在机器学习的方法存在局限的同时,药物-蛋白质相互作用网络的研究已经取得重大进展。利用网络推理(nbi)方法已经成功应用于药物新靶标的发现。社交网络的相似性算法被证明可以应用在药物-蛋白质的相互预测。异构网络中基于相似性的药物-蛋白质预测成功的定位药物新作用。基于二元网络投影推荐技术被应用在药物-蛋白质网络内的资源传输。基于网络的方法重新定位出特定疾病的特定目标。这些网络方法为预测药物和蛋白质作相互作用提供了新的思路。因此本发明中利用网络预测的方法研究药物-dbps结合位点的相互作用关系。
药物-dbps结合位点的预测方法的实施例1
本实施例中的药物-dbps结合位点的预测方法过程如图1所示,包括如下步骤:
1、从sc-pdb数据库(http://bioinfo-pharma.u-strasbg.fr/scpdb/)获得药物-dbps的复合物。截至2019年6月,在sc-pdb网站上发布了16034个条目,4782个蛋白质和6326个配体。在下载了所有蛋白质-小分子后,通过筛选获得110个药物-dbps复合物作为数据集。其中涉及到的dna结合蛋白质110个,药物97个。
1)dna结合蛋白质(110个)
1al71al81cd21gg51gg51jty1jup1jus1kbo1kbo1kbq1kbq1mq01ozq1p0b1p0e1qu21qu31qvt1s6q1s9e1s9g1sv51w131w5v1w5w1w5x1w5y2b5j2ban2be22brg2brh2brm2brn2bro2c3k2c3l2cci2cgu2cgv2cgw2cgx2gdo2h422hk92hk92hkj2j0d2vf02vg52vg72vuk2wkm2wkz2wl02x0v2x0w2x6o2x9d2xp22xye2xyf2ynf2ynh2zd12ze22zoz3bgr3bt93bti3btj3bvb3cku3cyw3d203d6y3d703ey43gbk3ha83k1o3ml83ml93nb53o8g3o8h3pm13qps3qqa3s2o3sfi3tdl3zv74agc4agd4agl4ago4agq4ase4c8e4cee4cef4ceo4duh4gqs4i224i234qt34u0i。
2)药物(97个)
x0w,1c9,017,e09,2tc,11d,hst,tch,rhq,p74,dfy,nnc,3sf,aza,g40,p84,tpb,dhp,3b3,3a3,cxg,mrc,atr,itc,3c3,whu,ery,ml9,fad,t27,deq,rdc,av9,2pq,vgh,et,via,f89,be3,c5p,brd,o8h,x0v,pfy,atp,sm1,dfw,nni,g0t,p83,5ah,idz,o8g,0xv,r21,chd,12c,adb,df1,b49,ber,poz,be5,340,r22,352,0li,357,dfz,ml8,ev6,3ac,pq0,p96,rap,imd,ire,b0t,d0t,be4,3d3,abo,prl,936,fol,be6,5jz,prf,df2,nhg,axi,65b,mgr,nap,rli,abz,eur。
2、结合位点片段的生成
药物-dbps结合是药物与dbps序列上的结合位点片段结合,因此使用氨基酸三聚体被用来表示结合位点片段。首先获取dbps的序列,并标注药物结合位点。尽管在dbps序列中药物结合位点不连续,但在空间结构中距离较近,因此认为这些结合位点近似连续,通过连接形成连续的结合位点序列。然后,3个氨基酸的距离作为窗口的长度,在该序列上滑动生成结合位点片段。例如,序列ngmgng生成两个结合位点片段ngm和gng。最后,dbps的结合位点生成3219个结合位点片段(如图2所示)。
3、结合位点片段的理化性质表示
目前,氨基酸被237种特征表示成向量的形式,这些特征来源于从公共数据库swissprot和dbget。为了降低维数并简化后续分析,对237种特征进行主成分分析,并保留前五个主成分(如表1所示)。氨基酸可以表示为5维载体。
具体是利用pca将237维降成5维。pca(principalcomponentanalysis),即主成分分析方法,是一种使用最广泛的数据降维算法。这五种主成分并不是对应单一的化学性质。与五种主要组分相关的性质是疏水性,氨基酸大小,氨基酸在α-螺旋中的偏好,简并三联体密码子的数量和β-链中氨基酸残基的出现频率。
表1前五个主成分的向量和特征值
结合位点片段由单个氨基酸组合表示。结合位点片段通过区分中间氨基酸和两侧氨基酸以强调中间氨基酸的核心位置(不区分两侧氨基酸的顺序)。结合位点片段的理化性质的计算方法为:
ɑtri(α01,α00,α10)=x(α00)+(x(α01)+x(α10))/k;
其中ɑtri表示结合位点片段的5维向量,x(*)代表氨基酸的5维向量,*为ɑ00、ɑ01或ɑ10,ɑ00是中心氨基酸;ɑ00是中心氨基酸(主要),ɑ01和ɑ10分别是左右氨基酸(从属);k为边侧氨基酸理化性质量化值修正系数,k=4。
其中x(*)的计算方法为:
其中λ代表不同氨基酸理化性质的权重,e代表不同氨基酸理化性质的特征值。
例如,丙氨酸的e1=0.008,e2=0.134,e3=-0.475,e4=-0.039,e5=-0.181,λ1=1961.504,λ2=788.2,λ3=539.776,λ4=276.624,λ5=244.10。
因此丙氨酸可以被五维向(0.354,3.762,-11.036,-0.649,-2.828)量表示。
4、结合位点片段的聚类
为了在网络中研究药物和dbps结合位点结合的理化性质。使用层次聚类的方法,将理化性质类似的结合位点片段聚成不同的簇,用簇表示结合位点片段的类别,在五维空间中进行聚类。由于药物具有97种,而通常情况下,希望每种药物聚集对应一类理化基团,因此将结合片段定义为97簇。
每个氨基酸被表示成五维向量,氨基酸三聚体用这些单个氨基酸也可以表示成五维向量。氨基酸三聚体由五维向量表示,放在五维空间中。根据层次聚类的算法把距离相近的氨基酸三聚体聚类为一类。层次聚类算法是一种聚类算法,对空间中的点进行考察并按照某种距离(在这里采用欧式距离)测度将他们聚成多个“簇”的过程(如图3所示)。聚类的目标是使得同一簇内的点之间的距离较短,而不同簇中点之间的距离较大。其计算过程根据已有的算法程序进行计算,其程序可在网上找到也可以提供代码。
层次聚类方法具体为:首先,将每个结合位点片段作为单独的类处理,并计算每2个片段的距离。然后,距离最小的两个片段合并到同一个类中。最后,循环迭代到预先设置的类别数。通过聚类的方式,3210个结合位点片段被表示成97簇。每一簇内的结合位点片段含有相似的物理化学性质。这样药物-结合位点片段的相互作用就被表示成了药物-簇的相互作用(1993个药物-簇相互作用),并根据这些相互作用关系构建药物-簇相互作用网络。
5、药物-簇相互作用网络的建立
首先将所有的结合位点片段聚类成97簇,每簇含有同类型的片段。然后查找每个dna结合蛋白质的结合位点片段属于哪一类簇。与之对应的药物就与相关的簇建立作用关系。例如,药物mrc中的结合位点片段为:tlg、ppy、hmg,而tlg、ppy、hmg分别位于簇2、簇45、簇74,这样药物mrc就和簇2、簇45、簇74建立连接(如图4所示)。
即是,首先找到蛋白质结合位点片段所在的簇,然后将药物与这些簇建立相互作用关系。还如蛋白质1cd2和药物fol结合的位点被划分成片段tsi、pfr、lkr……。通过查找这些片段在簇3、7、8中;将药物fol和相应的簇3、7、8建立作用关系。
网络描述:
将药物集合表示为d={d1,d2,...,dn},簇设为c={c1,c2,...,cn},dci描述为二分dc图g(d,c,e),其中e(eij:di∈d,cj∈c);当药物di与簇cj结合时,在di和cj之间存在连接;dc二分网络可以由相邻矩阵{aij}表示,其中如果di和cj被链接,则aij=1,否则aij=0。
6、基于结构的链路预测方法
由于只有药物-簇的相互作用并不知道其他的特征信息,在网络中基于结构相似性的链路预测方法来预测药物-簇的作用。这种方法基于网络的拓扑结构进行链路预测并不需要其他的特征信息。
基于在相关网络中的成功经验,选择了下面三种链路预测方法:
commonneighbor(cn)方法:
jaccard(ja)方法:
preferentialattachment(pa)方法:
其中
其中所述的cn算法是改进的commonneighbours算法。在单节点网络中该算法计算不连接的两个节点共同连接的节点数量,作为该两节点相似性的依据。药物和靶标相互作用网络有两类节点构成,对commonneighbours进行了改进(公式如
在实际的计算中,例如簇95连接药物chd,5jz,p83。chd连接的簇集合是{95,29,7,2};5jz连接的簇集合是{95,91,7,5};p83连接的簇集合是{95,75,12,1}。则
7、链路预测方法的评估
使用10倍交叉验证,已知它可以在子数据集中给出最低的偏差和方差。数据集被随机地划分为数量相等的10个非重叠子集。每次选取一个子集并且随机取样相同数量的非相互作用作为测试集(测试集包括199个相互作用和199个非相互作用)。剩余9个子集构建网络。该过程重复10次,并且每次迭代计算的假正例率和真正例率被平均以产生最终得分。在预测过程中,药物-簇相互作用的得分被计算。然后将得分用作阈值。当分数大于或等于该阈值时,预测的结果是存在相互作用,否则预测为不存在相互作用。
假正例率(fpr)定义为:
真正例率(tpr)定义为:
其中fp被预测存在相互作用,但事实上不存在相互作用;tn被预测不存在相互作用,事实上也不存在相互作用;fn被预测为不存在相互作用,但事实上存在相互作用;tp被预测为存在相互作用,事实上也存在相互作用。
结果与讨论
1、调查交互数据
x射线和其他生物学研究表明,许多蛋白质含有一个以上的药物结合位点,这些药物的结合位点存在局部重叠。分析结合位点片段,以检查不同蛋白质中结合位点片段的重叠程度(如图6所示)。图6中横坐标的数值表示:1代表仅存在于一簇的结合位点片段的数目;2代表存在于两簇的结合位点片段的数目;……;15表示存在于十五簇的结合位点片段的数目。从图中可以看出,超过65%的结合位点片段位于多个dbps上,这与蛋白质的药物结合位点部分重叠的事实一致。
蛋白质的疏水性和电荷强度在药物-蛋白质结合过程中起着重要的作用。因此分析簇的疏水性和电荷强度(如图7所示)。从图中可以看出,药物倾向作用疏水簇和带正电的簇。推测药物-dbp结合过程发生在蛋白质内部并且dbps倾向结合带负电荷的药物分子。这为研究药物-dbps结合过程提供指导方向。
网络中度的分布情况反映药物-簇连接的稀疏性。因此分析网络以检查簇和药物的度分布(如图8-9所示)。从图8可以看出,超过87%的药物与15至30种之间的簇相互作用。图9显示超过66%的结合位点簇与少于20种药物相互作用。这表明药物-簇二分图的连接是稀疏的。
2、链路预测方法比较
首先,评估三种链路预测方法的性能,然后简要分析网络中三种方法的预测机制,最后为网络预测选择最佳的预测方法。
在不同的网络中三种方法展现不同的性能,因此在该网络中进行性能比较以选择最佳的方法。可以通过随机创建无效的相互作用来误导预测结果,得到的曲线作为基线。通过三种方法和基线的比较,cn方法展现出最佳预测性能(如图10所示)。
在网络中三种方法的预测机制不同导致性能的差异。cn方法只考虑网络中节点的邻居。ja方法不仅考虑节点的共同邻居,还考虑节点的其他邻居,但是ja方法在网络中的表现并不如cn方法。分析ja方法可能是在二阶路径之后节点链接的不相关节点增加导致ja方法预测性能变差。pa方法只考虑节点的度,不能有效的利用网络中的结构信息。通过比较,cn方法在网络中的预测机制较为可靠。
通过性能和预测机制的分析,采用cn方法来预测网络中的药物-簇相互作用。cn方法计算出的得分作为网络预测的依据,根据得分判断药物-簇的相互作用的可能性。
3、网络预测
首先,根据链路预测得分创建药物-簇相互作用的预测矩阵,然后对预测矩阵进行分析,最后对预测结果进行验证。
通过方法比较,cn方法被用来进行链路预测。根据cn方法计算出的分数构建药物-簇相互作用的预测矩阵(如图11所示),矩阵中的值表示预测分数。在药物-簇相互作用的预测矩阵中有7416个非零元素,只有那些值大于101的元素才被认为是显著作用(平均标准误差为101)。结果,网络中有3468个显著的相互作用。在显著的相互作用中,值大于274(前20%)的相互作用被视为重要的相互作用。
目前通过统计方法交叉检验、随机检验及已发表的化学实验结果进行验证,对预测结果排名靠前的结果进行化学分析。根据已有的化学知识判断预测结果,例如氢原子与电负性大的原子n以共价键结合时,当电负性大、半径小的原子f接近时,在n和f之间以氢为媒介,生成n-h…f形式氢键。
根据分数确定药物-簇相互作用,有必要研究预测结果中的药物-簇是否相互作用。簇由结合位点片段组成,因此通过验证药物和簇内的片段相互作用来验证药物-簇的相互作用。可视化预测结果(如图12所示)。图12表示预测的药物-簇相互作用;簇由结合位点片段组成,结合位点片段的第一个字母代表片段的中心氨基酸,括号中的字母代表从属氨基酸。
4、预测结果中药物-结合位点片段的结合机制
分析预测结果中药物-结合位点片段的结合机制。例如,mrc(莫匹罗星)中的羟基和天冬氨酸的羧基反应生成脂基和水。在某些情况下,主要氨基酸不能与药物发生显著的化学作用,而是通过氢键与药物发生相互作用。例如,在甘氨酸中氢原子与电负性大的原子n以共价键结合,当vgh(赛可瑞)中电负性大、半径小的原子f接近时,在n和f之间以氢为媒介,生成n-h…f形式氢键。可以类似地分析其它的相互作用关系(如图13所示)。
本发明中从片段的化学性质和药物的化学性质来确定相互作用关系,结合机制是根据现有的化学反应机制进行的预测,例如氢离子和氢氧根发生反应。
结论
在本发明中,药物-dbps相互作用被描述为药物和dbps结合位点片段的相互作用。为了分析药物和结合位点片段结合的理化性质,将相似的结合位点片段聚类成簇,形成药物-簇相互作用关系。由于只知道药物-簇相互作用关系并没有其他的特征信息,因此选择基于网络结构的链路预测方法来预测新的药物-簇相互作用关系。通过比较三种链路预测方法,cn方法被用来进行链路预测。
此外,还分析了预测结果中5个药物-结合位点片段的结合机制。
与传统的药物-蛋白质网络相比,所提出的网络预测模型能够发现候选结合位点片段。从药物-dbps复合物中提取药物-dbps结合位点,并应用结合位点片段来描述结合位点。通过这种方式,可以清楚地知道药物和结合位点片段的相互作用机制。
- 还没有人留言评论。精彩留言会获得点赞!